
An Overview of the Molecular Mechanisms Associated with Myocardial Ischemic Injury: State of the Art and Translational Perspectives

 ,
,  , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
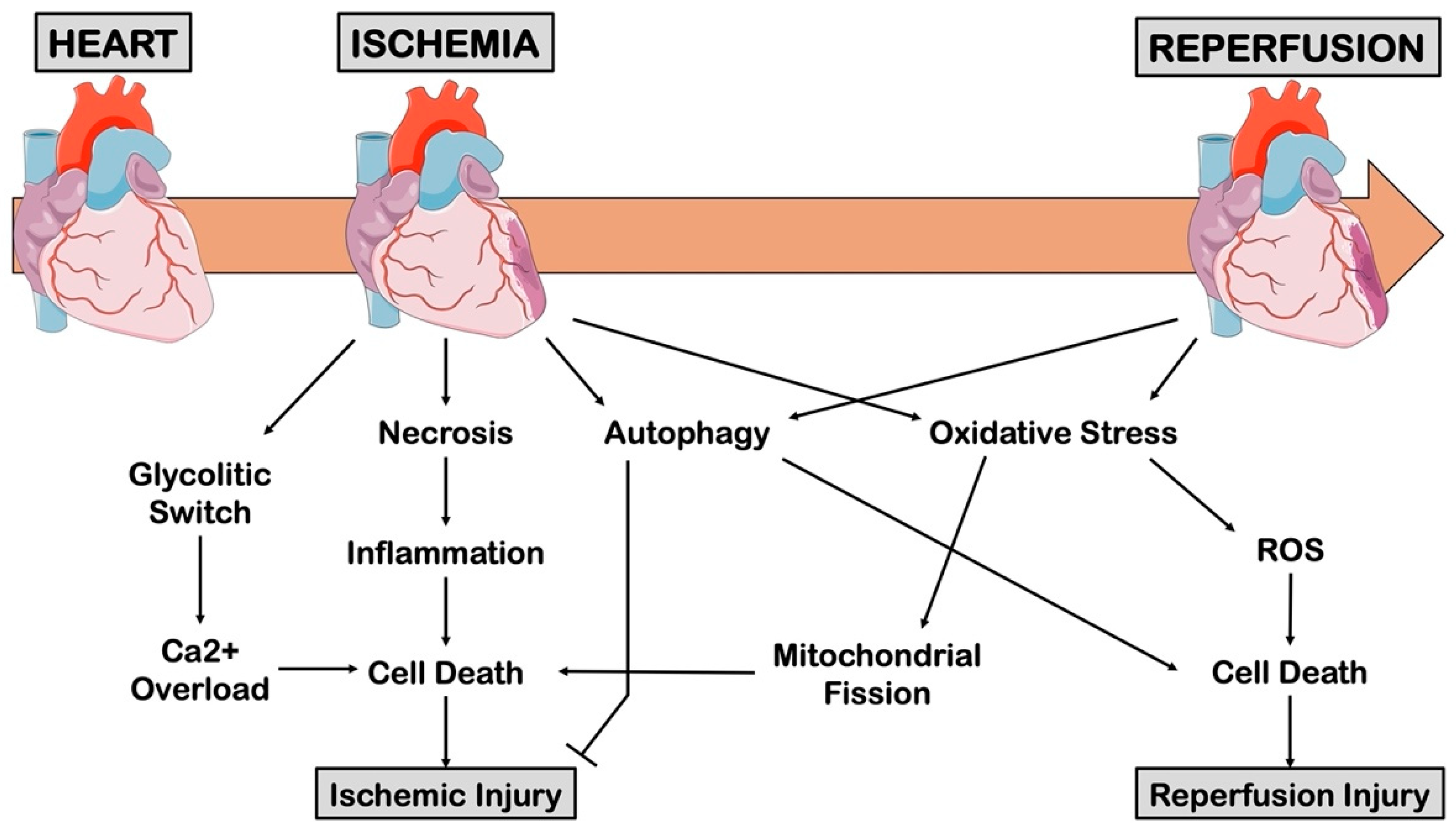
2. Pathophysiology of Myocardial Infarction
2.1. Causes of Ischemic Heart Disease
- -
- Coronary artery atherosclerosis
- -
- Coronary artery spasm
- -
- Microvascular dysfunction
- -
- Vascular remodeling
2.2. Ischemia/Reperfusion Injury
2.3. Chonic Post-Infarction Remodeling
2.4. Cell Death in Myocardial Ischemic Injury
3. The Role of Inflammation in Ischemic Injury
3.1. TNF-Alpha
3.2. IL-1
3.3. IL-6
4. The Role of Cell Metabolism in Myocardial Ischemia
4.1. Glycolytic Switch
4.2. Fatty Acids and Ischemic Injury
5. The Role of ROS in Myocardial Ischemia
5.1. Non-Reperfused Chronic Myocardial Infarction
5.2. Acute Myocardial Ischemia
6. The Role of Autophagy in Myocardial Ischemia
6.1. Acute and Chronic Ischemic Injury
6.2. Reperfusion Injury
6.3. The Specific Role of Mitophagy in I/R Injury
7. The Role of Mitochondrial Dynamics in I/R Injury
8. Clinical and Translational Perspectives
8.1. Targeting of Inflammatory Mediators
8.2. Pre and Post-Conditioning
8.3. Stem Cells and Exosome-Based Therapies
8.4. Bioengineering and Heart Reparation
8.4.1. Cell Sheets
8.4.2. Hydrogel Scaffolds
8.5. Mitochondrial Transplantation
8.6. Robotics and Nanotechnology
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hu, M.; Liu, H.; Zhang, X.; Li, H.; Zhou, F.; Liu, Y.M.; Lei, F.; Qin, J.J.; Zhao, Y.C.; et al. Global Burden of Disease Study 2019 suggests that metabolic risk factors are the leading drivers of the burden of ischemic heart disease. Cell Metab. 2021, 33, 1943–1956.e2. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Bray, F.; Ilbawi, A.; Soerjomataram, I. Effect on longevity of one-third reduction in premature mortality from non-communicable diseases by 2030: A global analysis of the Sustainable Development Goal health target. Lancet Glob. Health 2018, 6, e1288–e1296. [Google Scholar] [CrossRef]
- Lejay, A.; Fang, F.; John, R.; Van, J.A.; Barr, M.; Thaveau, F.; Chakfe, N.; Geny, B.; Scholey, J.W. Ischemia reperfusion injury, ischemic conditioning and diabetes mellitus. J. Mol. Cell. Cardiol. 2016, 91, 11–22. [Google Scholar] [CrossRef]
- Pepine, C.J.; Nichols, W.W. The pathophysiology of chronic ischemic heart disease. Clin. Cardiol. 2007, 30, I4–I9. [Google Scholar] [CrossRef]
- Depuydt, M.A.C.; Prange, K.H.M.; Slenders, L.; Örd, T.; Elbersen, D.; Boltjes, A.; de Jager, S.C.A.; Asselbergs, F.W.; de Borst, G.J.; Aavik, E.; et al. Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ. Res. 2020, 127, 1437–1455. [Google Scholar] [CrossRef]
- Sueda, S.; Sakaue, T. Coronary artery spasm-induced acute myocardial infarction in patients with myocardial infarction with non-obstructive coronary arteries. Heart Vessel. 2021, 36, 1804–1810. [Google Scholar] [CrossRef]
- Anjum, I. Calcium sensitization mechanisms in detrusor smooth muscles. J. Basic Clin. Physiol. Pharmacol. 2018, 29, 227–235. [Google Scholar] [CrossRef]
- Lanza, G.A.; Careri, G.; Crea, F. Mechanisms of coronary artery spasm. Circulation 2011, 124, 1774–1782. [Google Scholar] [CrossRef]
- Lanza, G.A.; Crea, F. Primary coronary microvascular dysfunction: Clinical presentation, pathophysiology, and management. Circulation 2010, 121, 2317–2325. [Google Scholar] [CrossRef]
- Biondi Zoccai, G.; Carnevale, R.; Vitali, M.; Tritapepe, L.; Martinelli, O.; Macrina, F.; Bullen, C.; Peruzzi, M.; Cavarretta, E.; Marullo, A.G.; et al. A randomized trial comparing the acute coronary, systemic, and environmental effects of electronic vaping cigarettes versus heat-not-burn cigarettes in smokers of combustible cigarettes undergoing invasive coronary assessment: Rationale and design of the SUR-VAPES 3 trial. Minerva Cardioangiol. 2020, 68, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Sciarretta, S.; Violi, F.; Nocella, C.; Loffredo, L.; Perri, L.; Peruzzi, M.; Marullo, A.G.; De Falco, E.; Chimenti, I.; et al. Acute Impact of Tobacco vs Electronic Cigarette Smoking on Oxidative Stress and Vascular Function. Chest 2016, 150, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.; Carnevale, R.; Battaglia, S.; Marti, R.; Pizzolo, S.; Bartimoccia, S.; Nocella, C.; Cammisotto, V.; Sciarretta, S.; Chimenti, I.; et al. Impact of chronic use of heat-not-burn cigarettes on oxidative stress, endothelial dysfunction and platelet activation: The SUR-VAPES Chronic Study. Thorax 2021, 76, 618–620. [Google Scholar] [CrossRef] [PubMed]
- Mastrangeli, S.; Carnevale, R.; Cavarretta, E.; Sciarretta, S.; Peruzzi, M.; Marullo, A.G.M.; De Falco, E.; Chimenti, I.; Valenti, V.; Bullen, C.; et al. Predictors of oxidative stress and vascular function in an experimental study of tobacco versus electronic cigarettes: A post hoc analysis of the SUR-VAPES 1 Study. Tob. Induc. Dis. 2018, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Camici, P.G.; Crea, F. Coronary microvascular dysfunction. N. Engl. J. Med. 2007, 356, 830–840. [Google Scholar] [CrossRef]
- Uriel, N.; Sayer, G.; Annamalai, S.; Kapur, N.K.; Burkhoff, D. Mechanical Unloading in Heart Failure. J. Am. Coll. Cardiol. 2018, 72, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.; Bonney, M.; Bonney, S.; Weitzel, L.; Koeppen, M.; Eckle, T. Myocardial ischemia reperfusion injury: From basic science to clinical bedside. Semin. Cardiothorac. Vasc. Anesth. 2012, 16, 123–132. [Google Scholar] [CrossRef]
- Jennings, R.B.; Reimer, K.A. The cell biology of acute myocardial ischemia. Annu. Rev. Med. 1991, 42, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [PubMed]
- Sanada, S.; Komuro, I.; Kitakaze, M. Pathophysiology of myocardial reperfusion injury: Preconditioning, postconditioning, and translational aspects of protective measures. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1723–H1741. [Google Scholar] [CrossRef]
- Haas, M.S.; Alicot, E.M.; Schuerpf, F.; Chiu, I.; Li, J.; Moore, F.D.; Carroll, M.C. Blockade of self-reactive IgM significantly reduces injury in a murine model of acute myocardial infarction. Cardiovasc. Res. 2010, 87, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, C.; Ladilov, Y.; Inserte, J.; Schäfer, M.; Haffner, S.; Garcia-Dorado, D.; Piper, H.M. Role of the reverse mode of the Na+/Ca2+ exchanger in reoxygenation-induced cardiomyocyte injury. Cardiovasc. Res. 2001, 51, 241–250. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Ruiz-Meana, M.; Inserte, J.; Rodriguez-Sinovas, A.; Piper, H.M. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc. Res. 2012, 94, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007, 9, 550–555. [Google Scholar] [CrossRef]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Bonora, M.; Campo, G.; Aquila, G.; Rizzo, P.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Mechanistic Role of mPTP in Ischemia-Reperfusion Injury. Adv. Exp. Med. Biol. 2017, 982, 169–189. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Molkentin, J.D. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef]
- Toldo, S.; Mauro, A.G.; Cutter, Z.; Abbate, A. Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1553–H1568. [Google Scholar] [CrossRef]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Javadov, S.; Margreiter, R.; Grimm, M.; Hagenbuchner, J.; Ausserlechner, M.J. The Role of Mitochondria in the Mechanisms of Cardiac Ischemia-Reperfusion Injury. Antioxidants 2019, 8, 454. [Google Scholar] [CrossRef] [PubMed]
- Schirone, L.; Forte, M.; Palmerio, S.; Yee, D.; Nocella, C.; Angelini, F.; Pagano, F.; Schiavon, S.; Bordin, A.; Carrizzo, A.; et al. A Review of the Molecular Mechanisms Underlying the Development and Progression of Cardiac Remodeling. Oxidative Med. Cell. Longev. 2017, 2017, 3920195. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.A.; Weinlich, R.; Brown, S.; Guy, C.; Fitzgerald, P.; Dillon, C.P.; Oberst, A.; Quarato, G.; Low, J.; Cripps, J.G.; et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016, 23, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef]
- Newton, K.; Dugger, D.L.; Maltzman, A.; Greve, J.M.; Hedehus, M.; Martin-McNulty, B.; Carano, R.A.; Cao, T.C.; van Bruggen, N.; Bernstein, L.; et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016, 23, 1565–1576. [Google Scholar] [CrossRef]
- Smith, C.C.; Davidson, S.M.; Lim, S.Y.; Simpkin, J.C.; Hothersall, J.S.; Yellon, D.M. Necrostatin: A potentially novel cardioprotective agent? Cardiovasc. Drugs Ther. 2007, 21, 227–233. [Google Scholar] [CrossRef]
- Kurrelmeyer, K.M.; Michael, L.H.; Baumgarten, G.; Taffet, G.E.; Peschon, J.J.; Sivasubramanian, N.; Entman, M.L.; Mann, D.L. Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. Proc. Natl. Acad. Sci. USA 2000, 97, 5456–5461. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef]
- Merkle, S.; Frantz, S.; Schön, M.P.; Bauersachs, J.; Buitrago, M.; Frost, R.J.; Schmitteckert, E.M.; Lohse, M.J.; Engelhardt, S. A role for caspase-1 in heart failure. Circ. Res. 2007, 100, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Higa, J.K.; Shimada, B.K.; Horiuchi, K.M.; Suhara, T.; Kobayashi, M.; Woo, J.D.; Aoyagi, H.; Marh, K.S.; Kitaoka, H.; et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H659–H668. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- Dabkowski, E.R.; Williamson, C.L.; Hollander, J.M. Mitochondria-specific transgenic overexpression of phospholipid hydroperoxide glutathione peroxidase (GPx4) attenuates ischemia/reperfusion-associated cardiac dysfunction. Free Radic. Biol. Med. 2008, 45, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Feng, G.; Gauthier, J.M.; Lokshina, I.; Higashikubo, R.; Evans, S.; Liu, X.; Hassan, A.; Tanaka, S.; Cicka, M.; et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Investig. 2019, 129, 2293–2304. [Google Scholar] [CrossRef]
- Jang, S.; Chapa-Dubocq, X.R.; Tyurina, Y.Y.; St Croix, C.M.; Kapralov, A.A.; Tyurin, V.A.; Bayır, H.; Kagan, V.E.; Javadov, S. Elucidating the contribution of mitochondrial glutathione to ferroptosis in cardiomyocytes. Redox Biol. 2021, 45, 102021. [Google Scholar] [CrossRef]
- Sparvero, L.J.; Tian, H.; Amoscato, A.A.; Sun, W.Y.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Kapralov, O.; Javadov, S.; He, R.R.; Watkins, S.C.; et al. Direct Mapping of Phospholipid Ferroptotic Death Signals in Cells and Tissues by Gas Cluster Ion Beam Secondary Ion Mass Spectrometry (GCIB-SIMS). Angew. Chem. 2021, 60, 11784–11788. [Google Scholar] [CrossRef]
- Ong, S.B.; Hernández-Reséndiz, S.; Crespo-Avilan, G.E.; Mukhametshina, R.T.; Kwek, X.Y.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol. Ther. 2018, 186, 73–87. [Google Scholar] [CrossRef]
- Andrassy, M.; Volz, H.C.; Igwe, J.C.; Funke, B.; Eichberger, S.N.; Kaya, Z.; Buss, S.; Autschbach, F.; Pleger, S.T.; Lukic, I.K.; et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation 2008, 117, 3216–3226. [Google Scholar] [CrossRef]
- Kitahara, T.; Takeishi, Y.; Harada, M.; Niizeki, T.; Suzuki, S.; Sasaki, T.; Ishino, M.; Bilim, O.; Nakajima, O.; Kubota, I. High-mobility group box 1 restores cardiac function after myocardial infarction in transgenic mice. Cardiovasc. Res. 2008, 80, 40–46. [Google Scholar] [CrossRef]
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; Van Tassell, B.W.; Salloum, F.N.; Kannan, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 19725–19730. [Google Scholar] [CrossRef] [PubMed]
- Silvis, M.J.M.; Demkes, E.J.; Fiolet, A.T.L.; Dekker, M.; Bosch, L.; van Hout, G.P.J.; Timmers, L.; de Kleijn, D.P.V. Immunomodulation of the NLRP3 Inflammasome in Atherosclerosis, Coronary Artery Disease, and Acute Myocardial Infarction. J. Cardiovasc. Transl. Res. 2021, 14, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Dawood, F.; Wen, W.H.; Chen, M.; Dixon, I.; Kirshenbaum, L.A.; Liu, P.P. Excessive tumor necrosis factor activation after infarction contributes to susceptibility of myocardial rupture and left ventricular dysfunction. Circulation 2004, 110, 3221–3228. [Google Scholar] [CrossRef]
- Maekawa, N.; Wada, H.; Kanda, T.; Niwa, T.; Yamada, Y.; Saito, K.; Fujiwara, H.; Sekikawa, K.; Seishima, M. Improved myocardial ischemia/reperfusion injury in mice lacking tumor necrosis factor-alpha. J. Am. Coll. Cardiol. 2002, 39, 1229–1235. [Google Scholar] [CrossRef]
- Monden, Y.; Kubota, T.; Inoue, T.; Tsutsumi, T.; Kawano, S.; Ide, T.; Tsutsui, H.; Sunagawa, K. Tumor necrosis factor-alpha is toxic via receptor 1 and protective via receptor 2 in a murine model of myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H743–H753. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, D.; Cifelli, G.; Mascio, G.; Madonna, M.; Sbroggiò, M.; Perrino, C.; Persico, M.G.; Frati, G.; Lembo, G. Placental growth factor regulates cardiac inflammation through the tissue inhibitor of metalloproteinases-3/tumor necrosis factor-α-converting enzyme axis: Crucial role for adaptive cardiac remodeling during cardiac pressure overload. Circulation 2011, 124, 1337–1350. [Google Scholar] [CrossRef]
- Suzuki, K.; Murtuza, B.; Smolenski, R.T.; Sammut, I.A.; Suzuki, N.; Kaneda, Y.; Yacoub, M.H. Overexpression of interleukin-1 receptor antagonist provides cardioprotection against ischemia-reperfusion injury associated with reduction in apoptosis. Circulation 2001, 104, I308–I313. [Google Scholar] [CrossRef]
- Bujak, M.; Dobaczewski, M.; Chatila, K.; Mendoza, L.H.; Li, N.; Reddy, A.; Frangogiannis, N.G. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am. J. Pathol. 2008, 173, 57–67. [Google Scholar] [CrossRef]
- Hwang, M.W.; Matsumori, A.; Furukawa, Y.; Ono, K.; Okada, M.; Iwasaki, A.; Hara, M.; Miyamoto, T.; Touma, M.; Sasayama, S. Neutralization of interleukin-1beta in the acute phase of myocardial infarction promotes the progression of left ventricular remodeling. J. Am. Coll. Cardiol. 2001, 38, 1546–1553. [Google Scholar] [CrossRef]
- Javaheri, A.; Bajpai, G.; Picataggi, A.; Mani, S.; Foroughi, L.; Evie, H.; Kovacs, A.; Weinheimer, C.J.; Hyrc, K.; Xiao, Q.; et al. TFEB activation in macrophages attenuates postmyocardial infarction ventricular dysfunction independently of ATG5-mediated autophagy. JCI Insight 2019, 4, e127312. [Google Scholar] [CrossRef]
- Fuchs, M.; Hilfiker, A.; Kaminski, K.; Hilfiker-Kleiner, D.; Guener, Z.; Klein, G.; Podewski, E.; Schieffer, B.; Rose-John, S.; Drexler, H. Role of interleukin-6 for LV remodeling and survival after experimental myocardial infarction. FASEB J. 2003, 17, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Jong, W.M.; Ten Cate, H.; Linnenbank, A.C.; de Boer, O.J.; Reitsma, P.H.; de Winter, R.J.; Zuurbier, C.J. Reduced acute myocardial ischemia-reperfusion injury in IL-6-deficient mice employing a closed-chest model. Inflamm. Res. 2016, 65, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Kamiński, K.A.; Kozuch, M.; Bonda, T.A.; Stepaniuk, M.M.; Waszkiewicz, E.; Chyczewski, L.; Musiał, W.J.; Winnicka, M.M. Effect of interleukin 6 deficiency on the expression of Bcl-2 and Bax in the murine heart. Pharmacol. Rep. 2009, 61, 504–513. [Google Scholar] [CrossRef]
- Hartman, M.H.; Vreeswijk-Baudoin, I.; Groot, H.E.; van de Kolk, K.W.; de Boer, R.A.; Mateo Leach, I.; Vliegenthart, R.; Sillje, H.H.; van der Harst, P. Inhibition of Interleukin-6 Receptor in a Murine Model of Myocardial Ischemia-Reperfusion. PLoS ONE 2016, 11, e0167195. [Google Scholar] [CrossRef]
- Nossuli, T.O.; Lakshminarayanan, V.; Baumgarten, G.; Taffet, G.E.; Ballantyne, C.M.; Michael, L.H.; Entman, M.L. A chronic mouse model of myocardial ischemia-reperfusion: Essential in cytokine studies. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1049–H1055. [Google Scholar] [CrossRef] [PubMed]
- Geraets, I.M.E.; Glatz, J.F.C.; Luiken, J.; Nabben, M. Pivotal role of membrane substrate transporters on the metabolic alterations in the pressure-overloaded heart. Cardiovasc. Res. 2019, 115, 1000–1012. [Google Scholar] [CrossRef]
- Wang, P.; Lloyd, S.G.; Chatham, J.C. Impact of high glucose/high insulin and dichloroacetate treatment on carbohydrate oxidation and functional recovery after low-flow ischemia and reperfusion in the isolated perfused rat heart. Circulation 2005, 111, 2066–2072. [Google Scholar] [CrossRef]
- Rousou, A.J.; Ericsson, M.; Federman, M.; Levitsky, S.; McCully, J.D. Opening of mitochondrial KATP channels enhances cardioprotection through the modulation of mitochondrial matrix volume, calcium accumulation, and respiration. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1967. [Google Scholar] [CrossRef]
- Burwell, L.S.; Nadtochiy, S.M.; Brookes, P.S. Cardioprotection by metabolic shut-down and gradual wake-up. J. Mol. Cell. Cardiol. 2009, 46, 804–810. [Google Scholar] [CrossRef]
- Valls-Lacalle, L.; Barba, I.; Miró-Casas, E.; Alburquerque-Béjar, J.J.; Ruiz-Meana, M.; Fuertes-Agudo, M.; Rodríguez-Sinovas, A.; García-Dorado, D. Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc. Res. 2016, 109, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yu, Y.; Zhang, Y.; Zhang, Z.; An, W.; Zhao, X. Treatment with D-β-hydroxybutyrate protects heart from ischemia/reperfusion injury in mice. Eur. J. Pharmacol. 2018, 829, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Sasaguri, S.; Rajesh, K.G.; Suzuki, R. dl-3-Hydroxybutyrate administration prevents myocardial damage after coronary occlusion in rat hearts. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1968–H1974. [Google Scholar] [CrossRef] [PubMed]
- Zuurbier, C.J.; Bertrand, L.; Beauloye, C.R.; Andreadou, I.; Ruiz-Meana, M.; Jespersen, N.R.; Kula-Alwar, D.; Prag, H.A.; Eric Botker, H.; Dambrova, M.; et al. Cardiac metabolism as a driver and therapeutic target of myocardial infarction. J. Cell. Mol. Med. 2020, 24, 5937–5954. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.R., 3rd; Li, J.; Coven, D.L.; Pypaert, M.; Zechner, C.; Palmeri, M.; Giordano, F.J.; Mu, J.; Birnbaum, M.J.; Young, L.H. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Investig. 2004, 114, 495–503. [Google Scholar] [CrossRef]
- Timmermans, A.D.; Balteau, M.; Gélinas, R.; Renguet, E.; Ginion, A.; de Meester, C.; Sakamoto, K.; Balligand, J.L.; Bontemps, F.; Vanoverschelde, J.L.; et al. A-769662 potentiates the effect of other AMP-activated protein kinase activators on cardiac glucose uptake. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1619–H1630. [Google Scholar] [CrossRef]
- Sun, L.; Shukair, S.; Naik, T.J.; Moazed, F.; Ardehali, H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol. Cell. Biol. 2008, 28, 1007–1017. [Google Scholar] [CrossRef]
- Sciarretta, S.; Zhai, P.; Shao, D.; Zablocki, D.; Nagarajan, N.; Terada, L.S.; Volpe, M.; Sadoshima, J. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2α/activating transcription factor 4 pathway. Circ. Res. 2013, 113, 1253–1264. [Google Scholar] [CrossRef]
- Ussher, J.R.; Wang, W.; Gandhi, M.; Keung, W.; Samokhvalov, V.; Oka, T.; Wagg, C.S.; Jaswal, J.S.; Harris, R.A.; Clanachan, A.S.; et al. Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc. Res. 2012, 94, 359–369. [Google Scholar] [CrossRef]
- van der Horst, I.C.; Timmer, J.R.; Ottervanger, J.P.; Bilo, H.J.; Gans, R.O.; de Boer, M.J.; Zijlstra, F. Glucose-insulin-potassium and reperfusion in acute myocardial infarction: Rationale and design of the Glucose-Insulin-Potassium Study-2 (GIPS-2). Am. Heart J. 2005, 149, 585–591. [Google Scholar] [CrossRef]
- Sabia, P.J.; Powers, E.R.; Ragosta, M.; Sarembock, I.J.; Burwell, L.R.; Kaul, S. An association between collateral blood flow and myocardial viability in patients with recent myocardial infarction. N. Engl. J. Med. 1992, 327, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Nederlof, R.; Eerbeek, O.; Baartscheer, A.; Schumacher, C.; Buchholtz, N.; Hollmann, M.W.; Coronel, R.; Weber, N.C.; Zuurbier, C.J. Delayed ischaemic contracture onset by empagliflozin associates with NHE1 inhibition and is dependent on insulin in isolated mouse hearts. Cardiovasc. Res. 2019, 115, 1533–1545. [Google Scholar] [CrossRef]
- Yurista, S.R.; Silljé, H.H.W.; Oberdorf-Maass, S.U.; Schouten, E.M.; Pavez Giani, M.G.; Hillebrands, J.L.; van Goor, H.; van Veldhuisen, D.J.; de Boer, R.A.; Westenbrink, B.D. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur. J. Heart Fail. 2019, 21, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Heitmeier, M.R.; Payne, M.A.; Weinheimer, C.; Kovacs, A.; Hresko, R.C.; Jay, P.Y.; Hruz, P.W. Metabolic and Cardiac Adaptation to Chronic Pharmacologic Blockade of Facilitative Glucose Transport in Murine Dilated Cardiomyopathy and Myocardial Ischemia. Sci. Rep. 2018, 8, 6475. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Zeng, J.; Yang, Y.X.; Qi, C.L.; Xiong, T.; Wu, G.Z.; Zeng, C.Y.; Wang, D.X. DRD4 Mitigates Myocardial Ischemia/Reperfusion Injury in Association With PI3K/AKT Mediated Glucose Metabolism. Front. Pharmacol. 2020, 11, 619426. [Google Scholar] [CrossRef]
- Ngoh, G.A.; Watson, L.J.; Facundo, H.T.; Dillmann, W.; Jones, S.P. Non-canonical glycosyltransferase modulates post-hypoxic cardiac myocyte death and mitochondrial permeability transition. J. Mol. Cell. Cardiol. 2008, 45, 313–325. [Google Scholar] [CrossRef]
- Jones, S.P.; Zachara, N.E.; Ngoh, G.A.; Hill, B.G.; Teshima, Y.; Bhatnagar, A.; Hart, G.W.; Marbán, E. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation 2008, 117, 1172–1182. [Google Scholar] [CrossRef]
- Liu, J.; Marchase, R.B.; Chatham, J.C. Glutamine-induced protection of isolated rat heart from ischemia/reperfusion injury is mediated via the hexosamine biosynthesis pathway and increased protein O-GlcNAc levels. J. Mol. Cell. Cardiol. 2007, 42, 177–185. [Google Scholar] [CrossRef]
- Liu, J.; Marchase, R.B.; Chatham, J.C. Increased O-GlcNAc levels during reperfusion lead to improved functional recovery and reduced calpain proteolysis. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1391–H1399. [Google Scholar] [CrossRef]
- Champattanachai, V.; Marchase, R.B.; Chatham, J.C. Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein O-GlcNAc and increased mitochondrial Bcl-2. Am. J. Physiol. Cell Physiol. 2008, 294, C1509–C1520. [Google Scholar] [CrossRef]
- Littlejohns, B.; Pasdois, P.; Duggan, S.; Bond, A.R.; Heesom, K.; Jackson, C.L.; Angelini, G.D.; Halestrap, A.P.; Suleiman, M.S. Hearts from mice fed a non-obesogenic high-fat diet exhibit changes in their oxidative state, calcium and mitochondria in parallel with increased susceptibility to reperfusion injury. PLoS ONE 2014, 9, e100579. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lloyd, S.G. High-fat, low-carbohydrate diet alters myocardial oxidative stress and impairs recovery of cardiac function after ischemia and reperfusion in obese rats. Nutr. Res. 2013, 33, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Rozanski, A.; Gransar, H.; Sharir, T.; Einstein, A.J.; Fish, M.B.; Ruddy, T.D.; Kaufmann, P.A.; Sinusas, A.J.; Miller, E.J.; et al. Myocardial Ischemic Burden and Differences in Prognosis Among Patients With and Without Diabetes: Results From the Multicenter International REFINE SPECT Registry. Diabetes Care 2020, 43, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Shinlapawittayatorn, K.; Chattipakorn, S.C.; Chattipakorn, N. The Influence of Obese Insulin-Resistance on the Outcome of the Ischemia/Reperfusion Insult to the Heart. Curr. Med. Chem. 2018, 25, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Frati, G.; Schirone, L.; Chimenti, I.; Yee, D.; Biondi-Zoccai, G.; Volpe, M.; Sciarretta, S. An overview of the inflammatory signalling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovasc. Res. 2017, 113, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.S.; Griffith, T.A.; Helman, T.; Du Toit, E.F.; Peart, J.N.; Headrick, J.P. Chronic type 2 but not type 1 diabetes impairs myocardial ischaemic tolerance and preconditioning in C57Bl/6 mice. Exp. Physiol. 2019, 104, 1868–1880. [Google Scholar] [CrossRef]
- Inserte, J.; Aluja, D.; Barba, I.; Ruiz-Meana, M.; Miró, E.; Poncelas, M.; Vilardosa, Ú.; Castellano, J.; Garcia-Dorado, D. High-fat diet improves tolerance to myocardial ischemia by delaying normalization of intracellular PH at reperfusion. J. Mol. Cell. Cardiol. 2019, 133, 164–173. [Google Scholar] [CrossRef]
- Dalgas, C.; Povlsen, J.A.; Løfgren, B.; Erichsen, S.B.; Bøtker, H.E. Effects of fatty acids on cardioprotection by pre-ischaemic inhibition of the malate-aspartate shuttle. Clin. Exp. Pharmacol. Physiol. 2012, 39, 878–885. [Google Scholar] [CrossRef]
- Forte, M.; Palmerio, S.; Yee, D.; Frati, G.; Sciarretta, S. Functional Role of Nox4 in Autophagy. Adv. Exp. Med. Biol. 2017, 982, 307–326. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schröder, K. NADPH oxidases in cardiovascular disease. Free Radic. Biol. Med. 2010, 49, 687–706. [Google Scholar] [CrossRef] [PubMed]
- de Vries, D.K.; Kortekaas, K.A.; Tsikas, D.; Wijermars, L.G.; van Noorden, C.J.; Suchy, M.T.; Cobbaert, C.M.; Klautz, R.J.; Schaapherder, A.F.; Lindeman, J.H. Oxidative damage in clinical ischemia/reperfusion injury: A reappraisal. Antioxid. Redox Signal. 2013, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, T.; Tsutsui, H.; Matsusaka, H.; Murakami, K.; Hayashidani, S.; Ikeuchi, M.; Wen, J.; Kubota, T.; Utsumi, H.; Takeshita, A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2004, 109, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Kinugawa, S.; Tsutsui, H.; Hayashidani, S.; Ide, T.; Suematsu, N.; Satoh, S.; Utsumi, H.; Takeshita, A. Treatment with dimethylthiourea prevents left ventricular remodeling and failure after experimental myocardial infarction in mice: Role of oxidative stress. Circ. Res. 2000, 87, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Nocella, C.; De Falco, E.; Palmerio, S.; Schirone, L.; Valenti, V.; Frati, G.; Carnevale, R.; Sciarretta, S. The Pathophysiological Role of NOX2 in Hypertension and Organ Damage. High Blood Press. Cardiovasc. Prev. 2016, 23, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Kuroda, J.; Matsushima, S.; Ago, T.; Sadoshima, J. Regulation of myocardial growth and death by NADPH oxidase. J. Mol. Cell. Cardiol. 2011, 50, 408–416. [Google Scholar] [CrossRef]
- Looi, Y.H.; Grieve, D.J.; Siva, A.; Walker, S.J.; Anilkumar, N.; Cave, A.C.; Marber, M.; Monaghan, M.J.; Shah, A.M. Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension 2008, 51, 319–325. [Google Scholar] [CrossRef]
- Sirker, A.; Murdoch, C.E.; Protti, A.; Sawyer, G.J.; Santos, C.X.; Martin, D.; Zhang, X.; Brewer, A.C.; Zhang, M.; Shah, A.M. Cell-specific effects of Nox2 on the acute and chronic response to myocardial infarction. J. Mol. Cell. Cardiol. 2016, 98, 11–17. [Google Scholar] [CrossRef]
- Sengupta, A.; Molkentin, J.D.; Paik, J.H.; DePinho, R.A.; Yutzey, K.E. FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J. Biol. Chem. 2011, 286, 7468–7478. [Google Scholar] [CrossRef]
- Matsushima, S.; Kuroda, J.; Ago, T.; Zhai, P.; Ikeda, Y.; Oka, S.; Fong, G.H.; Tian, R.; Sadoshima, J. Broad suppression of NADPH oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1α and upregulation of peroxisome proliferator-activated receptor-α. Circ. Res. 2013, 112, 1135–1149. [Google Scholar] [CrossRef]
- Siu, K.L.; Lotz, C.; Ping, P.; Cai, H. Netrin-1 abrogates ischemia/reperfusion-induced cardiac mitochondrial dysfunction via nitric oxide-dependent attenuation of NOX4 activation and recoupling of NOS. J. Mol. Cell. Cardiol. 2015, 78, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, X.; Liu, S.; Zeng, S.; Yu, L.; Yang, G.; Guo, J.; Xu, Y. BRG1 regulates NOX gene transcription in endothelial cells and contributes to cardiac ischemia-reperfusion injury. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2018, 1864, 3477–3486. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Lee, C.F.; Wang, W.; Karamanlidis, G.; Kuroda, J.; Matsushima, S.; Sadoshima, J.; Tian, R. Elimination of NADPH oxidase activity promotes reductive stress and sensitizes the heart to ischemic injury. J. Am. Heart Assoc. 2014, 3, e000555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.; Hafstad, A.D.; Beretta, M.; Zhang, M.; Molenaar, C.; Kopec, J.; Fotinou, D.; Murray, T.V.; Cobb, A.M.; Martin, D.; et al. Targeted redox inhibition of protein phosphatase 1 by Nox4 regulates eIF2α-mediated stress signaling. EMBO J. 2016, 35, 319–334. [Google Scholar] [CrossRef]
- Matsushima, S.; Tsutsui, H.; Sadoshima, J. Physiological and pathological functions of NADPH oxidases during myocardial ischemia-reperfusion. Trends Cardiovasc. Med. 2014, 24, 202–205. [Google Scholar] [CrossRef]
- Forte, M.; Palmerio, S.; Bianchi, F.; Volpe, M.; Rubattu, S. Mitochondrial complex I deficiency and cardiovascular diseases: Current evidence and future directions. J. Mol. Med. 2019, 97, 579–591. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Weiss, J.N.; Korge, P.; Honda, H.M.; Ping, P. Role of the mitochondrial permeability transition in myocardial disease. Circ. Res. 2003, 93, 292–301. [Google Scholar] [CrossRef]
- Zhang, Y.; Sano, M.; Shinmura, K.; Tamaki, K.; Katsumata, Y.; Matsuhashi, T.; Morizane, S.; Ito, H.; Hishiki, T.; Endo, J.; et al. 4-hydroxy-2-nonenal protects against cardiac ischemia-reperfusion injury via the Nrf2-dependent pathway. J. Mol. Cell. Cardiol. 2010, 49, 576–586. [Google Scholar] [CrossRef]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G. Autophagy: A druggable process that is deregulated in aging and human disease. J. Clin. Investig. 2015, 125, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Hall, M.N.; Lin, S.C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New Insights Into the Role of mTOR Signaling in the Cardiovascular System. Circ. Res. 2018, 122, 489–505. [Google Scholar] [CrossRef]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. The complex network of mTOR signaling in the heart. Cardiovasc. Res. 2021, 118, 424–439. [Google Scholar] [CrossRef]
- Sciarretta, S.; Zhai, P.; Volpe, M.; Sadoshima, J. Pharmacological modulation of autophagy during cardiac stress. J. Cardiovasc. Pharmacol. 2012, 60, 235–241. [Google Scholar] [CrossRef]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef]
- Sciarretta, S.; Zhai, P.; Shao, D.; Maejima, Y.; Robbins, J.; Volpe, M.; Condorelli, G.; Sadoshima, J. Rheb is a critical regulator of autophagy during myocardial ischemia: Pathophysiological implications in obesity and metabolic syndrome. Circulation 2012, 125, 1134–1146. [Google Scholar] [CrossRef]
- Kanamori, H.; Takemura, G.; Goto, K.; Maruyama, R.; Ono, K.; Nagao, K.; Tsujimoto, A.; Ogino, A.; Takeyama, T.; Kawaguchi, T.; et al. Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2261–H2271. [Google Scholar] [CrossRef] [PubMed]
- Buss, S.J.; Muenz, S.; Riffel, J.H.; Malekar, P.; Hagenmueller, M.; Weiss, C.S.; Bea, F.; Bekeredjian, R.; Schinke-Braun, M.; Izumo, S.; et al. Beneficial effects of Mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. J. Am. Coll. Cardiol. 2009, 54, 2435–2446. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Takemura, G.; Goto, K.; Maruyama, R.; Tsujimoto, A.; Ogino, A.; Takeyama, T.; Kawaguchi, T.; Watanabe, T.; Fujiwara, T.; et al. The role of autophagy emerging in postinfarction cardiac remodelling. Cardiovasc. Res. 2011, 91, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Frati, G.; Vecchione, C.; Sciarretta, S. Novel Beneficial Cardiovascular Effects of Natural Activators of Autophagy. Circ. Res. 2018, 123, 947–949. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Yee, D.; Nagarajan, N.; Bianchi, F.; Saito, T.; Valenti, V.; Tong, M.; Del Re, D.P.; Vecchione, C.; Schirone, L.; et al. Trehalose-Induced Activation of Autophagy Improves Cardiac Remodeling After Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 71, 1999–2010. [Google Scholar] [CrossRef]
- Carmona-Gutierrez, D.; Zimmermann, A.; Kainz, K.; Pietrocola, F.; Chen, G.; Maglioni, S.; Schiavi, A.; Nah, J.; Mertel, S.; Beuschel, C.B.; et al. The flavonoid 4,4′-dimethoxychalcone promotes autophagy-dependent longevity across species. Nat. Commun. 2019, 10, 651. [Google Scholar] [CrossRef]
- Feng, H.; Mou, S.Q.; Li, W.J.; Zhang, N.; Zhou, Z.Y.; Ding, W.; Bian, Z.Y.; Liao, H.H. Resveratrol Inhibits Ischemia-Induced Myocardial Senescence Signals and NLRP3 Inflammasome Activation. Oxidative Med. Cell. Longev. 2020, 2020, 2647807. [Google Scholar] [CrossRef]
- Wang, N.P.; Wang, Z.F.; Tootle, S.; Philip, T.; Zhao, Z.Q. Curcumin promotes cardiac repair and ameliorates cardiac dysfunction following myocardial infarction. Br. J. Pharmacol. 2012, 167, 1550–1562. [Google Scholar] [CrossRef]
- Maejima, Y.; Kyoi, S.; Zhai, P.; Liu, T.; Li, H.; Ivessa, A.; Sciarretta, S.; Del Re, D.P.; Zablocki, D.K.; Hsu, C.P.; et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 2013, 19, 1478–1488. [Google Scholar] [CrossRef]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef]
- Nah, J.; Zhai, P.; Huang, C.Y.; Fernández, Á.F.; Mareedu, S.; Levine, B.; Sadoshima, J. Upregulation of Rubicon promotes autosis during myocardial ischemia/reperfusion injury. J. Clin. Investig. 2020, 130, 2978–2991. [Google Scholar] [CrossRef] [PubMed]
- Zhai, P.; Sciarretta, S.; Galeotti, J.; Volpe, M.; Sadoshima, J. Differential roles of GSK-3β during myocardial ischemia and ischemia/reperfusion. Circ. Res. 2011, 109, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Cho, G.W.; Kong, Y.; Li, D.L.; Altamirano, F.; Luo, X.; Morales, C.R.; Jiang, N.; Schiattarella, G.G.; May, H.I.; et al. Activation of Autophagic Flux Blunts Cardiac Ischemia/Reperfusion Injury. Circ. Res. 2021, 129, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Andres, A.M.; Hernandez, G.; Lee, P.; Huang, C.; Ratliff, E.P.; Sin, J.; Thornton, C.A.; Damasco, M.V.; Gottlieb, R.A. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid. Redox Signal. 2014, 21, 1960–1973. [Google Scholar] [CrossRef]
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S.; et al. The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 2021, 178, 2060–2076. [Google Scholar] [CrossRef]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: Therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014, 28, 316–326. [Google Scholar] [CrossRef]
- Zepeda, R.; Kuzmicic, J.; Parra, V.; Troncoso, R.; Pennanen, C.; Riquelme, J.A.; Pedrozo, Z.; Chiong, M.; Sánchez, G.; Lavandero, S. Drp1 loss-of-function reduces cardiomyocyte oxygen dependence protecting the heart from ischemia-reperfusion injury. J. Cardiovasc. Pharmacol. 2014, 63, 477–487. [Google Scholar] [CrossRef]
- Maneechote, C.; Palee, S.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, S.C.; Chattipakorn, N. Balancing mitochondrial dynamics via increasing mitochondrial fusion attenuates infarct size and left ventricular dysfunction in rats with cardiac ischemia/reperfusion injury. Clin. Sci. 2019, 133, 497–513. [Google Scholar] [CrossRef]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabò, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef]
- Rodríguez-Graciani, K.M.; Chapa-Dubocq, X.R.; MacMillan-Crow, L.A.; Javadov, S. Association Between L-OPA1 Cleavage and Cardiac Dysfunction During Ischemia-Reperfusion Injury in Rats. Cell. Physiol. Biochem. 2020, 54, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Javadov, S. OPA1 regulates respiratory supercomplexes assembly: The role of mitochondrial swelling. Mitochondrion 2020, 51, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Huang, X.; Han, L.; Wang, X.; Cheng, H.; Zhao, Y.; Chen, Q.; Chen, J.; Cheng, H.; Xiao, R.; et al. Central role of mitofusin 2 in autophagosome-lysosome fusion in cardiomyocytes. J. Biol. Chem. 2012, 287, 23615–23625. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, K.N.; Khairallah, R.J.; Ngoh, G.A.; Chikando, A.; Luptak, I.; O’Shea, K.M.; Riley, D.D.; Lugus, J.J.; Colucci, W.S.; Lederer, W.J.; et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol. 2011, 31, 1309–1328. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Zheng, M.; Cao, C.; Chen, C.; Tang, J.; Zhang, W.; Cheng, H.; Chen, K.H.; Xiao, R.P. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J. Biol. Chem. 2007, 282, 23354–23361. [Google Scholar] [CrossRef]
- Zechner, C.; Lai, L.; Zechner, J.F.; Geng, T.; Yan, Z.; Rumsey, J.W.; Collia, D.; Chen, Z.; Wozniak, D.F.; Leone, T.C.; et al. Total skeletal muscle PGC-1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab. 2010, 12, 633–642. [Google Scholar] [CrossRef]
- Javadov, S.; Purdham, D.M.; Zeidan, A.; Karmazyn, M. NHE-1 inhibition improves cardiac mitochondrial function through regulation of mitochondrial biogenesis during postinfarction remodeling. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1722–H1730. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Duchen, M.R.; Yellon, D.M. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc. Res. 2003, 60, 617–625. [Google Scholar] [CrossRef]
- Lønborg, J.; Vejlstrup, N.; Kelbæk, H.; Bøtker, H.E.; Kim, W.Y.; Mathiasen, A.B.; Jørgensen, E.; Helqvist, S.; Saunamäki, K.; Clemmensen, P.; et al. Exenatide reduces reperfusion injury in patients with ST-segment elevation myocardial infarction. Eur. Heart J. 2012, 33, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Iop, L.; Dal Sasso, E.; Schirone, L.; Forte, M.; Peruzzi, M.; Cavarretta, E.; Palmerio, S.; Gerosa, G.; Sciarretta, S.; Frati, G. The Light and Shadow of Senescence and Inflammation in Cardiovascular Pathology and Regenerative Medicine. Mediat. Inflamm. 2017, 2017, 7953486. [Google Scholar] [CrossRef]
- Abbate, A.; Van Tassell, B.W.; Biondi-Zoccai, G.; Kontos, M.C.; Grizzard, J.D.; Spillman, D.W.; Oddi, C.; Roberts, C.S.; Melchior, R.D.; Mueller, G.H.; et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study]. Am. J. Cardiol. 2013, 111, 1394–1400. [Google Scholar] [CrossRef]
- Abbate, A.; Trankle, C.R.; Buckley, L.F.; Lipinski, M.J.; Appleton, D.; Kadariya, D.; Canada, J.M.; Carbone, S.; Roberts, C.S.; Abouzaki, N.; et al. Interleukin-1 Blockade Inhibits the Acute Inflammatory Response in Patients With ST-Segment-Elevation Myocardial Infarction. J. Am. Heart Assoc. 2020, 9, e014941. [Google Scholar] [CrossRef] [PubMed]
- Broch, K.; Anstensrud, A.K.; Woxholt, S.; Sharma, K.; Tøllefsen, I.M.; Bendz, B.; Aakhus, S.; Ueland, T.; Amundsen, B.H.; Damås, J.K.; et al. Randomized Trial of Interleukin-6 Receptor Inhibition in Patients With Acute ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2021, 77, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef]
- Ji, L.; Zhang, X.; Liu, W.; Huang, Q.; Yang, W.; Fu, F.; Ma, H.; Su, H.; Wang, H.; Wang, J.; et al. AMPK-regulated and Akt-dependent enhancement of glucose uptake is essential in ischemic preconditioning-alleviated reperfusion injury. PLoS ONE 2013, 8, e69910. [Google Scholar] [CrossRef]
- Nadtochiy, S.M.; Yao, H.; McBurney, M.W.; Gu, W.; Guarente, L.; Rahman, I.; Brookes, P.S. SIRT1-mediated acute cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1506–H1512. [Google Scholar] [CrossRef]
- Heusch, G. Molecular basis of cardioprotection: Signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef]
- Tamareille, S.; Mateus, V.; Ghaboura, N.; Jeanneteau, J.; Croué, A.; Henrion, D.; Furber, A.; Prunier, F. RISK and SAFE signaling pathway interactions in remote limb ischemic perconditioning in combination with local ischemic postconditioning. Basic Res. Cardiol. 2011, 106, 1329–1339. [Google Scholar] [CrossRef]
- Polshekan, M.; Khori, V.; Alizadeh, A.M.; Ghayour-Mobarhan, M.; Saeidi, M.; Jand, Y.; Rajaei, M.; Farnoosh, G.; Jamialahmadi, K. The SAFE pathway is involved in the postconditioning mechanism of oxytocin in isolated rat heart. Peptides 2019, 111, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Inserte, J.; Barba, I.; Poncelas-Nozal, M.; Hernando, V.; Agulló, L.; Ruiz-Meana, M.; Garcia-Dorado, D. cGMP/PKG pathway mediates myocardial postconditioning protection in rat hearts by delaying normalization of intracellular acidosis during reperfusion. J. Mol. Cell. Cardiol. 2011, 50, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, V.; Mudalagiri, N.R.; Di Salvo, C.; Kolvekar, S.; Hayward, M.; Yap, J.; Keogh, B.; Hausenloy, D.J.; Yellon, D.M. Postconditioning protects human atrial muscle through the activation of the RISK pathway. Basic Res. Cardiol. 2007, 102, 453–459. [Google Scholar] [CrossRef]
- Bolli, R.; Stein, A.B.; Guo, Y.; Wang, O.L.; Rokosh, G.; Dawn, B.; Molkentin, J.D.; Sanganalmath, S.K.; Zhu, Y.; Xuan, Y.T. A murine model of inducible, cardiac-specific deletion of STAT3: Its use to determine the role of STAT3 in the upregulation of cardioprotective proteins by ischemic preconditioning. J. Mol. Cell. Cardiol. 2011, 50, 589–597. [Google Scholar] [CrossRef]
- Phillips, D.; Reilley, M.J.; Aponte, A.M.; Wang, G.; Boja, E.; Gucek, M.; Balaban, R.S. Stoichiometry of STAT3 and mitochondrial proteins: Implications for the regulation of oxidative phosphorylation by protein-protein interactions. J. Biol. Chem. 2010, 285, 23532–23536. [Google Scholar] [CrossRef] [PubMed]
- Juhaszova, M.; Zorov, D.B.; Kim, S.H.; Pepe, S.; Fu, Q.; Fishbein, K.W.; Ziman, B.D.; Wang, S.; Ytrehus, K.; Antos, C.L.; et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Investig. 2004, 113, 1535–1549. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Wong, R.; Steenbergen, C. Signalosomes: Delivering cardioprotective signals from GPCRs to mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H920–H922. [Google Scholar] [CrossRef][Green Version]
- Javadov, S.A.; Clarke, S.; Das, M.; Griffiths, E.J.; Lim, K.H.; Halestrap, A.P. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J. Physiol. 2003, 549, 513–524. [Google Scholar] [CrossRef]
- Hausenloy, D.; Wynne, A.; Duchen, M.; Yellon, D. Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation 2004, 109, 1714–1717. [Google Scholar] [CrossRef]
- Zhao, Z.Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H579–H588. [Google Scholar] [CrossRef]
- Staat, P.; Rioufol, G.; Piot, C.; Cottin, Y.; Cung, T.T.; L’Huillier, I.; Aupetit, J.F.; Bonnefoy, E.; Finet, G.; André-Fouët, X.; et al. Postconditioning the human heart. Circulation 2005, 112, 2143–2148. [Google Scholar] [CrossRef]
- Cohen, M.V.; Yang, X.M.; Downey, J.M. The pH hypothesis of postconditioning: Staccato reperfusion reintroduces oxygen and perpetuates myocardial acidosis. Circulation 2007, 115, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Przyklenk, K.; Bauer, B.; Ovize, M.; Kloner, R.A.; Whittaker, P. Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 1993, 87, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G.; Bøtker, H.E.; Przyklenk, K.; Redington, A.; Yellon, D. Remote ischemic conditioning. J. Am. Coll. Cardiol. 2015, 65, 177–195. [Google Scholar] [CrossRef] [PubMed]
- Sloth, A.D.; Schmidt, M.R.; Munk, K.; Kharbanda, R.K.; Redington, A.N.; Schmidt, M.; Pedersen, L.; Sørensen, H.T.; Bøtker, H.E. Improved long-term clinical outcomes in patients with ST-elevation myocardial infarction undergoing remote ischaemic conditioning as an adjunct to primary percutaneous coronary intervention. Eur. Heart J. 2014, 35, 168–175. [Google Scholar] [CrossRef]
- Thielmann, M.; Kottenberg, E.; Kleinbongard, P.; Wendt, D.; Gedik, N.; Pasa, S.; Price, V.; Tsagakis, K.; Neuhäuser, M.; Peters, J.; et al. Cardioprotective and prognostic effects of remote ischaemic preconditioning in patients undergoing coronary artery bypass surgery: A single-centre randomised, double-blind, controlled trial. Lancet 2013, 382, 597–604. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Kharbanda, R.K.; Møller, U.K.; Ramlall, M.; Aarøe, J.; Butler, R.; Bulluck, H.; Clayton, T.; Dana, A.; Dodd, M.; et al. Effect of remote ischaemic conditioning on clinical outcomes in patients with acute myocardial infarction (CONDI-2/ERIC-PPCI): A single-blind randomised controlled trial. Lancet 2019, 394, 1415–1424. [Google Scholar] [CrossRef]
- Donato, M.; Buchholz, B.; Rodríguez, M.; Pérez, V.; Inserte, J.; García-Dorado, D.; Gelpi, R.J. Role of the parasympathetic nervous system in cardioprotection by remote hindlimb ischaemic preconditioning. Exp. Physiol. 2013, 98, 425–434. [Google Scholar] [CrossRef]
- Jones, W.K.; Fan, G.C.; Liao, S.; Zhang, J.M.; Wang, Y.; Weintraub, N.L.; Kranias, E.G.; Schultz, J.E.; Lorenz, J.; Ren, X. Peripheral nociception associated with surgical incision elicits remote nonischemic cardioprotection via neurogenic activation of protein kinase C signaling. Circulation 2009, 120, S1–S9. [Google Scholar] [CrossRef]
- Mastitskaya, S.; Marina, N.; Gourine, A.; Gilbey, M.P.; Spyer, K.M.; Teschemacher, A.G.; Kasparov, S.; Trapp, S.; Ackland, G.L.; Gourine, A.V. Cardioprotection evoked by remote ischaemic preconditioning is critically dependent on the activity of vagal pre-ganglionic neurones. Cardiovasc. Res. 2012, 95, 487–494. [Google Scholar] [CrossRef]
- Zhao, Y.; Zheng, Z.N.; Pi, Y.N.; Liang, X.; Jin, S.Q. Cardioprotective Effects of Transfusion of Late-Phase Preconditioned Plasma May Be Induced by Activating the Reperfusion Injury Salvage Kinase Pathway but Not the Survivor Activating Factor Enhancement Pathway in Rats. Oxidative Med. Cell. Longev. 2017, 2017, 8526561. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Dai, S.; Wu, W.J.; Tan, W.; Zhu, X.; Mu, J.; Guo, Y.; Bolli, R.; Rokosh, G. Stromal cell derived factor-1 alpha confers protection against myocardial ischemia/reperfusion injury: Role of the cardiac stromal cell derived factor-1 alpha CXCR4 axis. Circulation 2007, 116, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.P.; Parajuli, N.; Zheng, X.; Becker, L. Remote ischemic preconditioning confers late protection against myocardial ischemia-reperfusion injury in mice by upregulating interleukin-10. Basic Res. Cardiol. 2012, 107, 277. [Google Scholar] [CrossRef]
- Rassaf, T.; Totzeck, M.; Hendgen-Cotta, U.B.; Shiva, S.; Heusch, G.; Kelm, M. Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ. Res. 2014, 114, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, H.; Zhang, X.; Liu, Y.; Chen, J.; Medvedovic, M.; Li, H.; Weiss, M.J.; Ren, X.; Fan, G.C. Loss of the miR-144/451 cluster impairs ischaemic preconditioning-mediated cardioprotection by targeting Rac-1. Cardiovasc. Res. 2012, 94, 379–390. [Google Scholar] [CrossRef]
- Memon, I.A.; Sawa, Y.; Fukushima, N.; Matsumiya, G.; Miyagawa, S.; Taketani, S.; Sakakida, S.K.; Kondoh, H.; Aleshin, A.N.; Shimizu, T.; et al. Repair of impaired myocardium by means of implantation of engineered autologous myoblast sheets. J. Thorac. Cardiovasc. Surg. 2005, 130, 1333–1341. [Google Scholar] [CrossRef]
- Menasché, P.; Alfieri, O.; Janssens, S.; McKenna, W.; Reichenspurner, H.; Trinquart, L.; Vilquin, J.T.; Marolleau, J.P.; Seymour, B.; Larghero, J.; et al. The Myoblast Autologous Grafting in Ischemic Cardiomyopathy (MAGIC) trial: First randomized placebo-controlled study of myoblast transplantation. Circulation 2008, 117, 1189–1200. [Google Scholar] [CrossRef]
- Hassan, N.; Tchao, J.; Tobita, K. Concise review: Skeletal muscle stem cells and cardiac lineage: Potential for heart repair. Stem Cells Transl. Med. 2014, 3, 183–193. [Google Scholar] [CrossRef]
- Higuchi, A.; Ku, N.J.; Tseng, Y.C.; Pan, C.H.; Li, H.F.; Kumar, S.S.; Ling, Q.D.; Chang, Y.; Alarfaj, A.A.; Munusamy, M.A.; et al. Stem cell therapies for myocardial infarction in clinical trials: Bioengineering and biomaterial aspects. Lab. Investig. 2017, 97, 1167–1179. [Google Scholar] [CrossRef]
- Hirsch, A.; Nijveldt, R.; van der Vleuten, P.A.; Tijssen, J.G.; van der Giessen, W.J.; Tio, R.A.; Waltenberger, J.; ten Berg, J.M.; Doevendans, P.A.; Aengevaeren, W.R.; et al. Intracoronary infusion of mononuclear cells from bone marrow or peripheral blood compared with standard therapy in patients after acute myocardial infarction treated by primary percutaneous coronary intervention: Results of the randomized controlled HEBE trial. Eur. Heart J. 2011, 32, 1736–1747. [Google Scholar] [CrossRef]
- Tendera, M.; Wojakowski, W.; Ruzyłło, W.; Chojnowska, L.; Kepka, C.; Tracz, W.; Musiałek, P.; Piwowarska, W.; Nessler, J.; Buszman, P.; et al. Intracoronary infusion of bone marrow-derived selected CD34+CXCR4+ cells and non-selected mononuclear cells in patients with acute STEMI and reduced left ventricular ejection fraction: Results of randomized, multicentre Myocardial Regeneration by Intracoronary Infusion of Selected Population of Stem Cells in Acute Myocardial Infarction (REGENT) Trial. Eur. Heart J. 2009, 30, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Hare, J.M.; Traverse, J.H.; Henry, T.D.; Dib, N.; Strumpf, R.K.; Schulman, S.P.; Gerstenblith, G.; DeMaria, A.N.; Denktas, A.E.; Gammon, R.S.; et al. A randomized, double-blind, placebo-controlled, dose-escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. J. Am. Coll. Cardiol. 2009, 54, 2277–2286. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.R.; Chen, Y.; Zhang, N.K.; Yang, X.L.; Liu, H.L.; Wang, Z.G.; Yan, X.Y.; Wang, Y.; Zhu, Z.M.; Li, T.C.; et al. Intracoronary infusion of Wharton’s jelly-derived mesenchymal stem cells in acute myocardial infarction: Double-blind, randomized controlled trial. BMC Med. 2015, 13, 162. [Google Scholar] [CrossRef] [PubMed]
- Eschenhagen, T.; Bolli, R.; Braun, T.; Field, L.J.; Fleischmann, B.K.; Frisén, J.; Giacca, M.; Hare, J.M.; Houser, S.; Lee, R.T.; et al. Cardiomyocyte Regeneration: A Consensus Statement. Circulation 2017, 136, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Pagano, F.; Picchio, V.; Angelini, F.; Iaccarino, A.; Peruzzi, M.; Cavarretta, E.; Biondi-Zoccai, G.; Sciarretta, S.; De Falco, E.; Chimenti, I.; et al. The Biological Mechanisms of Action of Cardiac Progenitor Cell Therapy. Curr. Cardiol. Rep. 2018, 20, 84. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R.; Mitrani, R.D.; Hare, J.M.; Pepine, C.J.; Perin, E.C.; Willerson, J.T.; Traverse, J.H.; Henry, T.D.; Yang, P.C.; Murphy, M.P.; et al. A Phase II study of autologous mesenchymal stromal cells and c-kit positive cardiac cells, alone or in combination, in patients with ischaemic heart failure: The CCTRN CONCERT-HF trial. Eur. J. Heart Fail. 2021, 23, 661–674. [Google Scholar] [CrossRef]
- Williams, A.R.; Hatzistergos, K.E.; Addicott, B.; McCall, F.; Carvalho, D.; Suncion, V.; Morales, A.R.; Da Silva, J.; Sussman, M.A.; Heldman, A.W.; et al. Enhanced effect of combining human cardiac stem cells and bone marrow mesenchymal stem cells to reduce infarct size and to restore cardiac function after myocardial infarction. Circulation 2013, 127, 213–223. [Google Scholar] [CrossRef]
- Monsanto, M.M.; Wang, B.J.; Ehrenberg, Z.R.; Echeagaray, O.; White, K.S.; Alvarez, R., Jr.; Fisher, K.; Sengphanith, S.; Muliono, A.; Gude, N.A.; et al. Enhancing myocardial repair with CardioClusters. Nat. Commun. 2020, 11, 3955. [Google Scholar] [CrossRef]
- Chimenti, I.; Smith, R.R.; Li, T.S.; Gerstenblith, G.; Messina, E.; Giacomello, A.; Marbán, E. Relative roles of direct regeneration versus paracrine effects of human cardiosphere-derived cells transplanted into infarcted mice. Circ. Res. 2010, 106, 971–980. [Google Scholar] [CrossRef]
- Mazo, M.; Hernández, S.; Gavira, J.J.; Abizanda, G.; Araña, M.; López-Martínez, T.; Moreno, C.; Merino, J.; Martino-Rodríguez, A.; Uixeira, A.; et al. Treatment of reperfused ischemia with adipose-derived stem cells in a preclinical Swine model of myocardial infarction. Cell Transplant. 2012, 21, 2723–2733. [Google Scholar] [CrossRef]
- Zhao, J.; Li, X.; Hu, J.; Chen, F.; Qiao, S.; Sun, X.; Gao, L.; Xie, J.; Xu, B. Mesenchymal stromal cell-derived exosomes attenuate myocardial ischaemia-reperfusion injury through miR-182-regulated macrophage polarization. Cardiovasc. Res. 2019, 115, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Lin, C.; Guo, Y.; Chen, Y.; Du, Y.; Lau, W.B.; Xia, Y.; Zhang, F.; Su, R.; Gao, E.; et al. N-Cadherin Overexpression Mobilizes the Protective Effects of Mesenchymal Stromal Cells Against Ischemic Heart Injury Through a β-Catenin-Dependent Manner. Circ. Res. 2020, 126, 857–874. [Google Scholar] [CrossRef]
- Yue, Y.; Wang, C.; Benedict, C.; Huang, G.; Truongcao, M.; Roy, R.; Cimini, M.; Garikipati, V.N.S.; Cheng, Z.; Koch, W.J.; et al. Interleukin-10 Deficiency Alters Endothelial Progenitor Cell-Derived Exosome Reparative Effect on Myocardial Repair via Integrin-Linked Kinase Enrichment. Circ. Res. 2020, 126, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.M.; Yellon, D.M.; Sivaraman, V.; Das, D.; Boi-Doku, C.; Arjun, S.; Zheng, Y.; Riquelme, J.A.; Kearney, J.; Sharma, V.; et al. Plasma exosomes protect the myocardium from ischemia-reperfusion injury. J. Am. Coll. Cardiol. 2015, 65, 1525–1536. [Google Scholar] [CrossRef]
- Dai, Y.; Wang, S.; Chang, S.; Ren, D.; Shali, S.; Li, C.; Yang, H.; Huang, Z.; Ge, J. M2 macrophage-derived exosomes carry microRNA-148a to alleviate myocardial ischemia/reperfusion injury via inhibiting TXNIP and the TLR4/NF-κB/NLRP3 inflammasome signaling pathway. J. Mol. Cell. Cardiol. 2020, 142, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Wang, K.; Xu, Y.; Hu, H.; Zhang, N.; Wang, Y.; Zhong, Z.; Zhao, J.; Li, Q.; Zhu, D.; et al. Transplanted Mesenchymal Stem Cells Reduce Autophagic Flux in Infarcted Hearts via the Exosomal Transfer of miR-125b. Circ. Res. 2018, 123, 564–578. [Google Scholar] [CrossRef]
- de Couto, G.; Gallet, R.; Cambier, L.; Jaghatspanyan, E.; Makkar, N.; Dawkins, J.F.; Berman, B.P.; Marbán, E. Exosomal MicroRNA Transfer Into Macrophages Mediates Cellular Postconditioning. Circulation 2017, 136, 200–214. [Google Scholar] [CrossRef]
- Schena, G.J.; Murray, E.K.; Hildebrand, A.N.; Headrick, A.L.; Yang, Y.; Koch, K.A.; Kubo, H.; Eaton, D.; Johnson, J.; Berretta, R.; et al. Cortical bone stem cell-derived exosomes’ therapeutic effect on myocardial ischemia-reperfusion and cardiac remodeling. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H1014–H1029. [Google Scholar] [CrossRef]
- Khan, M.; Nickoloff, E.; Abramova, T.; Johnson, J.; Verma, S.K.; Krishnamurthy, P.; Mackie, A.R.; Vaughan, E.; Garikipati, V.N.; Benedict, C.; et al. Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ. Res. 2015, 117, 52–64. [Google Scholar] [CrossRef]
- Gao, L.; Wang, L.; Wei, Y.; Krishnamurthy, P.; Walcott, G.P.; Menasché, P.; Zhang, J. Exosomes secreted by hiPSC-derived cardiac cells improve recovery from myocardial infarction in swine. Sci. Transl. Med. 2020, 12, eaay1318. [Google Scholar] [CrossRef]
- Menasché, P.; Vanneaux, V.; Hagège, A.; Bel, A.; Cholley, B.; Parouchev, A.; Cacciapuoti, I.; Al-Daccak, R.; Benhamouda, N.; Blons, H.; et al. Transplantation of Human Embryonic Stem Cell-Derived Cardiovascular Progenitors for Severe Ischemic Left Ventricular Dysfunction. J. Am. Coll. Cardiol. 2018, 71, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Schirone, L.; Sadoshima, J. Editorial commentary: How to implement research studies on extracellular vesicle administration in myocardial infarction. Trends Cardiovasc. Med. 2021, 31, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, M.; Miyagawa, S.; Miki, K.; Saito, A.; Fukushima, S.; Higuchi, T.; Kawamura, T.; Kuratani, T.; Daimon, T.; Shimizu, T.; et al. Feasibility, safety, and therapeutic efficacy of human induced pluripotent stem cell-derived cardiomyocyte sheets in a porcine ischemic cardiomyopathy model. Circulation 2012, 126, S29–S37. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Gao, L.; Gregorich, Z.R.; Zhu, W.; Mattapally, S.; Oduk, Y.; Lou, X.; Kannappan, R.; Borovjagin, A.V.; Walcott, G.P.; Pollard, A.E.; et al. Large Cardiac Muscle Patches Engineered From Human Induced-Pluripotent Stem Cell-Derived Cardiac Cells Improve Recovery From Myocardial Infarction in Swine. Circulation 2018, 137, 1712–1730. [Google Scholar] [CrossRef]
- Kawamura, M.; Miyagawa, S.; Fukushima, S.; Saito, A.; Miki, K.; Funakoshi, S.; Yoshida, Y.; Yamanaka, S.; Shimizu, T.; Okano, T.; et al. Enhanced Therapeutic Effects of Human iPS Cell Derived-Cardiomyocyte by Combined Cell-Sheets with Omental Flap Technique in Porcine Ischemic Cardiomyopathy Model. Sci. Rep. 2017, 7, 8824. [Google Scholar] [CrossRef]
- Kawamura, M.; Miyagawa, S.; Fukushima, S.; Saito, A.; Miki, K.; Ito, E.; Sougawa, N.; Kawamura, T.; Daimon, T.; Shimizu, T.; et al. Enhanced survival of transplanted human induced pluripotent stem cell-derived cardiomyocytes by the combination of cell sheets with the pedicled omental flap technique in a porcine heart. Circulation 2013, 128, S87–S94. [Google Scholar] [CrossRef]
- Masumoto, H.; Ikuno, T.; Takeda, M.; Fukushima, H.; Marui, A.; Katayama, S.; Shimizu, T.; Ikeda, T.; Okano, T.; Sakata, R.; et al. Human iPS cell-engineered cardiac tissue sheets with cardiomyocytes and vascular cells for cardiac regeneration. Sci. Rep. 2014, 4, 6716. [Google Scholar] [CrossRef]
- Luo, L.; Tang, J.; Nishi, K.; Yan, C.; Dinh, P.U.; Cores, J.; Kudo, T.; Zhang, J.; Li, T.S.; Cheng, K. Fabrication of Synthetic Mesenchymal Stem Cells for the Treatment of Acute Myocardial Infarction in Mice. Circ. Res. 2017, 120, 1768–1775. [Google Scholar] [CrossRef]
- Hida, N.; Nishiyama, N.; Miyoshi, S.; Kira, S.; Segawa, K.; Uyama, T.; Mori, T.; Miyado, K.; Ikegami, Y.; Cui, C.; et al. Novel cardiac precursor-like cells from human menstrual blood-derived mesenchymal cells. Stem Cells 2008, 26, 1695–1704. [Google Scholar] [CrossRef]
- Chachques, J.C.; Lila, N.; Soler-Botija, C.; Martinez-Ramos, C.; Valles, A.; Autret, G.; Perier, M.C.; Mirochnik, N.; Monleon-Pradas, M.; Bayes-Genis, A.; et al. Elastomeric cardiopatch scaffold for myocardial repair and ventricular support. Eur. J. Cardio-Thorac. Surg. 2020, 57, 545–555. [Google Scholar] [CrossRef]
- Hamdi, H.; Planat-Benard, V.; Bel, A.; Puymirat, E.; Geha, R.; Pidial, L.; Nematalla, H.; Bellamy, V.; Bouaziz, P.; Peyrard, S.; et al. Epicardial adipose stem cell sheets results in greater post-infarction survival than intramyocardial injections. Cardiovasc. Res. 2011, 91, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, Y.; Miyagawa, S.; Maeda, N.; Fukushima, S.; Kitagawa-Sakakida, S.; Daimon, T.; Hirata, A.; Shimizu, T.; Okano, T.; Shimomura, I.; et al. Induced adipocyte cell-sheet ameliorates cardiac dysfunction in a mouse myocardial infarction model: A novel drug delivery system for heart failure. Circulation 2011, 124, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Joo, H.J.; Kim, M.; Choi, S.C.; Lee, J.I.; Hong, S.J.; Lim, D.S. Transplantation of Adipose-Derived Stem Cell Sheet Attenuates Adverse Cardiac Remodeling in Acute Myocardial Infarction. Tissue Eng. Part A 2017, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chachques, J.C.; Trainini, J.C.; Lago, N.; Cortes-Morichetti, M.; Schussler, O.; Carpentier, A. Myocardial Assistance by Grafting a New Bioartificial Upgraded Myocardium (MAGNUM trial): Clinical feasibility study. Ann. Thorac. Surg. 2008, 85, 901–908. [Google Scholar] [CrossRef]
- Itabashi, Y.; Miyoshi, S.; Yuasa, S.; Fujita, J.; Shimizu, T.; Okano, T.; Fukuda, K.; Ogawa, S. Analysis of the electrophysiological properties and arrhythmias in directly contacted skeletal and cardiac muscle cell sheets. Cardiovasc. Res. 2005, 67, 561–570. [Google Scholar] [CrossRef][Green Version]
- Roberts, E.G.; Piekarski, B.L.; Huang, K.; Emani, S.; Wong, J.Y.; Emani, S.M. Evaluation of Placental Mesenchymal Stem Cell Sheets for Myocardial Repair and Regeneration. Tissue Eng. Part A 2019, 25, 867–877. [Google Scholar] [CrossRef]
- Siltanen, A.; Kitabayashi, K.; Lakkisto, P.; Mäkelä, J.; Pätilä, T.; Ono, M.; Tikkanen, I.; Sawa, Y.; Kankuri, E.; Harjula, A. hHGF overexpression in myoblast sheets enhances their angiogenic potential in rat chronic heart failure. PLoS ONE 2011, 6, e19161. [Google Scholar] [CrossRef]
- Kirby, G.T.S.; Michelmore, A.; Smith, L.E.; Whittle, J.D.; Short, R.D. Cell sheets in cell therapies. Cytotherapy 2018, 20, 169–180. [Google Scholar] [CrossRef]
- Baraniak, P.R.; McDevitt, T.C. Stem cell paracrine actions and tissue regeneration. Regen. Med. 2010, 5, 121–143. [Google Scholar] [CrossRef]
- Guo, R.; Morimatsu, M.; Feng, T.; Lan, F.; Chang, D.; Wan, F.; Ling, Y. Stem cell-derived cell sheet transplantation for heart tissue repair in myocardial infarction. Stem Cell Res. Ther. 2020, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Masumoto, H.; Matsuo, T.; Yamamizu, K.; Uosaki, H.; Narazaki, G.; Katayama, S.; Marui, A.; Shimizu, T.; Ikeda, T.; Okano, T.; et al. Pluripotent stem cell-engineered cell sheets reassembled with defined cardiovascular populations ameliorate reduction in infarct heart function through cardiomyocyte-mediated neovascularization. Stem Cells 2012, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kim, R.Y.; Park, B.W.; Lee, S.; Choi, S.W.; Park, J.H.; Choi, J.J.; Kim, S.W.; Jang, J.; Cho, D.W.; et al. Dual stem cell therapy synergistically improves cardiac function and vascular regeneration following myocardial infarction. Nat. Commun. 2019, 10, 3123. [Google Scholar] [CrossRef] [PubMed]
- Hosoyama, T.; Samura, M.; Kudo, T.; Nishimoto, A.; Ueno, K.; Murata, T.; Ohama, T.; Sato, K.; Mikamo, A.; Yoshimura, K.; et al. Cardiosphere-derived cell sheet primed with hypoxia improves left ventricular function of chronically infarcted heart. Am. J. Transl. Res. 2015, 7, 2738–2751. [Google Scholar]
- Yamamoto, K.; Kurata, Y.; Inoue, Y.; Adachi, M.; Tsuneto, M.; Miake, J.; Ogino, K.; Ninomiya, H.; Yoshida, A.; Shirayoshi, Y.; et al. Pretreatment with an angiotensin II receptor blocker abolished ameliorating actions of adipose-derived stem cell sheets on cardiac dysfunction and remodeling after myocardial infarction. Regen. Ther. 2018, 9, 79–88. [Google Scholar] [CrossRef]
- Cortes-Morichetti, M.; Frati, G.; Schussler, O.; Duong Van Huyen, J.P.; Lauret, E.; Genovese, J.A.; Carpentier, A.F.; Chachques, J.C. Association between a cell-seeded collagen matrix and cellular cardiomyoplasty for myocardial support and regeneration. Tissue Eng. 2007, 13, 2681–2687. [Google Scholar] [CrossRef]
- Huang, K.; Ozpinar, E.W.; Su, T.; Tang, J.; Shen, D.; Qiao, L.; Hu, S.; Li, Z.; Liang, H.; Mathews, K.; et al. An off-the-shelf artificial cardiac patch improves cardiac repair after myocardial infarction in rats and pigs. Sci. Transl. Med. 2020, 12, eaat9683. [Google Scholar] [CrossRef]
- Lin, X.; Liu, Y.; Bai, A.; Cai, H.; Bai, Y.; Jiang, W.; Yang, H.; Wang, X.; Yang, L.; Sun, N.; et al. A viscoelastic adhesive epicardial patch for treating myocardial infarction. Nat. Biomed. Eng. 2019, 3, 632–643. [Google Scholar] [CrossRef]
- Gaetani, R.; Feyen, D.A.; Verhage, V.; Slaats, R.; Messina, E.; Christman, K.L.; Giacomello, A.; Doevendans, P.A.; Sluijter, J.P. Epicardial application of cardiac progenitor cells in a 3D-printed gelatin/hyaluronic acid patch preserves cardiac function after myocardial infarction. Biomaterials 2015, 61, 339–348. [Google Scholar] [CrossRef]
- Maiullari, F.; Costantini, M.; Milan, M.; Pace, V.; Chirivì, M.; Maiullari, S.; Rainer, A.; Baci, D.; Marei, H.E.; Seliktar, D.; et al. A multi-cellular 3D bioprinting approach for vascularized heart tissue engineering based on HUVECs and iPSC-derived cardiomyocytes. Sci. Rep. 2018, 8, 13532. [Google Scholar] [CrossRef]
- Qiu, T.; Jiang, W.; Yan, P.; Jiao, L.; Wang, X. Development of 3D-Printed Sulfated Chitosan Modified Bioresorbable Stents for Coronary Artery Disease. Front. Bioeng. Biotechnol. 2020, 8, 462. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H94–H105. [Google Scholar] [CrossRef] [PubMed]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef] [PubMed]
- Pacak, C.A.; Preble, J.M.; Kondo, H.; Seibel, P.; Levitsky, S.; Del Nido, P.J.; Cowan, D.B.; McCully, J.D. Actin-dependent mitochondrial internalization in cardiomyocytes: Evidence for rescue of mitochondrial function. Biol. Open 2015, 4, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Sun, L.; Ji, S.; Zhao, T.; Zhang, W.; Xu, J.; Zhang, J.; Wang, Y.; Wang, X.; Franzini-Armstrong, C.; et al. Kissing and nanotunneling mediate intermitochondrial communication in the heart. Proc. Natl. Acad. Sci. USA 2013, 110, 2846–2851. [Google Scholar] [CrossRef]
- Hayashida, K.; Takegawa, R.; Shoaib, M.; Aoki, T.; Choudhary, R.C.; Kuschner, C.E.; Nishikimi, M.; Miyara, S.J.; Rolston, D.M.; Guevara, S.; et al. Mitochondrial transplantation therapy for ischemia reperfusion injury: A systematic review of animal and human studies. J. Transl. Med. 2021, 19, 214. [Google Scholar] [CrossRef]
- Weisz, G.; Metzger, D.C.; Caputo, R.P.; Delgado, J.A.; Marshall, J.J.; Vetrovec, G.W.; Reisman, M.; Waksman, R.; Granada, J.F.; Novack, V.; et al. Safety and feasibility of robotic percutaneous coronary intervention: PRECISE (Percutaneous Robotically-Enhanced Coronary Intervention) Study. J. Am. Coll. Cardiol. 2013, 61, 1596–1600. [Google Scholar] [CrossRef]
- Dogan, S.; Aybek, T.; Andressen, E.; Byhahn, C.; Mierdl, S.; Westphal, K.; Matheis, G.; Moritz, A.; Wimmer-Greinecker, G. Totally endoscopic coronary artery bypass grafting on cardiopulmonary bypass with robotically enhanced telemanipulation: Report of forty-five cases. J. Thorac. Cardiovasc. Surg. 2002, 123, 1125–1131. [Google Scholar] [CrossRef]
- Bonatti, J.; Vetrovec, G.; Riga, C.; Wazni, O.; Stadler, P. Robotic technology in cardiovascular medicine. Nat. Rev. Cardiol. 2014, 11, 266–275. [Google Scholar] [CrossRef]
- Mihaljevic, T.; Jarrett, C.M.; Gillinov, A.M.; Williams, S.J.; DeVilliers, P.A.; Stewart, W.J.; Svensson, L.G.; Sabik, J.F., 3rd; Blackstone, E.H. Robotic repair of posterior mitral valve prolapse versus conventional approaches: Potential realized. J. Thorac. Cardiovasc. Surg. 2011, 141, 72–80.e4. [Google Scholar] [CrossRef]
- Gaudino, M.; Bakaeen, F.; Davierwala, P.; Di Franco, A.; Fremes, S.E.; Patel, N.; Puskas, J.D.; Ruel, M.; Torregrossa, G.; Vallely, M.; et al. New Strategies for Surgical Myocardial Revascularization. Circulation 2018, 138, 2160–2168. [Google Scholar] [CrossRef] [PubMed]
- Winter, P.M.; Neubauer, A.M.; Caruthers, S.D.; Harris, T.D.; Robertson, J.D.; Williams, T.A.; Schmieder, A.H.; Hu, G.; Allen, J.S.; Lacy, E.K.; et al. Endothelial alpha(v)beta3 integrin-targeted fumagillin nanoparticles inhibit angiogenesis in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2103–2109. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.R., Jr.; Abizaid, A.; Costa, R.; Feres, F.; Tanajura, L.F.; Abizaid, A.; Maldonado, G.; Staico, R.; Siqueira, D.; Sousa, A.G.; et al. 1-year results of the hydroxyapatite polymer-free sirolimus-eluting stent for the treatment of single de novo coronary lesions: The VESTASYNC I trial. JACC Cardiovasc. Interv. 2009, 2, 422–427. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, W.; Zhang, J.; Sun, W.; Zhang, R.; Gu, H. Fabrication of a novel polymer-free nanostructured drug-eluting coating for cardiovascular stents. ACS Appl. Mater. Interfaces 2013, 5, 10337–10345. [Google Scholar] [CrossRef] [PubMed]
- Acharya, G.; Lee, C.H.; Lee, Y. Optimization of cardiovascular stent against restenosis: Factorial design-based statistical analysis of polymer coating conditions. PLoS ONE 2012, 7, e43100. [Google Scholar] [CrossRef] [PubMed]
- Danenberg, H.D.; Golomb, G.; Groothuis, A.; Gao, J.; Epstein, H.; Swaminathan, R.V.; Seifert, P.; Edelman, E.R. Liposomal alendronate inhibits systemic innate immunity and reduces in-stent neointimal hyperplasia in rabbits. Circulation 2003, 108, 2798–2804. [Google Scholar] [CrossRef]
- Yin, R.X.; Yang, D.Z.; Wu, J.Z. Nanoparticle drug- and gene-eluting stents for the prevention and treatment of coronary restenosis. Theranostics 2014, 4, 175–200. [Google Scholar] [CrossRef]
- Bae, S.; Park, M.; Kang, C.; Dilmen, S.; Kang, T.H.; Kang, D.G.; Ke, Q.; Lee, S.U.; Lee, D.; Kang, P.M. Hydrogen Peroxide-Responsive Nanoparticle Reduces Myocardial Ischemia/Reperfusion Injury. J. Am. Heart Assoc. 2016, 5, e003697. [Google Scholar] [CrossRef]
- Lee, D.; Bae, S.; Hong, D.; Lim, H.; Yoon, J.H.; Hwang, O.; Park, S.; Ke, Q.; Khang, G.; Kang, P.M. H2O2-responsive molecularly engineered polymer nanoparticles as ischemia/reperfusion-targeted nanotherapeutic agents. Sci. Rep. 2013, 3, 2233. [Google Scholar] [CrossRef]
- Tashakori-Miyanroudi, M.; Rakhshan, K.; Ramez, M.; Asgarian, S.; Janzadeh, A.; Azizi, Y.; Seifalian, A.; Ramezani, F. Conductive carbon nanofibers incorporated into collagen bio-scaffold assists myocardial injury repair. Int. J. Biol. Macromol. 2020, 163, 1136–1146. [Google Scholar] [CrossRef]
- Nagase, K.; Nagumo, Y.; Kim, M.; Kim, H.J.; Kyung, H.W.; Chung, H.J.; Sekine, H.; Shimizu, T.; Kanazawa, H.; Okano, T.; et al. Local Release of VEGF Using Fiber Mats Enables Effective Transplantation of Layered Cardiomyocyte Sheets. Macromol. Biosci. 2017, 17, 1700073. [Google Scholar] [CrossRef]
- Malhotra, P.; Shahdadpuri, N. Nano-robotic based Thrombolysis: Dissolving Blood Clots using Nanobots. arXiv 2020, arXiv:2011.03534. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schirone, L.; Forte, M.; D’Ambrosio, L.; Valenti, V.; Vecchio, D.; Schiavon, S.; Spinosa, G.; Sarto, G.; Petrozza, V.; Frati, G.; et al. An Overview of the Molecular Mechanisms Associated with Myocardial Ischemic Injury: State of the Art and Translational Perspectives. Cells 2022, 11, 1165. https://doi.org/10.3390/cells11071165
Schirone L, Forte M, D’Ambrosio L, Valenti V, Vecchio D, Schiavon S, Spinosa G, Sarto G, Petrozza V, Frati G, et al. An Overview of the Molecular Mechanisms Associated with Myocardial Ischemic Injury: State of the Art and Translational Perspectives. Cells. 2022; 11(7):1165. https://doi.org/10.3390/cells11071165
Chicago/Turabian StyleSchirone, Leonardo, Maurizio Forte, Luca D’Ambrosio, Valentina Valenti, Daniele Vecchio, Sonia Schiavon, Giulia Spinosa, Gianmarco Sarto, Vincenzo Petrozza, Giacomo Frati, and et al. 2022. "An Overview of the Molecular Mechanisms Associated with Myocardial Ischemic Injury: State of the Art and Translational Perspectives" Cells 11, no. 7: 1165. https://doi.org/10.3390/cells11071165
APA StyleSchirone, L., Forte, M., D’Ambrosio, L., Valenti, V., Vecchio, D., Schiavon, S., Spinosa, G., Sarto, G., Petrozza, V., Frati, G., & Sciarretta, S. (2022). An Overview of the Molecular Mechanisms Associated with Myocardial Ischemic Injury: State of the Art and Translational Perspectives. Cells, 11(7), 1165. https://doi.org/10.3390/cells11071165