
NGLY1 Deficiency, a Congenital Disorder of Deglycosylation: From Disease Gene Function to Pathophysiology




Abstract

1. Introduction
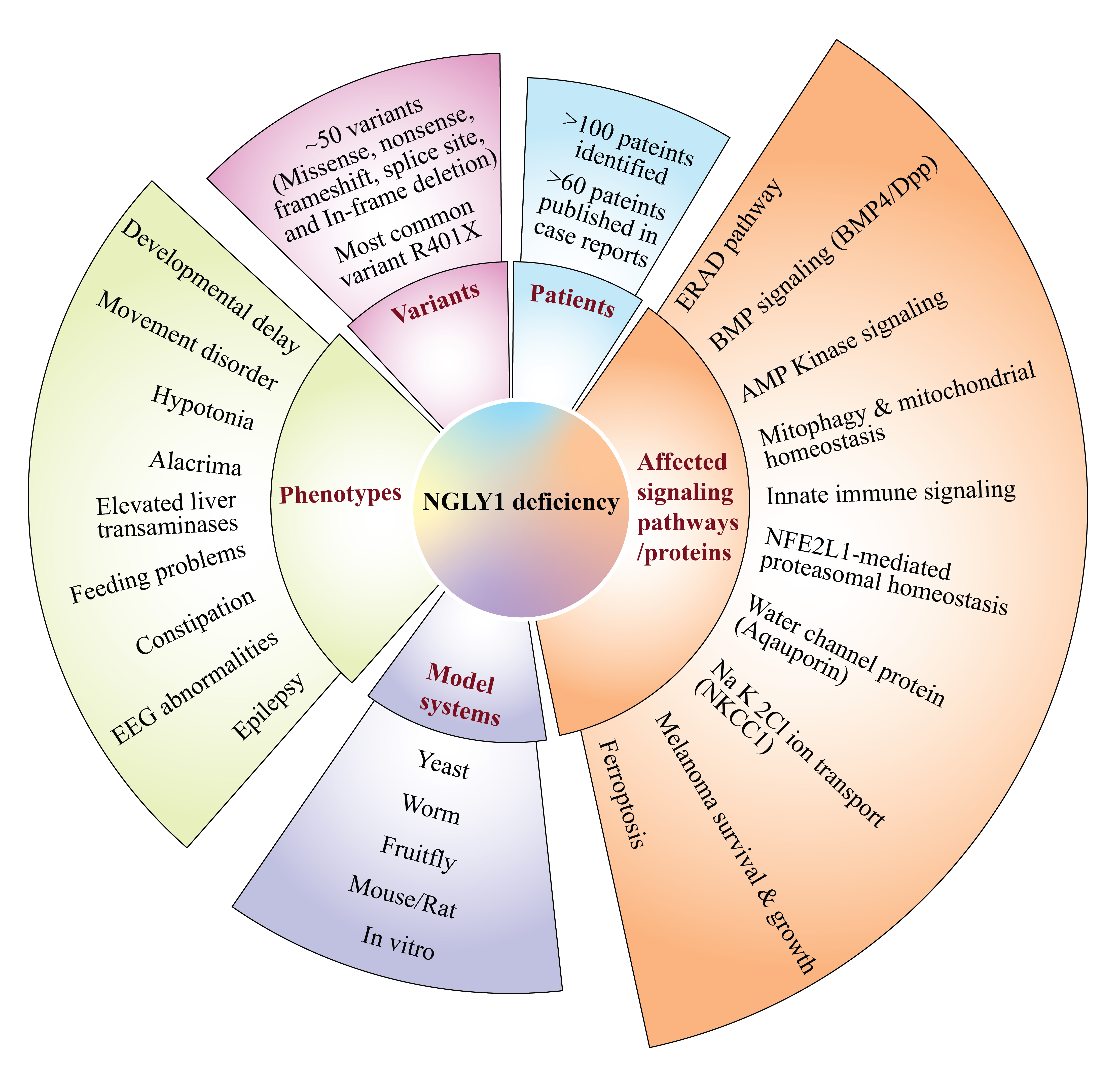
2. Patient Phenotypes in NGLY1 Deficiency
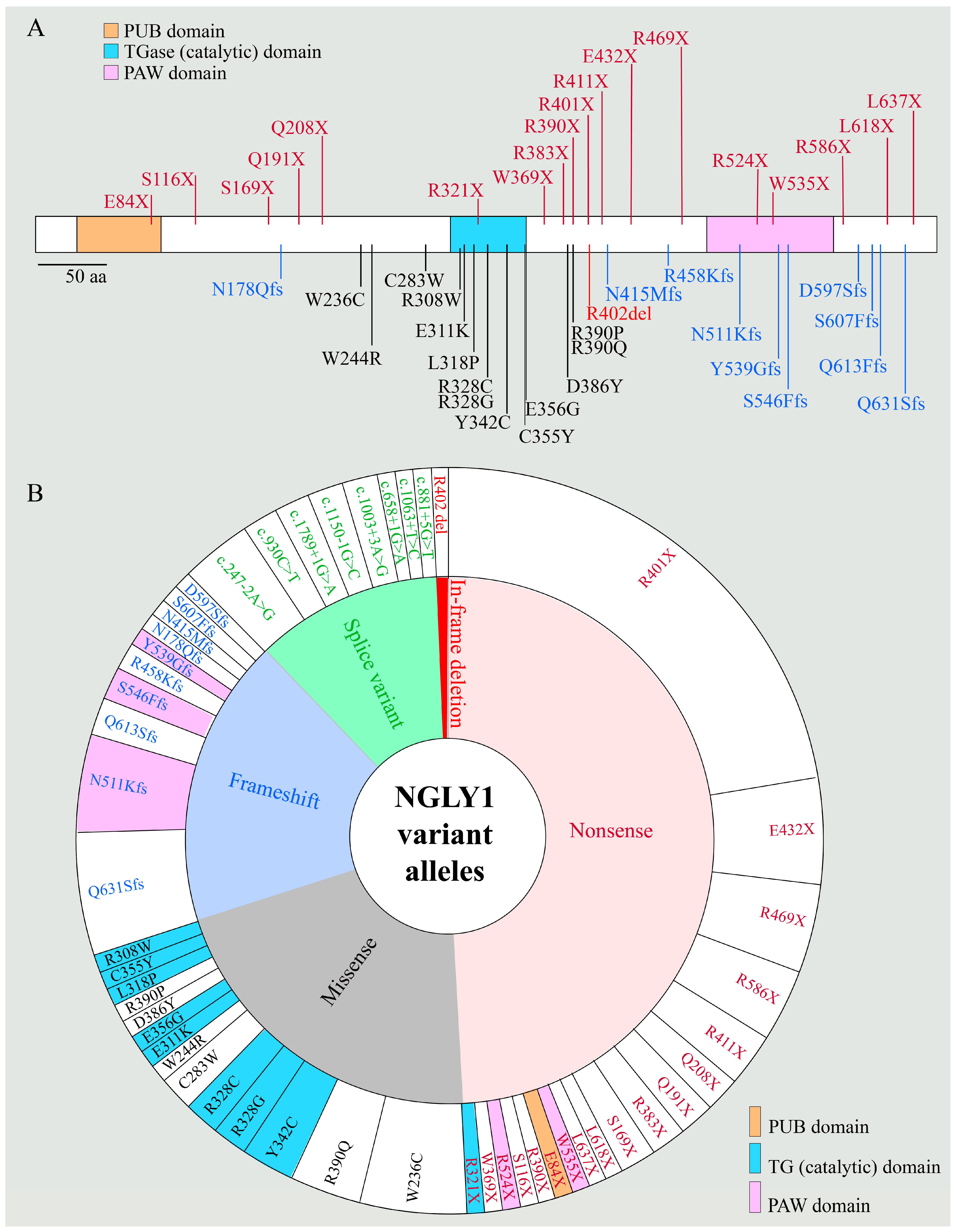
2.1. NGLY1 Deficiency Phenotypic Presentations Are Diverse
2.2. Biomarkers of NGLY1 Deficiency
3. Animal Models of NGLY1 Deficiency
3.1. Mammalian Models
3.2. Non-Mammalian Models
4. NGLY1 and the ERAD Pathway
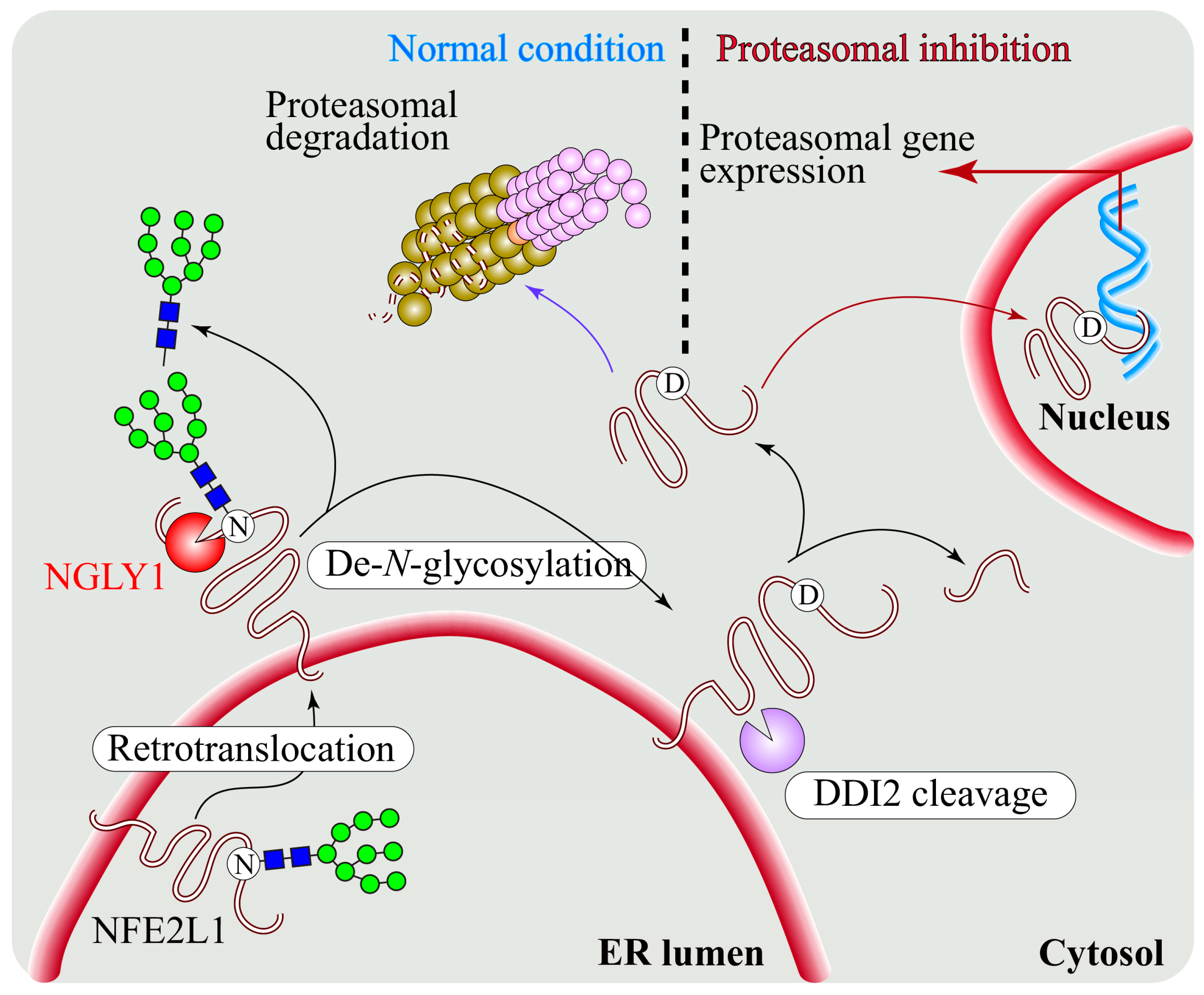
4.1. NGLY1 Is a Component of the ERAD Pathway
4.2. Engase Is A Modifier of NGLY1-Associated ERAD Dysfunction
4.3. The Ubiquitin Ligase Complex SCFFBS2 Mediates Proteasomal Dysfunction upon Loss of NGLY1
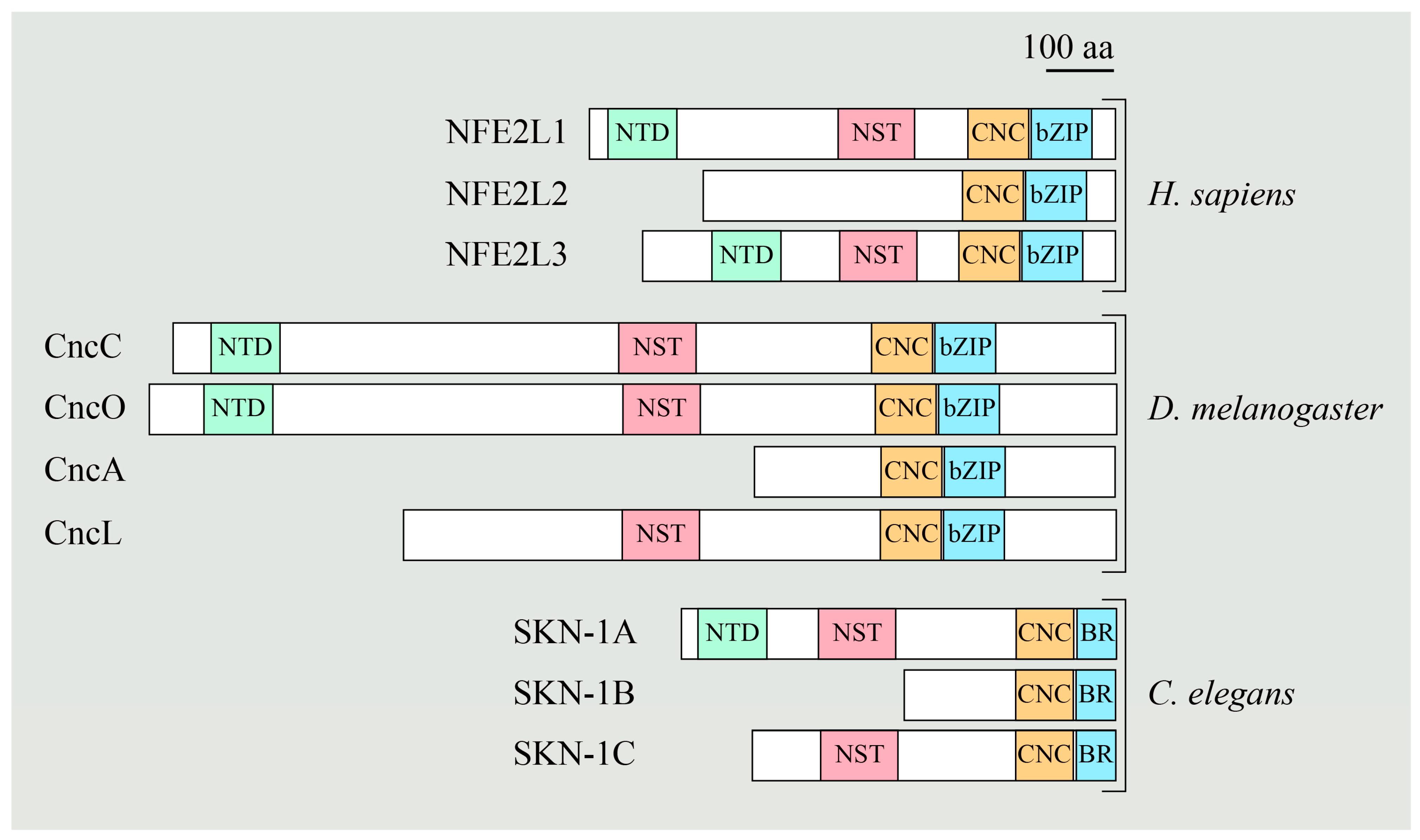
5. Regulation of NFE2L1 Activation by NGLY1
5.1. NGLY1 Regulates NFE2L1-Mediated Proteasome Bounce-Back Response
5.2. NFE2L1 Dysfunction and Pathogenesis of NGLY1 Deficiency
5.3. The NGLY1-NFE2L1 Axis as A Potential Therapeutic Target for Human Diseases
6. Regulation of Energy Homeostasis and Mitochondrial Structure and Function by NGLY1
6.1. Mitochondrial Abnormalities in Patients with NGLY1 Deficiency
6.2. Evidence for Abnormal Mitochondrial Structure and Function in Animal Models of NGLY1 Deficiency
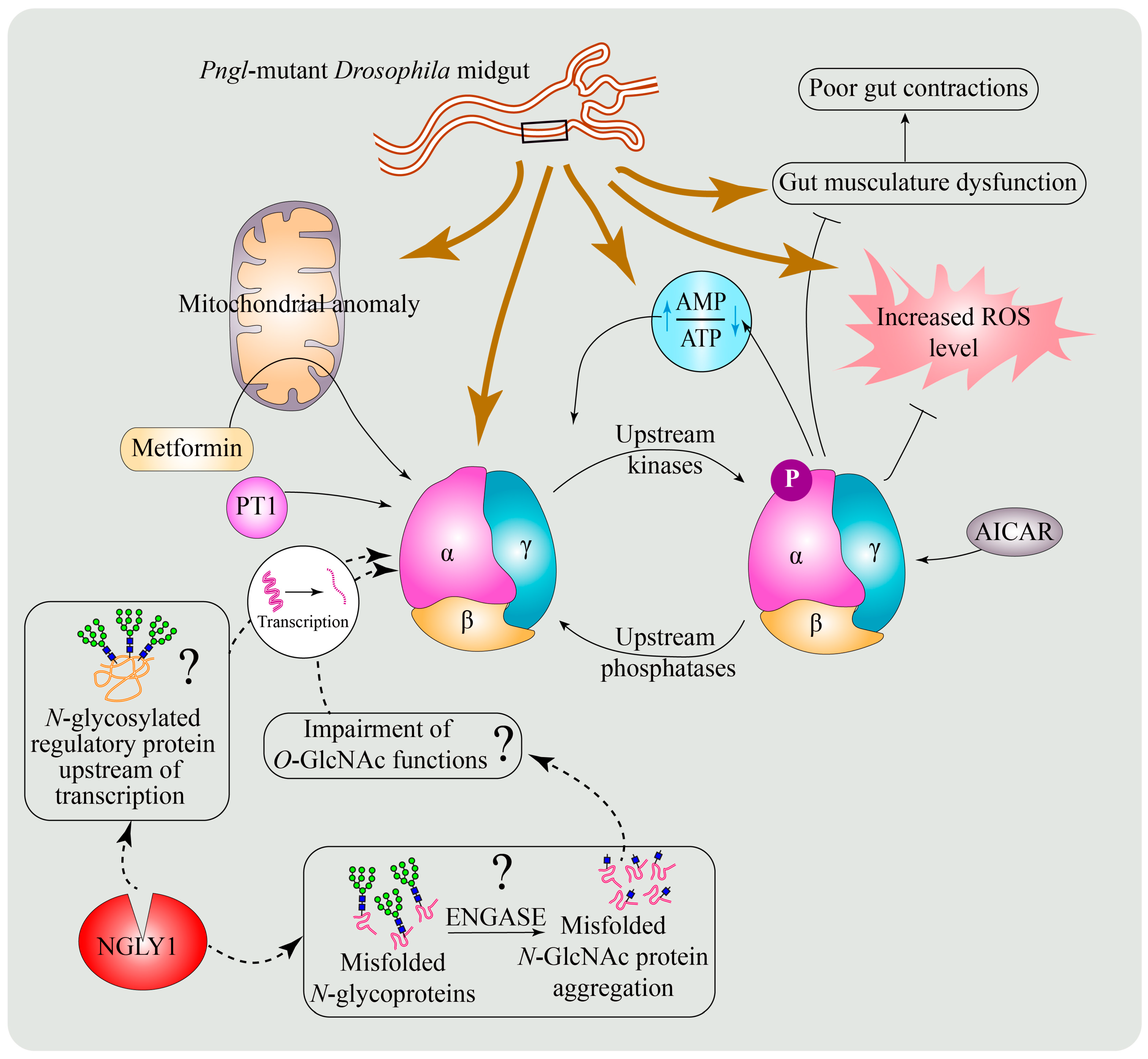
6.3. A Conserved Role for NGLY1 in the Regulation of AMP-Activated Protein Kinase (AMPK) Signaling
6.4. Potential Mechanisms for the Regulation of AMPKα by NGLY1
6.5. Enhancing AMPK Activation as a Potential Therapeutic Strategy for NGLY1 Deficiency
7. Regulation of BMP Signaling by NGLY1
7.1. Role of De-N-Glycosylation in the Regulation of BMP Signaling
7.2. Differential Regulatory Roles for NGLY1: BMP4 versus NFE2L1
7.3. Impaired BMP Signaling and Pathogenesis of NGLY1 Deficiency
8. Other Biological Processes and Pathways Regulated by NGLY1
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stanley, P.; Taniguchi, N.; Aebi, M. N-Glycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 99–111. [Google Scholar]
- Brodsky, J.L. Cleaning Up: ER-Associated Degradation to the Rescue. Cell 2012, 151, 1163–1167. [Google Scholar] [CrossRef]
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to Ruin: Targeting Proteins for Degradation in the Endoplasmic Reticulum. Science 2011, 334, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Park, H.; Hollingsworth, N.M.; Sternglanz, R.; Lennarz, W.J. PNG1, a yeast gene encoding a highly conserved peptide:N-glycanase. J. Cell Biol. 2000, 149, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T. The cytoplasmic peptide:N-glycanase (Ngly1)-basic science encounters a human genetic disorder. J. Biochem. 2015, 157, 23–34. [Google Scholar] [CrossRef]
- Katiyar, S.; Joshi, S.; Lennarz, W.J. The retrotranslocation protein Derlin-1 binds peptide:N-glycanase to the endoplasmic reticulum. Mol. Biol. Cell 2005, 16, 4584–4594. [Google Scholar] [CrossRef]
- Katiyar, S.; Li, G.; Lennarz, W.J. A complex between peptide:N-glycanase and two proteasome-linked proteins suggests a mechanism for the degradation of misfolded glycoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 13774–13779. [Google Scholar] [CrossRef]
- Li, G.; Zhao, G.; Zhou, X.; Schindelin, H.; Lennarz, W.J. The AAA ATPase p97 links peptide N-glycanase to the endoplasmic reticulum-associated E3 ligase autocrine motility factor receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 8348–8353. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Park, H.; Till, E.A.; Lennarz, W.J. The PUB domain: A putative protein-protein interaction domain implicated in the ubiquitin-proteasome pathway. Biochem. Biophys. Res. Commun. 2001, 287, 1083–1087. [Google Scholar] [CrossRef]
- Zhao, G.; Li, G.; Zhou, X.; Matsuo, I.; Ito, Y.; Suzuki, T.; Lennarz, W.J.; Schindelin, H. Structural and mutational studies on the importance of oligosaccharide binding for the activity of yeast PNGase. Glycobiology 2009, 19, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M.; Shashi, V.; Bainbridge, M.; Gambello, M.J.; Zahir, F.R.; Bast, T.; Crimian, R.; Schoch, K.; Platt, J.; Cox, R.; et al. Mutations in NGLY1 cause an inherited disorder of the endoplasmic reticulum-associated degradation pathway. Genet. Med. 2014, 16, 751–758. [Google Scholar] [CrossRef]
- Lam, C.; Ferreira, C.; Krasnewich, D.; Toro, C.; Latham, L.; Zein, W.M.; Lehky, T.; Brewer, C.; Baker, E.H.; Thurm, A.; et al. Prospective phenotyping of NGLY1-CDDG, the first congenital disorder of deglycosylation. Genet. Med. 2017, 19, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.J.; Frater, C.H.; Gallentine, W.B.; Phillips, J.M.; Ruzhnikov, M.R. Delineating the Epilepsy Phenotype of NGLY1 Deficiency. J. Inherit. Metab. Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
- Need, A.C.; Shashi, V.; Hitomi, Y.; Schoch, K.; Shianna, K.V.; McDonald, M.T.; Meisler, M.H.; Goldstein, D.B. Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 2012, 49, 353–361. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Grotzke, J.E.; Ng, B.G.; Gunel, M.; Jafar-Nejad, H.; Cresswell, P.; Enns, G.M.; Freeze, H.H. A congenital disorder of deglycosylation: Biochemical characterization of N-glycanase 1 deficiency in patient fibroblasts. Glycobiology 2015, 25, 836–844. [Google Scholar] [CrossRef]
- Might, M.; Wilsey, M. The shifting model in clinical diagnostics: How next-generation sequencing and families are altering the way rare diseases are discovered, studied, and treated. Genet. Med. 2014, 16, 736–737. [Google Scholar] [CrossRef] [PubMed]
- Mueller, W.F.; Zhu, L.; Tan, B.; Dwight, S.; Beahm, B.; Wilsey, M.; Wechsler, T.; Mak, J.; Cowan, T.; Pritchett, J.; et al. GlcNAc-Asn (GNA) is a biomarker for NGLY1 deficiency. J. Biochem. 2022, 171, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Kalfon, L.; Baydany, M.; Samra, N.; Heno, N.; Segal, Z.; Eran, A.; Yulevich, A.; Fellig, Y.; Mandel, H.; Falik-Zaccai, T.C. Congenital Hypotonia: Cracking a SAGA of consanguineous kindred harboring four genetic variants. Mol. Genet. Genomic Med. 2021, 10, e1849. [Google Scholar] [CrossRef] [PubMed]
- Lipiński, P.; Bogdanska, A.; Rozdzynska-Swiatkowska, A.; Wierzbicka-Rucinska, A.; Tylki-Szymanska, A. NGLY1 deficiency: Novel patient, review of the literature and diagnostic algorithm. JIMD Rep. 2020, 51, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Abuduxikuer, K.; Zou, L.; Wang, L.; Chen, L.; Wang, J.S. Novel NGLY1 gene variants in Chinese children with global developmental delay, microcephaly, hypotonia, hypertransaminasemia, alacrimia, and feeding difficulty. J. Hum. Genet. 2020, 65, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Kariminejad, A.; Shakiba, M.; Shams, M.; Namiranian, P.; Eghbali, M.; Talebi, S.; Makvand, M.; Jaeken, J.; Najmabadi, H.; Hennekam, R.C. NGLY1 deficiency: Novel variants and literature review. Eur J. Med. Genet. 2021, 64, 104146. [Google Scholar] [CrossRef]
- Stuut, T.; Popescu, O.; Oviedo, A. N-Glycanase 1 Deficiency Is a Rare Cause of Pediatric Neurodegeneration With Neuronal Inclusions and Liver Steatosis. Cureus 2021, 13, e19126. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Peng, M.; Ostrovsky, J.; Kwon, Y.J.; Oretsky, O.; McCormick, E.M.; He, M.; Argon, Y.; Falk, M.J. Mitochondrial function requires NGLY1. Mitochondrion 2018, 38, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Caglayan, A.O.; Comu, S.; Baranoski, J.F.; Parman, Y.; Kaymakcalan, H.; Akgumus, G.T.; Caglar, C.; Dolen, D.; Erson-Omay, E.Z.; Harmanci, A.S.; et al. NGLY1 mutation causes neuromotor impairment, intellectual disability, and neuropathy. Eur J. Med. Genet. 2015, 58, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Heeley, J.; Shinawi, M. Multi-systemic involvement in NGLY1-related disorder caused by two novel mutations. Am. J. Med. Genet. A 2015, 167A, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Bosch, D.G.; Boonstra, F.N.; de Leeuw, N.; Pfundt, R.; Nillesen, W.M.; de Ligt, J.; Gilissen, C.; Jhangiani, S.; Lupski, J.R.; Cremers, F.P.; et al. Novel genetic causes for cerebral visual impairment. Eur J. Hum. Genet. 2016, 24, 660–665. [Google Scholar] [CrossRef]
- van Keulen, B.J.; Rotteveel, J.; Finken, M.J.J. Unexplained death in patients with NGLY1 mutations may be explained by adrenal insufficiency. Physiol. Rep. 2019, 7, e13979. [Google Scholar] [CrossRef]
- Panneman, D.M.; Wortmann, S.B.; Haaxma, C.A.; van Hasselt, P.M.; Wolf, N.I.; Hendriks, Y.; Kusters, B.; van Emst-de Vries, S.; van de Westerlo, E.; Koopman, W.J.H.; et al. Variants in NGLY1 lead to intellectual disability, myoclonus epilepsy, sensorimotor axonal polyneuropathy and mitochondrial dysfunction. Clin. Genet. 2020, 97, 556–566. [Google Scholar] [CrossRef]
- Chang, C.A.; Wei, X.C.; Martin, S.R.; Sinasac, D.S.; Al-Hertani, W. Transiently elevated plasma methionine, S-adenosylmethionine and S-adenosylhomocysteine: Unreported laboratory findings in a patient with NGLY1 deficiency, a congenital disorder of deglycosylation. JIMD Rep. 2019, 49, 21–29. [Google Scholar] [CrossRef]
- Haijes, H.A.; de Sain-van der Velden, M.G.M.; Prinsen, H.; Willems, A.P.; van der Ham, M.; Gerrits, J.; Couse, M.H.; Friedman, J.M.; van Karnebeek, C.D.M.; Selby, K.A.; et al. Aspartylglycosamine is a biomarker for NGLY1-CDDG, a congenital disorder of deglycosylation. Mol. Genet. Metab. 2019, 127, 368–372. [Google Scholar] [CrossRef]
- Lipiński, P.; Cielecka-Kuszyk, J.; Socha, P.; Tylki-Szymanska, A. Liver involvement in NGLY1 congenital disorder of deglycosylation. Pol. J. Pathol. 2020, 71, 66–68. [Google Scholar] [CrossRef]
- Ge, H.; Wu, Q.; Lu, H.; Huang, Y.; Zhou, T.; Tan, D.; Jin, Z. Two novel compound heterozygous mutations in NGLY1as a cause of congenital disorder of deglycosylation: A case presentation. BMC Med. Genet. 2020, 21, 135. [Google Scholar] [CrossRef] [PubMed]
- Rios-Flores, I.M.; Bonal-Perez, M.A.; Castellanos-Gonzalez, A.; Velez-Gomez, E.; Bertoli-Avella, A.M.; Bobadilla-Morales, L.; Pena-Padilla, C.; Appendini-Andrade, V.; Corona-Rivera, A.; Romero-Valenzuela, I.; et al. Acute liver failure in a male patient with NGLY1-congenital disorder of deglycosylation. Eur. J. Med. Genet. 2020, 63, 103952. [Google Scholar] [CrossRef] [PubMed]
- Lipari Pinto, P.; Machado, C.; Janeiro, P.; Dupont, J.; Quintas, S.; Sousa, A.B.; Gaspar, A. NGLY1 deficiency—A rare congenital disorder of deglycosylation. JIMD Rep. 2020, 53, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Dabaj, I.; Sudrie-Arnaud, B.; Lecoquierre, F.; Raymond, K.; Ducatez, F.; Guerrot, A.M.; Snanoudj, S.; Coutant, S.; Saugier-Veber, P.; Marret, S.; et al. NGLY1 Deficiency: A Rare Newly Described Condition with a Typical Presentation. Life 2021, 11, 187. [Google Scholar] [CrossRef] [PubMed]
- Marques-da-Silva, D.; Francisco, R.; Webster, D.; Dos Reis Ferreira, V.; Jaeken, J.; Pulinilkunnil, T. Cardiac complications of congenital disorders of glycosylation (CDG): A systematic review of the literature. J. Inherit. Metab. Dis. 2017, 40, 657–672. [Google Scholar] [CrossRef]
- Cahan, E.M.; Frick, S.L. Orthopaedic phenotyping of NGLY1 deficiency using an international, family-led disease registry. Orphanet J. Rare Dis. 2019, 14, 148. [Google Scholar] [CrossRef]
- Habibi-Babadi, N.; Su, A.; de Carvalho, C.E.; Colavita, A. The N-glycanase png-1 acts to limit axon branching during organ formation in Caenorhabditis elegans. J. Neurosci. 2010, 30, 1766–1776. [Google Scholar] [CrossRef]
- Fujihira, H.; Masahara-Negishi, Y.; Tamura, M.; Huang, C.; Harada, Y.; Wakana, S.; Takakura, D.; Kawasaki, N.; Taniguchi, N.; Kondoh, G.; et al. Lethality of mice bearing a knockout of the Ngly1-gene is partially rescued by the additional deletion of the Engase gene. PLoS Genet. 2017, 13, e1006696. [Google Scholar] [CrossRef]
- Galeone, A.; Adams, J.M.; Matsuda, S.; Presa, M.F.; Pandey, A.; Han, S.Y.; Tachida, Y.; Hirayama, H.; Vaccari, T.; Suzuki, T.; et al. Regulation of BMP4/Dpp retrotranslocation and signaling by deglycosylation. Elife 2020, 9, e55596. [Google Scholar] [CrossRef]
- Hall, P.L.; Lam, C.; Alexander, J.J.; Asif, G.; Berry, G.T.; Ferreira, C.; Freeze, H.H.; Gahl, W.A.; Nickander, K.K.; Sharer, J.D.; et al. Urine oligosaccharide screening by MALDI-TOF for the identification of NGLY1 deficiency. Mol. Genet. Metab. 2018, 124, 82–86. [Google Scholar] [CrossRef]
- Asahina, M.; Fujinawa, R.; Fujihira, H.; Masahara-Negishi, Y.; Andou, T.; Tozawa, R.; Suzuki, T. JF1/B6F1 Ngly1(-/-) mouse as an isogenic animal model of NGLY1 deficiency. Proc. Jpn Acad. Ser. B Phys. Biol. Sci. 2021, 97, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Harada, Y.; Hosomi, A.; Masahara-Negishi, Y.; Seino, J.; Fujihira, H.; Funakoshi, Y.; Suzuki, T.; Dohmae, N.; Suzuki, T. Endo-beta-N-acetylglucosaminidase forms N-GlcNAc protein aggregates during ER-associated degradation in Ngly1-defective cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Huang, R.; Fujihira, H.; Suzuki, T.; Yan, N. N-glycanase NGLY1 regulates mitochondrial homeostasis and inflammation through NRF1. J. Exp. Med. 2018, 215, 2600–2616. [Google Scholar] [CrossRef] [PubMed]
- Fujihira, H.; Masahara-Negishi, Y.; Akimoto, Y.; Hirayama, H.; Lee, H.C.; Story, B.A.; Mueller, W.F.; Jakob, P.; Clauder-Munster, S.; Steinmetz, L.M.; et al. Liver-specific deletion of Ngly1 causes abnormal nuclear morphology and lipid metabolism under food stress. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165588. [Google Scholar] [CrossRef]
- Asahina, M.; Fujinawa, R.; Nakamura, S.; Yokoyama, K.; Tozawa, R.; Suzuki, T. Ngly1−/− rats develop neurodegenerative phenotypes and pathological abnormalities in their peripheral and central nervous systems. Hum. Mol. Genet. 2020, 29, 1635–1647. [Google Scholar] [CrossRef]
- Asahina, M.; Fujinawa, R.; Hirayama, H.; Tozawa, R.; Kajii, Y.; Suzuki, T. Reversibility of motor dysfunction in the rat model of NGLY1 deficiency. Mol. Brain 2021, 14, 91. [Google Scholar] [CrossRef]
- Wang, J.; Lozier, J.; Johnson, G.; Kirshner, S.; Verthelyi, D.; Pariser, A.; Shores, E.; Rosenberg, A. Neutralizing antibodies to therapeutic enzymes: Considerations for testing, prevention and treatment. Nat. Biotechnol 2008, 26, 901–908. [Google Scholar] [CrossRef]
- Rauscher, B.; Mueller, W.F.; Clauder-Münster, S.; Jakob, P.; Islam, M.S.; Sun, H.; Ghidelli-Disse, S.; Boesche, M.; Bantscheff, M.; Pflaumer, H.; et al. Patient-derived gene and protein expression signatures of NGLY1 deficiency. J. Biochem. 2022, 171, 187–199. [Google Scholar] [CrossRef]
- Lehrbach, N.J. NGLY1: Insights from C. elegans. J. Biochem. 2022, 171, 145–154. [Google Scholar] [CrossRef]
- Pandey, A.; Jafar-Nejad, H. Tracing the NGLY1 footprints: Insights from Drosophila. J. Biochem. 2022, 171, 153–160. [Google Scholar] [CrossRef]
- Seiler, S.; Plamann, M. The genetic basis of cellular morphogenesis in the filamentous fungus Neurospora crassa. Mol. Biol. Cell 2003, 14, 4352–4364. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Aravind, L.; Koonin, E.V. A superfamily of archaeal, bacterial, and eukaryotic proteins homologous to animal transglutaminases. Protein Sci. 1999, 8, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, Y.; Negishi, Y.; Gergen, J.P.; Seino, J.; Ishii, K.; Lennarz, W.J.; Matsuo, I.; Ito, Y.; Taniguchi, N.; Suzuki, T. Evidence for an essential deglycosylation-independent activity of PNGase in Drosophila melanogaster. PLoS ONE 2010, 5, e10545. [Google Scholar] [CrossRef] [PubMed]
- Galeone, A.; Han, S.Y.; Huang, C.; Hosomi, A.; Suzuki, T.; Jafar-Nejad, H. Tissue-specific regulation of BMP signaling by Drosophila N-glycanase 1. eLife 2017, 6, e27612. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, T.P.; Mast, J.D.; Hartl, T.; Lee, T.; Sand, P.; Perlstein, E.O. Defects in the Neuroendocrine Axis Contribute to Global Development Delay in a Drosophila Model of NGLY1 Deficiency. G3 (Bethesda) 2018, 8, 2193–2204. [Google Scholar] [CrossRef]
- Owings, K.G.; Lowry, J.B.; Bi, Y.; Might, M.; Chow, C.Y. Transcriptome and functional analysis in a Drosophila model of NGLY1 deficiency provides insight into therapeutic approaches. Hum. Mol. Genet. 2018, 27, 1055–1066. [Google Scholar] [CrossRef]
- Kato, T.; Kawahara, A.; Ashida, H.; Yamamoto, K. Unique peptide:N-glycanase of Caenorhabditis elegans has activity of protein disulphide reductase as well as of deglycosylation. J. Biochem. 2007, 142, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Lehrbach, N.J.; Ruvkun, G. Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. eLife 2016, 5, e17721. [Google Scholar] [CrossRef]
- Iyer, S.; Mast, J.D.; Tsang, H.; Rodriguez, T.P.; DiPrimio, N.; Prangley, M.; Sam, F.S.; Parton, Z.; Perlstein, E.O. Drug screens of NGLY1 deficiency in worm and fly models reveal catecholamine, NRF2 and anti-inflammatory-pathway activation as potential clinical approaches. Dis. Model. Mech. 2019, 12, dmm040576. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Retzlaff, M.; Roos, T.; Frydman, J. Cellular strategies of protein quality control. Cold Spring Harb. Perspect. Biol. 2011, 3, a004374. [Google Scholar] [CrossRef] [PubMed]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, C.J.; Brodsky, J.L. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 2012, 92, 537–576. [Google Scholar] [CrossRef]
- Helenius, A.; Aebi, M. Intracellular functions of N-linked glycans. Science 2001, 291, 2364–2369. [Google Scholar] [CrossRef] [PubMed]
- Blom, D.; Hirsch, C.; Stern, P.; Tortorella, D.; Ploegh, H.L. A glycosylated type I membrane protein becomes cytosolic when peptide: N-glycanase is compromised. EMBO J. 2004, 23, 650–658. [Google Scholar] [CrossRef]
- Hirsch, C.; Blom, D.; Ploegh, H.L. A role for N-glycanase in the cytosolic turnover of glycoproteins. EMBO J. 2003, 22, 1036–1046. [Google Scholar] [CrossRef]
- Hosomi, A.; Suzuki, T. Cytoplasmic peptide:N-glycanase cleaves N-glycans on a carboxypeptidase Y mutant during ERAD in Saccharomyces cerevisiae. Biochim. Biophys. Acta 2015, 1850, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Ahn, J.; Liu, C.; Tanabe, K.; Apodaca, J.; Suzuki, T.; Rao, H. The Png1-Rad23 complex regulates glycoprotein turnover. J. Cell Biol. 2006, 172, 211–219. [Google Scholar] [CrossRef]
- Masahara-Negishi, Y.; Hosomi, A.; Della Mea, M.; Serafini-Fracassini, D.; Suzuki, T. A plant peptide: N-glycanase orthologue facilitates glycoprotein ER-associated degradation in yeast. Biochim. Biophys. Acta 2012, 1820, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Grotzke, J.E.; Lu, Q.; Cresswell, P. Deglycosylation-dependent fluorescent proteins provide unique tools for the study of ER-associated degradation. Proc. Natl. Acad. Sci. USA 2013, 110, 3393–3398. [Google Scholar] [CrossRef] [PubMed]
- Misaghi, S.; Pacold, M.E.; Blom, D.; Ploegh, H.L.; Korbel, G.A. Using a small molecule inhibitor of peptide: N-glycanase to probe its role in glycoprotein turnover. Chem. Biol. 2004, 11, 1677–1687. [Google Scholar] [CrossRef]
- Kario, E.; Tirosh, B.; Ploegh, H.L.; Navon, A. N-linked glycosylation does not impair proteasomal degradation but affects class I major histocompatibility complex presentation. J. Biol. Chem. 2008, 283, 244–254. [Google Scholar] [CrossRef]
- Hosomi, A.; Fujita, M.; Tomioka, A.; Kaji, H.; Suzuki, T. Identification of PNGase-dependent ERAD substrates in Saccharomyces cerevisiae. Biochem. J. 2016, 473, 3001–3012. [Google Scholar] [CrossRef]
- Needs, S.H.; Bootman, M.D.; Grotzke, J.E.; Kramer, H.B.; Allman, S.A. Off-target inhibition of NGLY1 by the polycaspase inhibitor Z-VAD-fmk induces cellular autophagy. FEBS J. 2022. [Google Scholar] [CrossRef]
- Park, S.; Jang, I.; Zuber, C.; Lee, Y.; Cho, J.W.; Matsuo, I.; Ito, Y.; Roth, J. ERADication of EDEM1 occurs by selective autophagy and requires deglycosylation by cytoplasmic peptide N-glycanase. Histochem. Cell Biol. 2014, 142, 153–169. [Google Scholar] [CrossRef]
- Suzuki, T.; Funakoshi, Y. Free N-linked oligosaccharide chains: Formation and degradation. Glycoconj. J. 2006, 23, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Might, M.; Vankayalapati, H.; Kuberan, B. Repurposing of Proton Pump Inhibitors as first identified small molecule inhibitors of endo-beta-N-acetylglucosaminidase (ENGase) for the treatment of NGLY1 deficiency, a rare genetic disease. Bioorg. Med. Chem. Lett. 2017, 27, 2962–2966. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Asahina, M.; Murakami, A.; Kawawaki, J.; Yoshida, M.; Fujinawa, R.; Iwai, K.; Tozawa, R.; Matsuda, N.; Tanaka, K.; et al. Loss of peptide:N-glycanase causes proteasome dysfunction mediated by a sugar-recognizing ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2021, 118, e2102902118. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Adachi, E.; Fukiya, K.; Iwai, K.; Tanaka, K. Glycoprotein-specific ubiquitin ligases recognize N-glycans in unfolded substrates. EMBO Rep. 2005, 6, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Chiba, T.; Tokunaga, F.; Kawasaki, H.; Iwai, K.; Suzuki, T.; Ito, Y.; Matsuoka, K.; Yoshida, M.; Tanaka, K.; et al. E3 ubiquitin ligase that recognizes sugar chains. Nature 2002, 418, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, B. Role of proteasomes in disease. BMC Biochem. 2007, 8 (Suppl. 1), S3. [Google Scholar] [CrossRef]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 2014, 1843, 13–25. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef]
- Suraweera, A.; Munch, C.; Hanssum, A.; Bertolotti, A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol. Cell 2012, 48, 242–253. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef]
- Steffen, J.; Seeger, M.; Koch, A.; Kruger, E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef]
- Grimberg, K.B.; Beskow, A.; Lundin, D.; Davis, M.M.; Young, P. Basic leucine zipper protein Cnc-C is a substrate and transcriptional regulator of the Drosophila 26S proteasome. Mol. Cell Biol. 2011, 31, 897–909. [Google Scholar] [CrossRef]
- Li, X.; Matilainen, O.; Jin, C.; Glover-Cutter, K.M.; Holmberg, C.I.; Blackwell, T.K. Specific SKN-1/Nrf stress responses to perturbations in translation elongation and proteasome activity. PLoS Genet. 2011, 7, e1002119. [Google Scholar] [CrossRef]
- Zhang, Y.; Lucocq, J.M.; Hayes, J.D. The Nrf1 CNC/bZIP protein is a nuclear envelope-bound transcription factor that is activated by t-butyl hydroquinone but not by endoplasmic reticulum stressors. Biochem. J. 2009, 418, 293–310. [Google Scholar] [CrossRef]
- Suzuki, T.; Seko, A.; Kitajima, K.; Inoue, Y.; Inoue, S. Identification of peptide:N-glycanase activity in mammalian-derived cultured cells. Biochem. Biophys. Res. Commun. 1993, 194, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Irie, T.; Hirayama, S.; Sakurai, Y.; Yashiroda, H.; Naguro, I.; Ichijo, H.; Hamazaki, J.; Murata, S. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. Elife 2016, 5, e18357. [Google Scholar] [CrossRef] [PubMed]
- Tomlin, F.M.; Gerling-Driessen, U.I.M.; Liu, Y.C.; Flynn, R.A.; Vangala, J.R.; Lentz, C.S.; Clauder-Muenster, S.; Jakob, P.; Mueller, W.F.; Ordonez-Rueda, D.; et al. Inhibition of NGLY1 Inactivates the Transcription Factor Nrf1 and Potentiates Proteasome Inhibitor Cytotoxicity. ACS Cent. Sci. 2017, 3, 1143–1155. [Google Scholar] [CrossRef]
- Sha, Z.; Goldberg, A.L. Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr. Biol. 2014, 24, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Lehrbach, N.J.; Breen, P.C.; Ruvkun, G. Protein Sequence Editing of SKN-1A/Nrf1 by Peptide:N-Glycanase Controls Proteasome Gene Expression. Cell 2019, 177, 737–750.e15. [Google Scholar] [CrossRef]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef]
- Misra, J.R.; Horner, M.A.; Lam, G.; Thummel, C.S. Transcriptional regulation of xenobiotic detoxification in Drosophila. Genes Dev. 2011, 25, 1796–1806. [Google Scholar] [CrossRef]
- Han, S.Y.; Pandey, A.; Moore, T.; Galeone, A.; Duraine, L.; Cowan, T.M.; Jafar-Nejad, H. A conserved role for AMP-activated protein kinase in NGLY1 deficiency. PLoS Genet. 2020, 16, e1009258. [Google Scholar] [CrossRef]
- Forcina, G.C.; Pope, L.; Murray, M.; Dong, W.; Abu-Remaileh, M.; Bertozzi, C.R.; Dixon, S.J. Ferroptosis regulation by the NGLY1/NFE2L1 pathway. Proc. Natl. Acad. Sci. USA 2022, 119, e2118646119. [Google Scholar] [CrossRef]
- Chan, J.Y.; Kwong, M.; Lu, R.; Chang, J.; Wang, B.; Yen, T.S.; Kan, Y.W. Targeted disruption of the ubiquitous CNC-bZIP transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO J. 1998, 17, 1779–1787. [Google Scholar] [CrossRef]
- Kobayashi, A.; Tsukide, T.; Miyasaka, T.; Morita, T.; Mizoroki, T.; Saito, Y.; Ihara, Y.; Takashima, A.; Noguchi, N.; Fukamizu, A.; et al. Central nervous system-specific deletion of transcription factor Nrf1 causes progressive motor neuronal dysfunction. Genes Cells 2011, 16, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Lee, C.; Hu, T.; Nguyen, J.M.; Zhang, J.; Martin, M.V.; Vawter, M.P.; Huang, E.J.; Chan, J.Y. Loss of nuclear factor E2-related factor 1 in the brain leads to dysregulation of proteasome gene expression and neurodegeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 8408–8413. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chen, L.; Leung, L.; Yen, T.S.; Lee, C.; Chan, J.Y. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc. Natl. Acad. Sci. USA 2005, 102, 4120–4125. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Bauer, I.; Schafmayer, C.; Tepel, J.; Muerkoster, S.S.; Brosch, M.; Roder, C.; Kalthoff, H.; Hampe, J.; Moyer, M.P.; et al. Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2). Oncogene 2009, 28, 3983–3996. [Google Scholar] [CrossRef]
- Pajares, M.; Jimenez-Moreno, N.; Garcia-Yague, A.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rabano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol. Cell Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef]
- Deshaies, R.J. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014, 12, 94. [Google Scholar] [CrossRef]
- Zolekar, A.; Lin, V.J.T.; Mishra, N.M.; Ho, Y.Y.; Hayatshahi, H.S.; Parab, A.; Sampat, R.; Liao, X.; Hoffmann, P.; Liu, J.; et al. Stress and interferon signalling-mediated apoptosis contributes to pleiotropic anticancer responses induced by targeting NGLY1. Br. J. Cancer 2018, 119, 1538–1551. [Google Scholar] [CrossRef]
- Zhang, Z.; Falk, M.J. Integrated transcriptome analysis across mitochondrial disease etiologies and tissues improves understanding of common cellular adaptations to respiratory chain dysfunction. Int. J. Biochem. Cell Biol. 2014, 50, 106–111. [Google Scholar] [CrossRef][Green Version]
- Alston, C.L.; Rocha, M.C.; Lax, N.Z.; Turnbull, D.M.; Taylor, R.W. The genetics and pathology of mitochondrial disease. J. Pathol. 2017, 241, 236–250. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Widenmaier, S.B.; Schlein, C.; Johann, K.; Goncalves, R.L.S.; Eguchi, K.; Fischer, A.W.; Parlakgul, G.; Snyder, N.A.; Nguyen, T.B.; et al. Brown adipose tissue thermogenic adaptation requires Nrf1-mediated proteasomal activity. Nat. Med. 2018, 24, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, D.; Mitchelhill, K.I.; Gao, G.; Widmer, J.; Michell, B.J.; Teh, T.; House, C.M.; Fernandez, C.S.; Cox, T.; Witters, L.A.; et al. Mammalian AMP-activated protein kinase subfamily. J. Biol. Chem. 1996, 271, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Loson, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, B.; Li, T.; Zhu, Y.; Luo, G.; Jiang, Y.; Tang, F.; Jian, Z.; Xiao, Y. AMPK activation serves a critical role in mitochondria quality control via modulating mitophagy in the heart under chronic hypoxia. Int. J. Mol. Med. 2018, 41, 69–76. [Google Scholar] [CrossRef]
- Bland, M.L.; Lee, R.J.; Magallanes, J.M.; Foskett, J.K.; Birnbaum, M.J. AMPK supports growth in Drosophila by regulating muscle activity and nutrient uptake in the gut. Dev. Biol. 2010, 344, 293–303. [Google Scholar] [CrossRef]
- Hart, G.W. Nutrient regulation of signaling and transcription. J. Biol. Chem. 2019, 294, 2211–2231. [Google Scholar] [CrossRef]
- Cokorinos, E.C.; Delmore, J.; Reyes, A.R.; Albuquerque, B.; Kjobsted, R.; Jorgensen, N.O.; Tran, J.L.; Jatkar, A.; Cialdea, K.; Esquejo, R.M.; et al. Activation of Skeletal Muscle AMPK Promotes Glucose Disposal and Glucose Lowering in Non-human Primates and Mice. Cell Metab. 2017, 25, 1147–1159.e10. [Google Scholar] [CrossRef]
- Hardie, D.G. Regulation of AMP-activated protein kinase by natural and synthetic activators. Acta Pharm. Sin. B 2016, 6, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.; Yanes, R.E.; Calton, M.A.; Vollrath, D.; Enns, G.M.; Cowan, T.M. AMP-independent activator of AMPK for treatment of mitochondrial disorders. PLoS ONE 2020, 15, e0240517. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.W.; Guan, H.P.; Ehrhart, J.; Petrov, A.; Prahalada, S.; Tozzo, E.; Yang, X.; Kurtz, M.M.; Trujillo, M.; Gonzalez Trotter, D.; et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 2017, 357, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Pang, T.; Zhang, Z.S.; Gu, M.; Qiu, B.Y.; Yu, L.F.; Cao, P.R.; Shao, W.; Su, M.B.; Li, J.Y.; Nan, F.J.; et al. Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. J. Biol. Chem. 2008, 283, 16051–16060. [Google Scholar] [CrossRef]
- Steneberg, P.; Lindahl, E.; Dahl, U.; Lidh, E.; Straseviciene, J.; Backlund, F.; Kjellkvist, E.; Berggren, E.; Lundberg, I.; Bergqvist, I.; et al. PAN-AMPK activator O304 improves glucose homeostasis and microvascular perfusion in mice and type 2 diabetes patients. JCI Insight 2018, 3, e99114. [Google Scholar] [CrossRef]
- Xiao, B.; Sanders, M.J.; Carmena, D.; Bright, N.J.; Haire, L.F.; Underwood, E.; Patel, B.R.; Heath, R.B.; Walker, P.A.; Hallen, S.; et al. Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 2013, 4, 3017. [Google Scholar] [CrossRef]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef]
- Wang, R.N.; Green, J.; Wang, Z.; Deng, Y.; Qiao, M.; Peabody, M.; Zhang, Q.; Ye, J.; Yan, Z.; Denduluri, S.; et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis. 2014, 1, 87–105. [Google Scholar] [CrossRef]
- Bragdon, B.; Moseychuk, O.; Saldanha, S.; King, D.; Julian, J.; Nohe, A. Bone morphogenetic proteins: A critical review. Cell. Signal. 2011, 23, 609–620. [Google Scholar] [CrossRef]
- O’Connor, M.B.; Umulis, D.; Othmer, H.G.; Blair, S.S. Shaping BMP morphogen gradients in the Drosophila embryo and pupal wing. Development 2006, 133, 183–193. [Google Scholar] [CrossRef]
- Aono, A.; Hazama, M.; Notoya, K.; Taketomi, S.; Yamasaki, H.; Tsukuda, R.; Sasaki, S.; Fujisawa, Y. Potent Ectopic Bone-Inducing Activity of Bone Morphogenetic Protein-4/7 Heterodimer. Biochem. Biophys. Res. Commun. 1995, 210, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Butler, S.J.; Dodd, J. A Role for BMP Heterodimers in Roof Plate-Mediated Repulsion of Commissural Axons. Neuron 2003, 38, 389–401. [Google Scholar] [CrossRef]
- Israel, D.I.; Nove, J.; Kerns, K.M.; Kaufman, R.J.; Rosen, V.; Cox, K.A.; Wozney, J.M. Heterodimeric Bone Morphogenetic Proteins Show Enhanced Activity In Vitro and In Vivo. Growth Factors 1996, 13, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Little, S.C.; Mullins, M.C. Bone morphogenetic protein heterodimers assemble heteromeric type I receptor complexes to pattern the dorsoventral axis. Nat. Cell Biol. 2009, 11, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, T.; Kaito, T.; Matsuo, Y.; Sugiura, T.; Kashii, M.; Makino, T.; Iwasaki, M.; Yoshikawa, H. The bone morphogenetic protein-2/7 heterodimer is a stronger inducer of bone regeneration than the individual homodimers in a rat spinal fusion model. Spine J. 2015, 15, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Valera, E.; Isaacs, M.J.; Kawakami, Y.; Izpisúa Belmonte, J.C.; Choe, S. BMP-2/6 Heterodimer Is More Effective than BMP-2 or BMP-6 Homodimers as Inductor of Differentiation of Human Embryonic Stem Cells. PLoS ONE 2010, 5, e11167. [Google Scholar] [CrossRef]
- Groppe, J.; Rumpel, K.; Economides, A.N.; Stahl, N.; Sebald, W.; Affolter, M. Biochemical and biophysical characterization of refolded Drosophila DPP, a homolog of bone morphogenetic proteins 2 and 4. J. Biol. Chem. 1998, 273, 29052–29065. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Heldin, C.-H. Role for carbohydrate structures inTGF-beta1 latency. Nature 1989, 338, 158–160. [Google Scholar] [CrossRef]
- Tauscher, P.M.; Gui, J.; Shimmi, O. Adaptive protein divergence of BMP ligands takes place under developmental and evolutionary constraints. Development 2016, 143, 3742–3750. [Google Scholar] [CrossRef] [PubMed]
- Antenos, M.; Stemler, M.; Boime, I.; Woodruff, T.K. N-Linked Oligosaccharides Direct the Differential Assembly and Secretion of Inhibin α- and βA-Subunit Dimers. Mol. Endocrinol. 2007, 21, 1670–1684. [Google Scholar] [CrossRef] [PubMed]
- Saremba, S.; Nickel, J.; Seher, A.; Kotzsch, A.; Sebald, W.; Mueller, T.D. Type I receptor binding of bone morphogenetic protein 6 is dependent on N-glycosylation of the ligand. FEBS J. 2008, 275, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Newfeld, S.J.; Chartoff, E.H.; Graff, J.M.; Melton, D.A.; Gelbart, W.M. Mothers against dpp encodes a conserved cytoplasmic protein required in DPP/TGF-beta responsive cells. Development 1996, 122, 2099–2108. [Google Scholar] [CrossRef]
- Panganiban, G.E.; Reuter, R.; Scott, M.P.; Hoffmann, F.M. A Drosophila growth factor homolog, decapentaplegic, regulates homeotic gene expression within and across germ layers during midgut morphogenesis. Development 1990, 110, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Kondratyev, M.; Avezov, E.; Shenkman, M.; Groisman, B.; Lederkremer, G.Z. PERK-dependent compartmentalization of ERAD and unfolded protein response machineries during ER stress. Exp. Cell Res. 2007, 313, 3395–3407. [Google Scholar] [CrossRef]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 2001, 414, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Huang, C.; Fujihira, H. The cytoplasmic peptide:N-glycanase (NGLY1)—Structure, expression and cellular functions. Gene 2016, 577, 1–7. [Google Scholar] [CrossRef]
- Hebert, J.M.; Hayhurst, M.; Marks, M.E.; Kulessa, H.; Hogan, B.L.; McConnell, S.K. BMP ligands act redundantly to pattern the dorsal telencephalic midline. Genesis 2003, 35, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Hebert, J.M.; Mishina, Y.; McConnell, S.K. BMP signaling is required locally to pattern the dorsal telencephalic midline. Neuron 2002, 35, 1029–1041. [Google Scholar] [CrossRef]
- Morrell, N.W.; Bloch, D.B.; ten Dijke, P.; Goumans, M.J.; Hata, A.; Smith, J.; Yu, P.B.; Bloch, K.D. Targeting BMP signalling in cardiovascular disease and anaemia. Nat. Rev. Cardiol. 2016, 13, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Kang, Y.J.; Davies, L.M.; Meghpara, S.; Lau, K.; Chung, C.Y.; Kathiriya, J.; Hadjantonakis, A.K.; Monuki, E.S. BMP4 sufficiency to induce choroid plexus epithelial fate from embryonic stem cell-derived neuroepithelial progenitors. J. Neurosci 2012, 32, 15934–15945. [Google Scholar] [CrossRef] [PubMed]
- Talsness, D.M.; Owings, K.G.; Coelho, E.; Mercenne, G.; Pleinis, J.M.; Partha, R.; Hope, K.A.; Zuberi, A.R.; Clark, N.L.; Lutz, C.M.; et al. A Drosophila screen identifies NKCC1 as a modifier of NGLY1 deficiency. Elife 2020, 9, e57831. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Forbush, B., 3rd. The Na-K-Cl cotransporter of secretory epithelia. Annu. Rev. Physiol. 2000, 62, 515–534. [Google Scholar] [CrossRef]
- Tambe, M.A.; Ng, B.G.; Freeze, H.H. N-Glycanase 1 Transcriptionally Regulates Aquaporins Independent of Its Enzymatic Activity. Cell Rep. 2019, 29, 4620–4631.e4. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Age/ Sex | NGLY1 Genotype | Phenotypes |
|---|---|---|---|
| Need et al., 2012 [14] Enns et al., 2014 [11] Lam et al., 2017 [12] Levy et al., 2022 [13] | 12 y/M * | Q631Sfs/R401X | Developmental delay, movement disorder, hypotonia, seizures, EEG abnormalities, epilepsy, corneal ulcerations, liver fibrosis, microcephaly, dysmorphic features, small hands and feet, peripheral neuropathy, alacrima, elevated liver transaminases, constipation |
| Enns et al., 2014 [11] | 5 y/M * | R401X/R401X | Developmental delay, epilepsy, intrauterine growth retardation, movement disorder, hypotonia, dysmorphic features, EEG abnormalities, seizures, alacrima, scoliosis, small hands and feet, elevated liver transaminases, constipation |
| Enns et al., 2014 [11] Lam et al., 2017 [12] Levy et al., 2022 [13] | 9 y/M | R401X/R401X | Developmental delay, epilepsy, intrauterine growth retardation, movement disorder, hypotonia, seizure, corneal ulcerations, EEG abnormalities, alacrima, strabismus, elevated liver transaminases, scoliosis, constipation, liver fibrosis, small hands and feet, neuropathy |
| Enns et al., 2014 [11] Lam et al., 2017 [12] | 4 y/F | R401X/R401X | Developmental delay, hypotonia, microcephaly, movement disorder, elevated liver transaminases, alacrima, strabismus, constipation, dysmorphic features |
| Enns et al., 2014 [11] Lam et al., 2017 [12] | 18 y/F | R401X/R401X | Developmental delay, movement disorder, microcephaly, intrauterine growth retardation, hypotonic, EEG abnormalities, seizure, corneal ulcerations, alacrima, strabismus, elevated lactate, elevated liver transaminases, scoliosis, constipation, dysmorphic features, small hands and feet |
| Enns et al., 2014 [11] | 9 m/F * | R401X/R401X | Developmental delay, microcephaly, hypotonia, movement disorder, intrauterine growth retardation, microcephaly, EEG abnormalities, seizure, alacrima, dysmorphic features |
| Enns et al., 2014 [11] Lam et al., 2017 [12] Levy et al., 2022 [13] | 27 y/F | R458Kfs/R458Kfs | Developmental delay, intrauterine growth retardation, microcephaly, movement disorder, EEG abnormalities, alacrima, corneal ulceration, hypotonia, elevated liver transaminases, elevated lactate, peripheral neuropathy, constipation, scoliosis |
| Enns et al., 2014 [11] Kong et al., 2018 [23] Levy et al., 2022 [13] | 11 y/F | R402del/R524X | Developmental delay, movement disorder, microcephaly, hypotonia, EEG abnormalities, alacrima/hypolacrima, strabismus, elevated lactate, elevated liver transaminases, constipation, small hands and feet, neuropathy |
| Caglayan et al., 2015 [24] | 16 y/M * | N511Kfs/N511Kfs | Developmental delay, hypotonia, feeding problems, peripheral neuropathy, speech impairment, corneal ulcerations, alacrima |
| Caglayan et al., 2015 [24] | 9 y/F | N511Kfs/N511Kfs | Developmental delay, movement disorder, hypotonia, EEG abnormalities, feeding problems, epilepsy, peripheral neuropathy, seizure, strabismus, speech impairment, dysmorphic features, elevated liver transaminase, scoliosis |
| Heeley et al., 2015 [25] Lam et al., 2017 [12] Levy et al., 2022 [13] | 21 y/M | S116X/c.881+5G>T | Developmental delay, movement disorder, hypotonia, seizure, epilepsy, dysmorphic features, alacrima/hypolacrima, strabismus, elevated liver transaminases, liver fibrosis, constipation, scoliosis, neuropathy |
| Bosch et al., 2016 [26] | 3 y/M | R401X/R401X | Developmental delay, movement disorder, alacrima/hypolacrima, hypotonia, peripheral neuropathy, microcephaly, strabismus |
| Lam et al., 2017 [12] | 3 y/M | L318P/R390P | Developmental delay, movement disorder, feeding problems, EEG abnormalities, epilepsy, dysmorphic features, alacrima/hypolacrima, microcephaly, elevated liver transaminases, elevated lactate |
| Lam et al., 2017 [12] Levy et al., 2022 [13] | 10 y/F | E311K/W244R | Developmental delay, movement disorder, feeding problems, EEG abnormalities, dysmorphic features, alacrima/hypolacrima, microcephaly, elevated liver transaminases, elevated lactate, neuropathy |
| Lam et al., 2017 [12] Kong et al., 2018 [23] Levy et al., 2022 [13] | 11 y/M | W535X/L637X | Developmental delay, movement disorder, feeding problems, hypotonia, epilepsy, EEG abnormalities, dysmorphic features, alacrima/hypolacrima, microcephaly, elevated liver transaminases, elevated lactate, neuropathy |
| Lam et al., 2017 [12] Levy et al., 2022 [13] | 13 y/F # | Q208X/c.930C>T (G310G; splice site) | Developmental delay, movement disorder, feeding problems, EEG abnormalities, epilepsy, dysmorphic features, alacrima/hypolacrima, microcephaly, elevated liver transaminases, elevated lactate |
| Lam et al., 2017 [12] Levy et al., 2022 [13] | 15 y/M # | Q208X/c.930C>T (G310G; splice site) | Developmental delay, movement disorder, feeding problems, EEG abnormalities, epilepsy, dysmorphic features, alacrima/hypolacrima, microcephaly, elevated liver transaminases, elevated lactate |
| Lam et al., 2017 [12] Levy et al., 2022 [13] | 22 y/F * | R401X/R401X | Developmental delay, movement disorder, feeding problems, EEG abnormalities, epilepsy, dysmorphic features, corneal ulcerations, alacrima/hypolacrima, microcephaly, elevated liver transaminases, elevated lactate, neuropathy |
| Van Keulen et al., 2019 [27] Panneman et al., 2020 [28] | 9 y/F | Q613fs/Q613fs | Developmental delay, movement disorder, seizures, scoliosis, adrenal insufficiency |
| Chang et al., 2019 [29] Levy et al., 2022 [13] | 7 y/F | R469X/R469X | Developmental delay, microcephaly, dysmorphic features, feeding problems, constipation, hypotonia, alacrima/hypolacrima, elevated liver transaminases, liver fibrosis, neuropathy, movement disorder |
| Haijes et al., 2019 [30] | 18 y/M | c.247-2A>G/c.247-2A>G | Developmental delay, movement disorder |
| Haijes et al., 2019 [30] | 26 y/F | c.247-2A>G/c.247-2A>G | Developmental delay, movement disorder, alacrima/hypolacrima, corneal ulcerations |
| Haijes et al., 2019 [30] | 11 y/M | R586X/R586X | Developmental delay, intrauterine growth retardation, EEG abnormalities, alacrima, corneal ulceration, elevated liver transaminases, constipation, scoliosis |
| Haijes et al., 2019 [30] | 6 y/F | R586X/R586X | Developmental delay, epilepsy, alacrima, EEG abnormalities, corneal ulceration, elevated liver transaminases, constipation |
| Haijes et al., 2019 [30] | 15 y/F | L618X/Y539Gfs | Developmental delay, epilepsy, EEG abnormalities, intrauterine growth retardation, microcephaly, constipation, elevated liver transaminases, strabismus |
| Panneman et al., 2020 [28] | 8 yr */? | R401X/C283W | Developmental delay, movement disorder, hypotonia, seizures, EEG abnormalities, peripheral neuropathy, dysmorphic features, small hands and feet, corneal ulceration, liver fibrosis, elevated liver transaminases, elevated lactate, epilepsy |
| Panneman et al., 2020 [28] | ?/? | R401X/R401X | Developmental delay, hypotonia, peripheral neuropathy, dysmorphic features, small hands and feet, alacrima/hypolacrima, strabismus, elevated liver transaminases |
| Panneman et al., 2020 [28] | ?/F | R401X/E356G | Developmental delay, movement disorder, hypotonia, seizure, EEG abnormalities, peripheral neuropathy, dysmorphic features, small hands and feet, strabismus, elevated lactate |
| Abuduxikuer et al., 2020 [20] | 17 m/M | Y342C/R411X | Developmental delay, movement disorder, hypotonia, alacrima/hypolacrima, microcephaly, epilepsy, feeding problems, elevated liver transaminases, seizures, speech impairment |
| Abuduxikuer et al., 2020 [20] | 5 y/F | Y342C/R411X | Developmental delay, movement disorder, hypotonia, alacrima/hypolacrima, epilepsy, microcephaly, feeding problems, elevated liver transaminases, constipation, seizures, speech impairment |
| Abuduxikuer et al., 2020 [20] | 19 m/F | Y342C/R411X | Developmental delay, movement disorder, hypotonia, intrauterine growth retardation, epilepsy, alacrima/hypolacrima, feeding problems, microcephaly, elevated liver transaminases, seizures, speech impairment, dysmorphic features, elevated lactate |
| Abuduxikuer et al., 2020 [20] | 8 m/F | S546Ffs/c.1003+3A>G | Developmental delay, hypotonia, intrauterine growth retardation, alacrima/hypolacrima, strabismus, microcephaly, small hands and feet, feeding problems, elevated liver transaminases, speech impairment, dysmorphic features |
| Abuduxikuer et al., 2020 [20] | 4 y/F | S546Ffs/c.1003+3A>G | Developmental delay, movement disorder, hypotonia, intrauterine growth retardation, alacrima/hypolacrima, microcephaly, scoliosis, small hands and feet, feeding problems, elevated liver transaminases, speech impairment, dysmorphic features |
| Abuduxikuer et al., 2020 [20] | 10 m/M | R328G/R328G | Developmental delay, movement disorder, hypotonia, intrauterine growth retardation, alacrima/hypolacrima, microcephaly, small hands and feet, feeding problems, elevated liver transaminases, constipation, peripheral neuropathy, dysmorphic features |
| Lipiński et al., 2020 [31] | 7 y/M | c.1789+1G>A/c.1063T>C | Elevated liver transaminases, liver steatosis, global developmental delay, movement disorder, hypolacrima |
| Lipiński et al., 2020 [19] | 1.5 y/? | E84X/R401X | Developmental delay, movement disorder, alacrima, elevated liver transaminases, hypotonia, hypolipidemia |
| Ge et al., 2020 [32] | 10 m/F | R390X/D386Y | Developmental delay, intrauterine growth retardation, alacrima/hypolacrima, elevated liver transaminases, elevated lactate, EEG abnormalities, seizures, constipation |
| Rios-Flores et al., 2020 [33] Levy et al., 2022 [13] | 8 y/M | Q631Sfs/N178Qfs | Developmental delay, movement disorder, hypotonia, intrauterine growth retardation, alacrima/hypolacrima, constipation, dysmorphic features, elevated liver transaminases, liver fibrosis, feeding problems, epilepsy |
| Lipari-Pinto et al., 2020 [34] | 8 y/M | Q631Sfs/Q631Sfs | Developmental delay, hypotonia, elevated liver transaminases, small hands and feet |
| Kariminejad et al., 2021 [21] | 30 y/M | W236C/W236C | Developmental delay, hypotonia, scoliosis, EEG abnormalities |
| Kariminejad et al., 2021 [21] | 34 y/M | W236C/W236C | Developmental delay, hypotonia, movement disorder, seizures, scoliosis, constipation |
| Kariminejad et al., 2021 [21] | 35 y/F | W236C/W236C | Developmental delay, hypotonia, scoliosis, constipation |
| Kariminejad et al., 2021 [21] | 14 y/F | R390Q/R390Q | Developmental delay, movement disorder, epilepsy, liver fibrosis, EEG abnormalities, seizures, elevated liver transaminases, scoliosis |
| Kariminejad et al., 2021 [21] | 29 y/F | R390Q/R390Q | Developmental delay, movement disorder, liver fibrosis, elevated liver transaminases, scoliosis |
| Stuut et al., 2021 [22] | 5 y/M * | R401X/R401X | Developmental delay, alacrima/hypolacrima, movement disorder, epilepsy, intrauterine growth retardation, feeding problems, elevated liver transaminases |
| Dabaj et al., 2021 [35] | 6.5 y/F * | R328C/R328C | Developmental delay, alacrima/hypolacrima, feeding problems, hypotonia, dysmorphic features, seizures, microcephaly, intrauterine growth retardation, elevated liver transaminases |
| Kalfon et al., 2022 [18] | 6 m/F * | E432X/E432X | Developmental delay, feeding problems, hypotonia, alacrima/hypolacrima |
| Kalfon et al., 2022 [18] | 3 y/M * | E432X/E432X | Developmental delay, hypotonia, movement disorder, EEG abnormalities, elevated lactate, elevated liver transaminases, peripheral neuropathy, seizure, feeding problems |
| Kalfon et al., 2022 [18] | 12 y/F | E432X/E432X | Developmental delay, alacrima, movement disorder, hypotonia, EEG abnormalities, elevated lactate, elevated liver transaminases, seizure, feeding problems, peripheral neuropathy, microcephaly, scoliosis |
| Levy et al., 2022 [13] | 17 y/F | N415Mfs/c.658+1G>A | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy, EEG abnormalities |
| Levy et al., 2022 [13] | 15 y/M | R401X/deletion of at least exon 1–3 | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy, EEG abnormalities, epilepsy |
| Levy et al., 2022 [13] | 17 y/M | S169X/R383X | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy |
| Levy et al., 2022 [13] | 8 y/M | S169X/R383X | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy |
| Levy et al., 2022 [13] | 16 y/M * | R321X/Q631Sfs | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy, EEG abnormalities, epilepsy |
| Levy et al., 2022 [13] | 4 y/M | R308W/c.1789+1G>T | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy, EEG abnormalities, epilepsy |
| Levy et al., 2022 [13] | 7 y/F | R401X/c.1150-1G>C | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy, EEG abnormalities, epilepsy |
| Levy et al., 2022 [13] | 3 y/F | R401X/c.1150-1G>C | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy, EEG abnormalities, epilepsy |
| Levy et al., 2022 [13] | 6 y/F | R401X/C283W | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy, EEG abnormalities, epilepsy |
| Levy et al., 2022 [13] | 5 y/M | R401X/deletion of intron3 and exon 3 splice junction | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy, EEG abnormalities |
| Levy et al., 2022 [13] | 4 y/F | R401X/S607Ffs | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy, no EEG abnormalities |
| Levy et al., 2022 [13] | 17 y/F | W369X/R469X | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy, EEG abnormalities, epilepsy |
| Levy et al., 2022 [13] | 9 y/M | C355Y/R469X | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy |
| Levy et al., 2022 [13] | 11 y/M | Q191X/Q191X | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy, epilepsy |
| Levy et al., 2022 [13] | 9 y/F | Q631Sfs/Q631Sfs | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy, epilepsy |
| Levy et al., 2022 [13] | 3 y/M | R401X/N511Kfs | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy |
| Levy et al., 2022 [13] | 13 y/F | R469X/D597Sfs | Developmental delay, elevated liver transaminases, alacrima/hypolacrima, movement disorder, neuropathy, EEG abnormalities |
| Associated Proteins or Signaling Pathways | Role of NGLY1 | References |
|---|---|---|
| ERAD pathway | Deglycosylation of misfolded proteins | Hirsch et al., 2003 [67] Grotzke et al., 2013 [71] Hosomi & Suzuki, 2015 [68] Hosomi et al., 2016 [74] |
| BMP signaling (BMP4/Dpp) | Deglycosylation and retrotranslocation of misfolded BMP4/Dpp | Galeone et al., 2017 [55] Galeone et al., 2020 [40] |
| AMP kinase signaling and mitochondrial structural integrity | Regulation of AMPKα mRNA level | Han et al., 2020 [99] |
| Proteasomal homeostasis | Regulation of proteasomal bounce-back response by deglycosylation and activation of NFE2L1 | Lehrbach et. al., 2016 [60] Tomlin et al., 2018 [94] |
| Resistance to hypotonic cell lysis | Non-enzymatic transcriptional regulation of aquaporin expression, in part through ATF1/CREB1 | Tambe et al., 2019 [154] |
| Na+, K+, 2Cl– ion transport | Regulation of NKCC1 function, potentially through NKCC1 deglycosylation | Talsness et al., 2020 [152] |
| Mitophagy and mitochondrial homeostasis | Regulation of mitochondrial biogenesis and mitophagy-related gene expression by deglycosylation and activation of NFE2L1 | Yang et al., 2018 [44] |
| Innate immune signaling | Suppression of interferon signaling, at least in part through the DNA-sensing cGAS-STING pathway | Yang et al., 2018 [44] Zolekar et al., 2018 [110] |
| Melanoma survival and growth | Suppression of stress-signaling-associated apoptosis and cytokine surge | Zolekar et al., 2018 [110] |
| Ferroptosis | Resistance to ferroptosis through NFE2L1 deglycosylation | Forcina et al., 2022 [100] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, A.; Adams, J.M.; Han, S.Y.; Jafar-Nejad, H. NGLY1 Deficiency, a Congenital Disorder of Deglycosylation: From Disease Gene Function to Pathophysiology. Cells 2022, 11, 1155. https://doi.org/10.3390/cells11071155
Pandey A, Adams JM, Han SY, Jafar-Nejad H. NGLY1 Deficiency, a Congenital Disorder of Deglycosylation: From Disease Gene Function to Pathophysiology. Cells. 2022; 11(7):1155. https://doi.org/10.3390/cells11071155
Chicago/Turabian StylePandey, Ashutosh, Joshua M. Adams, Seung Yeop Han, and Hamed Jafar-Nejad. 2022. "NGLY1 Deficiency, a Congenital Disorder of Deglycosylation: From Disease Gene Function to Pathophysiology" Cells 11, no. 7: 1155. https://doi.org/10.3390/cells11071155
APA StylePandey, A., Adams, J. M., Han, S. Y., & Jafar-Nejad, H. (2022). NGLY1 Deficiency, a Congenital Disorder of Deglycosylation: From Disease Gene Function to Pathophysiology. Cells, 11(7), 1155. https://doi.org/10.3390/cells11071155