
Specific NLRP3 Inflammasome Assembling and Regulation in Neutrophils: Relevance in Inflammatory and Infectious Diseases


Abstract
:1. The NLRP3 Inflammasome
2. Neutrophils: From Phagocytosis Sentinel to Immunoregulation Commander
3. Neutrophilic NLRP3 Inflammasome: A Novel Relevant Regulatory Pathway
3.1. Emerging Shreds of Evidence
3.2. Activation Mechanisms
3.2.1. Canonical Pathway
Priming Step
Activation Step
3.2.2. Non-Canonical and Alternative Pathways
3.3. Does NLRP3 Inflammasome-Mediated Cell Death Occur in Neutrophils?
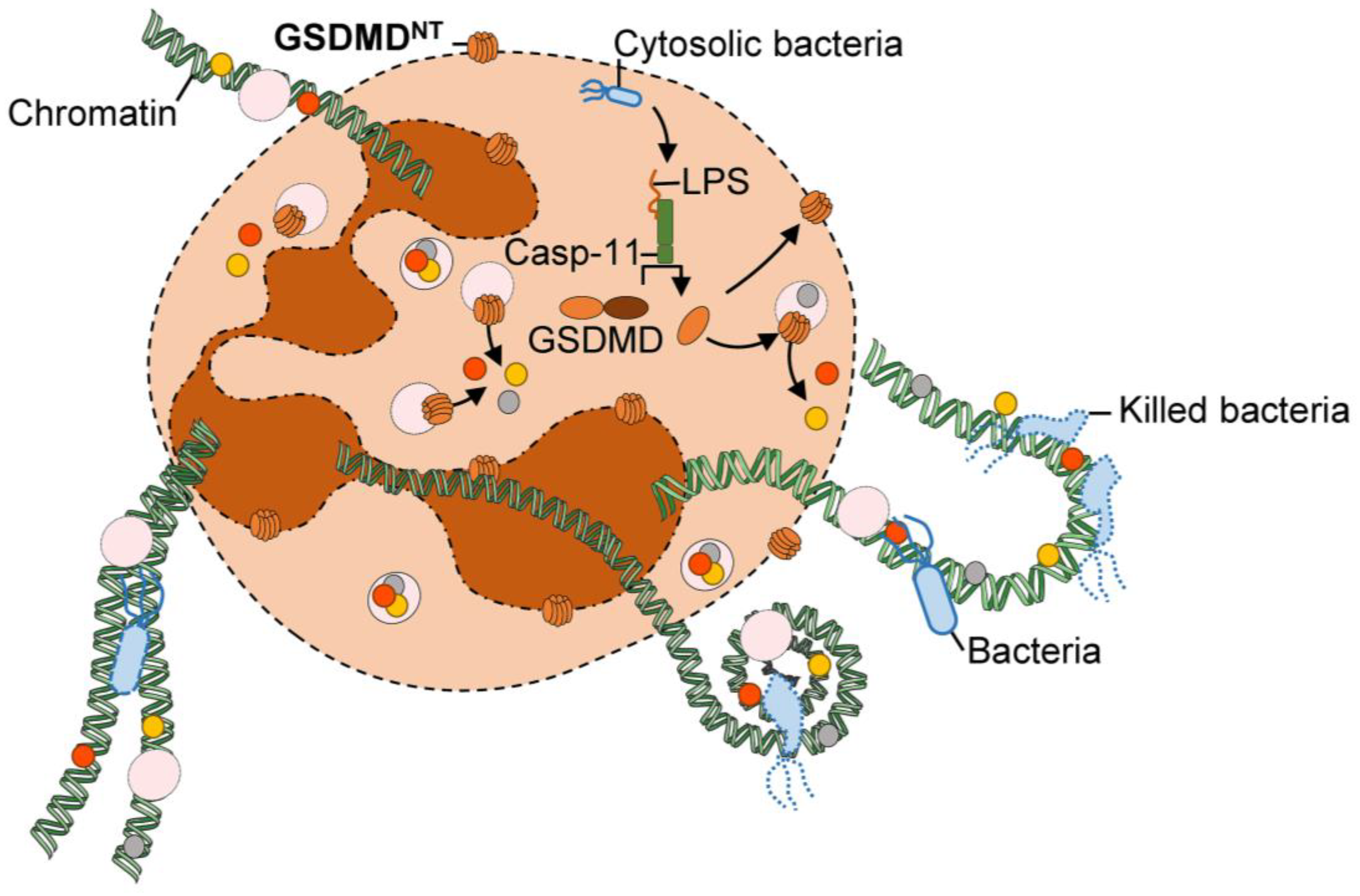
3.4. Relevant Functions of the NLRP3 Inflammasome Activation by Neutrophils in Diseases
3.4.1. (Auto)-Inflammation
3.4.2. Infections
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riera Romo, M.; Pérez-Martínez, D.; Castillo Ferrer, C. Innate Immunity in Vertebrates: An Overview. Immunology 2016, 148, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Briard, B.; Place, D.E.; Kanneganti, T.-D. DNA Sensing in the Innate Immune Response. Physiology 2020, 35, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.-D. Intracellular Innate Immune Receptors: Life inside the Cell. Immunol. Rev. 2020, 297, 5–12. [Google Scholar] [CrossRef]
- Xue, Y.; Enosi Tuipulotu, D.; Tan, W.H.; Kay, C.; Man, S.M. Emerging Activators and Regulators of Inflammasomes and Pyroptosis. Trends Immunol. 2019, 40, 1035–1052. [Google Scholar] [CrossRef]
- Kanneganti, T.-D. The Inflammasome: Firing up Innate Immunity. Immunol. Rev. 2015, 265, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Man, S.M.; Kanneganti, T.-D. Converging Roles of Caspases in Inflammasome Activation, Cell Death and Innate Immunity. Nat. Rev. Immunol. 2016, 16, 7–21. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Christgen, S.; Place, D.E.; Kanneganti, T.-D. Toward Targeting Inflammasomes: Insights into Their Regulation and Activation. Cell Res. 2020, 30, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; von Moltke, J.; Jones, J.W.; Vance, R.E.; Monack, D.M. Differential Requirement for Caspase-1 Autoproteolysis in Pathogen-Induced Cell Death and Cytokine Processing. Cell Host Microbe 2010, 8, 471–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Yang, J.; Shi, J.; Gong, Y.-N.; Lu, Q.; Xu, H.; Liu, L.; Shao, F. The NLRC4 Inflammasome Receptors for Bacterial Flagellin and Type III Secretion Apparatus. Nature 2011, 477, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Van Opdenbosch, N.; Gurung, P.; Vande Walle, L.; Fossoul, A.; Kanneganti, T.-D.; Lamkanfi, M. Activation of the NLRP1b Inflammasome Independently of ASC-Mediated Caspase-1 Autoproteolysis and Speck Formation. Nat. Commun. 2014, 5, 3209. [Google Scholar] [CrossRef] [Green Version]
- Chavarría-Smith, J.; Vance, R.E. Direct Proteolytic Cleavage of NLRP1B Is Necessary and Sufficient for Inflammasome Activation by Anthrax Lethal Factor. PLoS Pathog. 2013, 9, e1003452. [Google Scholar] [CrossRef] [Green Version]
- Levinsohn, J.L.; Newman, Z.L.; Hellmich, K.A.; Fattah, R.; Getz, M.A.; Liu, S.; Sastalla, I.; Leppla, S.H.; Moayeri, M. Anthrax Lethal Factor Cleavage of Nlrp1 Is Required for Activation of the Inflammasome. PLoS Pathog. 2012, 8, e1002638. [Google Scholar] [CrossRef]
- Sandstrom, A.; Mitchell, P.S.; Goers, L.; Mu, E.W.; Lesser, C.F.; Vance, R.E. Functional Degradation: A Mechanism of NLRP1 Inflammasome Activation by Diverse Pathogen Enzymes. Science 2019, 364, eaau1330. [Google Scholar] [CrossRef]
- Franchi, L.; Amer, A.; Body-Malapel, M.; Kanneganti, T.-D.; Özören, N.; Jagirdar, R.; Inohara, N.; Vandenabeele, P.; Bertin, J.; Coyle, A.; et al. Cytosolic Flagellin Requires Ipaf for Activation of Caspase-1 and Interleukin 1β in Salmonella-Infected Macrophages. Nat. Immunol. 2006, 7, 576–582. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Yu, J.-W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 Activates the Inflammasome and Cell Death in Response to Cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.-N.; Peng, X.; Xi, J.J.; Chen, S.; et al. Innate Immune Sensing of Bacterial Modifications of Rho GTPases by the Pyrin Inflammasome. Nature 2014, 513, 237–241. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.-D. NLRP3 Inflammasome in Cancer and Metabolic Diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, J.; Blander, J.M. Increasing Complexity of NLRP3 Inflammasome Regulation. J. Leukoc. Biol. 2021, 109, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Zhao, Y.; Shao, F. Innate Immunity to Intracellular LPS. Nat. Immunol. 2019, 20, 527–533. [Google Scholar] [CrossRef]
- Briard, B.; Fontaine, T.; Samir, P.; Place, D.E.; Muszkieta, L.; Malireddi, R.K.S.; Karki, R.; Christgen, S.; Bomme, P.; Vogel, P.; et al. Galactosaminogalactan Activates the Inflammasome to Provide Host Protection. Nature 2020, 588, 688–692. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory Caspases Are Innate Immune Receptors for Intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Chen, K.W.; Demarco, B.; Broz, P. Beyond Inflammasomes: Emerging Function of Gasdermins during Apoptosis and NETosis. EMBO J. 2020, 39, e103397. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C.W. Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Yipp, B.G.; Kim, J.H.; Lima, R.; Zbytnuik, L.D.; Petri, B.; Swanlund, N.; Ho, M.; Szeto, V.G.; Tak, T.; Koenderman, L.; et al. The Lung Is a Host Defense Niche for Immediate Neutrophil-Mediated Vascular Protection. Sci. Immunol. 2017, 2, eaam8929. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Hodgson, G.; Katz, M.; Dunn, A.R. Evaluation of Role of G-CSF in the Production, Survival, and Release of Neutrophils from Bone Marrow into Circulation. Blood 2002, 100, 854–861. [Google Scholar] [CrossRef] [Green Version]
- Pillay, J.; den Braber, I.; Vrisekoop, N.; Kwast, L.M.; de Boer, R.J.; Borghans, J.A.M.; Tesselaar, K.; Koenderman, L. In Vivo Labeling with 2H2O Reveals a Human Neutrophil Lifespan of 5.4 Days. Blood 2010, 116, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Borregaard, N.; Wynn, T.A. Phenotypic and Functional Plasticity of Cells of Innate Immunity: Macrophages, Mast Cells and Neutrophils. Nat. Immunol. 2011, 12, 1035–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summers, C.; Rankin, S.M.; Condliffe, A.M.; Singh, N.; Peters, A.M.; Chilvers, E.R. Neutrophil Kinetics in Health and Disease. Trends Immunol. 2010, 31, 318–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colotta, F.; Re, F.; Polentarutti, N.; Sozzani, S.; Mantovani, A. Modulation of Granulocyte Survival and Programmed Cell Death by Cytokines and Bacterial Products. Blood 1992, 80, 2012–2020. [Google Scholar] [CrossRef] [Green Version]
- Silvestre-Roig, C.; Hidalgo, A.; Soehnlein, O. Neutrophil Heterogeneity: Implications for Homeostasis and Pathogenesis. Blood 2016, 127, 2173–2181. [Google Scholar] [CrossRef] [Green Version]
- Deniset, J.F.; Surewaard, B.G.; Lee, W.-Y.; Kubes, P. Splenic Ly6Ghigh Mature and Ly6Gint Immature Neutrophils Contribute to Eradication of S. Pneumoniae. J. Exp. Med. 2017, 214, 1333–1350. [Google Scholar] [CrossRef]
- Segal, A.W. How Neutrophils Kill Microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef] [Green Version]
- Buvelot, H.; Posfay-Barbe, K.M.; Linder, P.; Schrenzel, J.; Krause, K.-H. Staphylococcus Aureus, Phagocyte NADPH Oxidase and Chronic Granulomatous Disease. FEMS Microbiol. Rev. 2017, 41, 139–157. [Google Scholar] [CrossRef] [Green Version]
- Song, E.; Jaishankar, G.B.; Saleh, H.; Jithpratuck, W.; Sahni, R.; Krishnaswamy, G. Chronic Granulomatous Disease: A Review of the Infectious and Inflammatory Complications. Clin. Mol. Allergy 2011, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Kutter, D.; Devaquet, P.; Vanderstocken, G.; Paulus, J.M.; Marchal, V.; Gothot, A. Consequences of Total and Subtotal Myeloperoxidase Deficiency: Risk or Benefit? AHA 2000, 104, 10–15. [Google Scholar] [CrossRef]
- Borregaard, N. Neutrophils, from Marrow to Microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannopoulos, V. Neutrophil Extracellular Traps in Immunity and Disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Megens, R.T.A.; Vijayan, S.; Lievens, D.; Döring, Y.; van Zandvoort, M.A.M.J.; Grommes, J.; Weber, C.; Soehnlein, O. Presence of Luminal Neutrophil Extracellular Traps in Atherosclerosis. Thromb. Haemost. 2012, 107, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs Are a Source of Citrullinated Autoantigens and Stimulate Inflammatory Responses in Rheumatoid Arthritis. Sci. Transl. Med. 2013, 5, 178ra40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting Neutrophils Are Major Inducers of Type I IFN Production in Pediatric Systemic Lupus Erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef] [Green Version]
- Radermecker, C.; Louis, R.; Bureau, F.; Marichal, T. Role of Neutrophils in Allergic Asthma. Curr. Opin. Immunol. 2018, 54, 28–34. [Google Scholar] [CrossRef]
- Radermecker, C.; Detrembleur, N.; Guiot, J.; Cavalier, E.; Henket, M.; d’Emal, C.; Vanwinge, C.; Cataldo, D.; Oury, C.; Delvenne, P.; et al. Neutrophil Extracellular Traps Infiltrate the Lung Airway, Interstitial, and Vascular Compartments in Severe COVID-19. J. Exp. Med. 2020, 217, e20201012. [Google Scholar] [CrossRef]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-Induced NETosis Is a Dynamic Process Involving Neutrophil Multitasking in Vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V.; Staab, D.; Zychlinsky, A. Neutrophil Elastase Enhances Sputum Solubilization in Cystic Fibrosis Patients Receiving DNase Therapy. PLoS ONE 2011, 6, e28526. [Google Scholar] [CrossRef]
- Moreira-Teixeira, L.; Stimpson, P.J.; Stavropoulos, E.; Hadebe, S.; Chakravarty, P.; Ioannou, M.; Aramburu, I.V.; Herbert, E.; Priestnall, S.L.; Suarez-Bonnet, A.; et al. Type I IFN Exacerbates Disease in Tuberculosis-Susceptible Mice by Inducing Neutrophil-Mediated Lung Inflammation and NETosis. Nat. Commun. 2020, 11, 5566. [Google Scholar] [CrossRef]
- de Melo, M.G.M.; Mesquita, E.D.D.; Oliveira, M.M.; da Silva-Monteiro, C.; Silveira, A.K.A.; Malaquias, T.S.; Dutra, T.C.P.; Galliez, R.M.; Kritski, A.L.; Silva, E.C.; et al. Imbalance of NET and Alpha-1-Antitrypsin in Tuberculosis Patients Is Related With Hyper Inflammation and Severe Lung Tissue Damage. Front. Immunol. 2019, 9, 3147. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular Mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218. [Google Scholar] [CrossRef] [PubMed]
- Vorobjeva, N.V.; Chernyak, B.V. NETosis: Molecular Mechanisms, Role in Physiology and Pathology. Biochem. Mosc. 2020, 85, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical Inflammasome Signaling Elicits Gasdermin D–Dependent Neutrophil Extracellular Traps. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D Plays a Vital Role in the Generation of Neutrophil Extracellular Traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [Green Version]
- Phillipson, M.; Kubes, P. The Healing Power of Neutrophils. Trends Immunol. 2019, 40, 635–647. [Google Scholar] [CrossRef]
- Puga, I.; Cols, M.; Barra, C.M.; He, B.; Cassis, L.; Gentile, M.; Comerma, L.; Chorny, A.; Shan, M.; Xu, W.; et al. B Cell–Helper Neutrophils Stimulate the Diversification and Production of Immunoglobulin in the Marginal Zone of the Spleen. Nat. Immunol. 2012, 13, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Tecchio, C.; Cassatella, M.A. Neutrophil-Derived Chemokines on the Road to Immunity. Semin. Immunol. 2016, 28, 119–128. [Google Scholar] [CrossRef]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the Activation and Regulation of Innate and Adaptive Immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef]
- Sagiv, J.Y.; Voels, S.; Granot, Z. Isolation and Characterization of Low- vs. High-Density Neutrophils in Cancer. Methods Mol. Biol. 2016, 1458, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, J.Y.; Michaeli, J.; Assi, S.; Mishalian, I.; Kisos, H.; Levy, L.; Damti, P.; Lumbroso, D.; Polyansky, L.; Sionov, R.V.; et al. Phenotypic Diversity and Plasticity in Circulating Neutrophil Subpopulations in Cancer. Cell Rep. 2015, 10, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.; Sagar, D.; Hanna, R.N.; Lightfoot, Y.L.; Mistry, P.; Smith, C.K.; Manna, Z.; Hasni, S.; Siegel, R.M.; Sanjuan, M.A.; et al. Low-Density Granulocytes Activate T Cells and Demonstrate a Non-Suppressive Role in Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2019, 78, 957–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandau, S.; Trellakis, S.; Bruderek, K.; Schmaltz, D.; Steller, G.; Elian, M.; Suttmann, H.; Schenck, M.; Welling, J.; Zabel, P.; et al. Myeloid-Derived Suppressor Cells in the Peripheral Blood of Cancer Patients Contain a Subset of Immature Neutrophils with Impaired Migratory Properties. J. Leukoc. Biol. 2011, 89, 311–317. [Google Scholar] [CrossRef]
- Deng, Y.; Ye, J.; Luo, Q.; Huang, Z.; Peng, Y.; Xiong, G.; Guo, Y.; Jiang, H.; Li, J. Low-Density Granulocytes Are Elevated in Mycobacterial Infection and Associated with the Severity of Tuberculosis. PLoS ONE 2016, 11, e0153567. [Google Scholar] [CrossRef]
- Rocha, B.C.; Marques, P.E.; Leoratti, F.M.D.S.; Junqueira, C.; Pereira, D.B.; Antonelli, L.R.d.V.; Menezes, G.B.; Golenbock, D.T.; Gazzinelli, R.T. Type I Interferon Transcriptional Signature in Neutrophils and Low-Density Granulocytes Are Associated with Tissue Damage in Malaria. Cell Rep. 2015, 13, 2829–2841. [Google Scholar] [CrossRef] [Green Version]
- Herteman, N.; Vargas, A.; Lavoie, J.-P. Characterization of Circulating Low-Density Neutrophils Intrinsic Properties in Healthy and Asthmatic Horses. Sci. Rep. 2017, 7, 7743. [Google Scholar] [CrossRef] [Green Version]
- Grecian, R.; Whyte, M.K.B.; Walmsley, S.R. The Role of Neutrophils in Cancer. Br. Med. Bull. 2018, 128, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Almand, B.; Clark, J.I.; Nikitina, E.; van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased Production of Immature Myeloid Cells in Cancer Patients: A Mechanism of Immunosuppression in Cancer. J. Immunol. 2001, 166, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.-I.; Collazo, M.; Shalova, I.N.; Biswas, S.K.; Gabrilovich, D.I. Characterization of the Nature of Granulocytic Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. J. Leukoc. Biol. 2012, 91, 167–181. [Google Scholar] [CrossRef] [Green Version]
- Condamine, T.; Dominguez, G.A.; Youn, J.-I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C.; et al. Lectin-Type Oxidized LDL Receptor-1 Distinguishes Population of Human Polymorphonuclear Myeloid-Derived Suppressor Cells in Cancer Patients. Sci. Immunol. 2016, 1, aaf8943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorhoi, A.; Glaría, E.; Garcia-Tellez, T.; Nieuwenhuizen, N.E.; Zelinskyy, G.; Favier, B.; Singh, A.; Ehrchen, J.; Gujer, C.; Münz, C.; et al. MDSCs in Infectious Diseases: Regulation, Roles, and Readjustment. Cancer Immunol. Immunother. 2019, 68, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.D.; McGarry Houghton, A. Tumor-Associated Neutrophils: New Targets for Cancer Therapy. Cancer Res. 2011, 71, 2411–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Albelda, S.M. Tumor-Associated Neutrophils: Friend or Foe? Carcinogenesis 2012, 33, 949–955. [Google Scholar] [CrossRef] [Green Version]
- Yajuk, O.; Baron, M.; Toker, S.; Zelter, T.; Fainsod-Levi, T.; Granot, Z. The PD-L1/PD-1 Axis Blocks Neutrophil Cytotoxicity in Cancer. Cells 2021, 10, 1510. [Google Scholar] [CrossRef]
- Thanabalasuriar, A.; Chiang, A.J.; Morehouse, C.; Camara, M.; Hawkins, S.; Keller, A.E.; Koksal, A.C.; Caceres, C.S.; Berlin, A.A.; Holoweckyj, N.; et al. PD-L1+ Neutrophils Contribute to Injury-Induced Infection Susceptibility. Sci. Adv. 2021, 7, eabd9436. [Google Scholar] [CrossRef]
- Luo, Q.; Huang, Z.; Ye, J.; Deng, Y.; Fang, L.; Li, X.; Guo, Y.; Jiang, H.; Ju, B.; Huang, Q.; et al. PD-L1-Expressing Neutrophils as a Novel Indicator to Assess Disease Activity and Severity of Systemic Lupus Erythematosus. Arthritis Res. Ther. 2016, 18, 47. [Google Scholar] [CrossRef] [Green Version]
- da Fonseca-Martins, A.M.; de Souza Lima-Gomes, P.; Antunes, M.M.; de Moura, R.G.; Covre, L.P.; Calôba, C.; Rocha, V.G.; Pereira, R.M.; Menezes, G.B.; Gomes, D.C.O.; et al. Leishmania Parasites Drive PD-L1 Expression in Mice and Human Neutrophils With Suppressor Capacity. Front. Immunol. 2021, 12, 598943. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Xie, J.; Zhao, Z.; Gupta, S.; Guo, Y.; Jia, S.; Parodo, J.; Marshall, J.C.; Deng, X. Upregulated PD-L1 Delays Human Neutrophil Apoptosis and Promotes Lung Injury in an Experimental Mouse Model of Sepsis. Blood 2021, 138, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, T.-C.; Schattgen, S.; Monks, B.; Wang, D.; Cerny, A.; Latz, E.; Fitzgerald, K.; Golenbock, D.T. A Fluorescent Reporter Mouse for Inflammasome Assembly Demonstrates an Important Role for Cell-Bound and Free ASC Specks during In Vivo Infection. Cell Rep. 2016, 16, 571–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarda, G.; Zenger, M.; Yazdi, A.S.; Schroder, K.; Ferrero, I.; Menu, P.; Tardivel, A.; Mattmann, C.; Tschopp, J. Differential Expression of NLRP3 among Hematopoietic Cells. J. Immunol. 2011, 186, 2529–2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coeshott, C.; Ohnemus, C.; Pilyavskaya, A.; Ross, S.; Wieczorek, M.; Kroona, H.; Leimer, A.H.; Cheronis, J. Converting Enzyme-Independent Release of Tumor Necrosis Factor α and IL-1β from a Stimulated Human Monocytic Cell Line in the Presence of Activated Neutrophils or Purified Proteinase 3. Proc. Natl. Acad. Sci. USA 1999, 96, 6261–6266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karmakar, M.; Sun, Y.; Hise, A.G.; Rietsch, A.; Pearlman, E. Cutting Edge: IL-1β Processing during Pseudomonas Aeruginosa Infection Is Mediated by Neutrophil Serine Proteases and Is Independent of NLRC4 and Caspase-1. J. Immunol. 2012, 189, 4231–4235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joosten, L.A.B.; Netea, M.G.; Fantuzzi, G.; Koenders, M.I.; Helsen, M.M.A.; Sparrer, H.; Pham, C.T.; van der Meer, J.W.M.; Dinarello, C.A.; van den Berg, W.B. Inflammatory Arthritis in Caspase 1 Gene-Deficient Mice: Contribution of Proteinase 3 to Caspase 1-Independent Production of Bioactive Interleukin-1beta. Arthritis Rheum. 2009, 60, 3651–3662. [Google Scholar] [CrossRef] [Green Version]
- Bakele, M.; Joos, M.; Burdi, S.; Allgaier, N.; Pöschel, S.; Fehrenbacher, B.; Schaller, M.; Marcos, V.; Kümmerle-Deschner, J.; Rieber, N.; et al. Localization and Functionality of the Inflammasome in Neutrophils*. J. Biol. Chem. 2014, 289, 5320–5329. [Google Scholar] [CrossRef] [Green Version]
- Mankan, A.K.; Dau, T.; Jenne, D.; Hornung, V. The NLRP3/ASC/Caspase-1 Axis Regulates IL-1β Processing in Neutrophils. Eur. J. Immunol. 2012, 42, 710–715. [Google Scholar] [CrossRef]
- Son, S.; Yoon, S.-H.; Chae, B.J.; Hwang, I.; Shim, D.-W.; Choe, Y.H.; Hyun, Y.-M.; Yu, J.-W. Neutrophils Facilitate Prolonged Inflammasome Response in the DAMP-Rich Inflammatory Milieu. Front. Immunol. 2021, 12, 746032. [Google Scholar] [CrossRef]
- Cho, J.S.; Guo, Y.; Ramos, R.I.; Hebroni, F.; Plaisier, S.B.; Xuan, C.; Granick, J.L.; Matsushima, H.; Takashima, A.; Iwakura, Y.; et al. Neutrophil-Derived IL-1β Is Sufficient for Abscess Formation in Immunity against Staphylococcus Aureus in Mice. PLoS Pathog. 2012, 8, e1003047. [Google Scholar] [CrossRef]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 Inflammasome in Inflammatory Diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, M.; Katsnelson, M.A.; Dubyak, G.R.; Pearlman, E. Neutrophil P2X 7 Receptors Mediate NLRP3 Inflammasome-Dependent IL-1β Secretion in Response to ATP. Nat. Commun. 2016, 7, 10555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karmakar, M.; Katsnelson, M.; Malak, H.A.; Greene, N.G.; Howell, S.J.; Hise, A.G.; Camilli, A.; Kadioglu, A.; Dubyak, G.R.; Pearlman, E. Neutrophil IL-1β Processing Induced by Pneumolysin Is Mediated by the NLRP3/ASC Inflammasome and Caspase-1 Activation and Is Dependent on K+ Efflux. J. Immunol. 2015, 194, 1763–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassane, M.; Demon, D.; Soulard, D.; Fontaine, J.; Keller, L.E.; Patin, E.C.; Porte, R.; Prinz, I.; Ryffel, B.; Kadioglu, A.; et al. Neutrophilic NLRP3 Inflammasome-Dependent IL-1β Secretion Regulates the ΓδT17 Cell Response in Respiratory Bacterial Infections. Mucosal Immunol. 2017, 10, 1056–1068. [Google Scholar] [CrossRef] [Green Version]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A Small-Molecule Inhibitor of the NLRP3 Inflammasome for the Treatment of Inflammatory Diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Passegué, E.; Wagner, E.F.; Weissman, I.L. JunB Deficiency Leads to a Myeloproliferative Disorder Arising from Hematopoietic Stem Cells. Cell 2004, 119, 431–443. [Google Scholar] [CrossRef] [Green Version]
- Hasenberg, A.; Hasenberg, M.; Männ, L.; Neumann, F.; Borkenstein, L.; Stecher, M.; Kraus, A.; Engel, D.R.; Klingberg, A.; Seddigh, P.; et al. Catchup: A Mouse Model for Imaging-Based Tracking and Modulation of Neutrophil Granulocytes. Nat. Methods 2015, 12, 445–452. [Google Scholar] [CrossRef]
- Stackowicz, J.; Gaudenzio, N.; Serhan, N.; Conde, E.; Godon, O.; Marichal, T.; Starkl, P.; Balbino, B.; Roers, A.; Bruhns, P.; et al. Neutrophil-Specific Gain-of-Function Mutations in Nlrp3 Promote Development of Cryopyrin-Associated Periodic Syndrome. J. Exp. Med. 2021, 218, e20201466. [Google Scholar] [CrossRef]
- Demirel, I.; Persson, A.; Brauner, A.; Särndahl, E.; Kruse, R.; Persson, K. Activation of NLRP3 by Uropathogenic Escherichia Coli Is Associated with IL-1β Release and Regulation of Antimicrobial Properties in Human Neutrophils. Sci. Rep. 2020, 10, 21837. [Google Scholar] [CrossRef]
- Cirl, C.; Wieser, A.; Yadav, M.; Duerr, S.; Schubert, S.; Fischer, H.; Stappert, D.; Wantia, N.; Rodriguez, N.; Wagner, H.; et al. Subversion of Toll-like Receptor Signaling by a Unique Family of Bacterial Toll/Interleukin-1 Receptor Domain–Containing Proteins. Nat. Med. 2008, 14, 399–406. [Google Scholar] [CrossRef]
- Pérez-Figueroa, E.; Torres, J.; Sánchez-Zauco, N.; Contreras-Ramos, A.; Alvarez-Arellano, L.; Maldonado-Bernal, C. Activation of NLRP3 Inflammasome in Human Neutrophils by Helicobacter Pylori Infection. Innate Immun. 2016, 22, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, A.-R.; Kang, M.-J.; Shin, J.-I.; Kwon, S.-W.; Park, J.-Y.; Ahn, J.-H.; Lee, T.-S.; Kim, D.-Y.; Choi, B.-G.; Seo, M.-W.; et al. Unveiling the Crucial Role of Type IV Secretion System and Motility of Helicobacter Pylori in IL-1β Production via NLRP3 Inflammasome Activation in Neutrophils. Front. Immunol. 2020, 11, 1121. [Google Scholar] [CrossRef] [PubMed]
- Bezbradica, J.S.; Coll, R.C.; Schroder, K. Sterile Signals Generate Weaker and Delayed Macrophage NLRP3 Inflammasome Responses Relative to Microbial Signals. Cell. Mol. Immunol. 2017, 14, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokose, K.; Sato, S.; Asano, T.; Yashiro, M.; Kobayashi, H.; Watanabe, H.; Suzuki, E.; Sato, C.; Kozuru, H.; Yatsuhashi, H.; et al. TNF-α Potentiates Uric Acid-Induced Interleukin-1β (IL-1β) Secretion in Human Neutrophils. Mod. Rheumatol. 2018, 28, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.T.; Duret, H.; Andrews, D.M.; Faveeuw, C.; Möller, A.; Smyth, M.J.; Paget, C. Type I NKT-Cell-Mediated TNF-α Is a Positive Regulator of NLRP3 Inflammasome Priming. Eur. J. Immunol. 2014, 44, 2111–2120. [Google Scholar] [CrossRef]
- Ericson, J.A.; Duffau, P.; Yasuda, K.; Ortiz-Lopez, A.; Rothamel, K.; Rifkin, I.R.; Monach, P.A. ImmGen Consortium Gene Expression during the Generation and Activation of Mouse Neutrophils: Implication of Novel Functional and Regulatory Pathways. PLoS ONE 2014, 9, e108553. [Google Scholar] [CrossRef]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-Expression Profiles and Transcriptional Regulatory Pathways That Underlie the Identity and Diversity of Mouse Tissue Macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, N.; Midiri, A.; Mancuso, G.; Patanè, F.; Venza, M.; Venza, I.; Passantino, A.; Galbo, R.; Teti, G.; Beninati, C.; et al. Neutrophils Directly Recognize Group B Streptococci and Contribute to Interleukin-1β Production during Infection. PLoS ONE 2016, 11, e0160249. [Google Scholar] [CrossRef]
- Hou, F.; Peng, L.; Jiang, J.; Chen, T.; Xu, D.; Huang, Q.; Ye, C.; Peng, Y.; Hu, D.-L.; Fang, R. ATP Facilitates Staphylococcal Enterotoxin O Induced Neutrophil IL-1β Secretion via NLRP3 Inflammasome Dependent Pathways. Front. Immunol. 2021, 12, 1640. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica Crystals and Aluminum Salts Activate the NALP3 Inflammasome through Phagosomal Destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Bezbradica, J.S.; Groß, C.J.; Wall, A.A.; Sweet, M.J.; Stow, J.L.; Schroder, K. The Murine Neutrophil NLRP3 Inflammasome Is Activated by Soluble but Not Particulate or Crystalline Agonists. Eur. J. Immunol. 2016, 46, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, E.L.; Asher, J.L.; Molony, R.D.; Shaw, A.C.; Zeiss, C.J.; Wang, C.; Morozova-Roche, L.A.; Herzog, R.I.; Iwasaki, A.; Dixit, V.D. β-Hydroxybutyrate Deactivates Neutrophil NLRP3 Inflammasome to Relieve Gout Flares. Cell Rep. 2017, 18, 2077–2087. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Matsuoka, N.; Temmoku, J.; Furuya, M.Y.; Asano, T.; Sato, S.; Kobayashi, H.; Watanabe, H.; Suzuki, E.; Urano, T.; et al. Hydroxychloroquine Inhibits IL-1β Production from Amyloid-Stimulated Human Neutrophils. Arthritis Res. Ther. 2019, 21, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migita, K.; Izumi, Y.; Jiuchi, Y.; Kozuru, H.; Kawahara, C.; Nakamura, M.; Nakamura, T.; Agematsu, K.; Masumoto, J.; Yasunami, M.; et al. Serum Amyloid A Induces NLRP-3-Mediated IL-1β Secretion in Neutrophils. PLoS ONE 2014, 9, e96703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, S.B.; Oh, C.; Maltez, V.I.; McGlaughon, B.D.; Verma, A.; Miao, E.A.; Aachoui, Y. Neutrophil Caspase-11 Is Essential to Defend against a Cytosol-Invasive Bacterium. Cell Rep. 2020, 32, 107967. [Google Scholar] [CrossRef]
- Sun, Y.; Abbondante, S.; Karmakar, M.; de Jesus Carrion, S.; Che, C.; Hise, A.G.; Pearlman, E. Neutrophil Caspase-11 Is Required for Cleavage of Caspase-1 and Secretion of IL-1β in Aspergillus Fumigatus Infection. J. Immunol. 2018, 201, 2767–2775. [Google Scholar] [CrossRef] [Green Version]
- Karki, R.; Man, S.M.; Malireddi, R.K.S.; Gurung, P.; Vogel, P.; Lamkanfi, M.; Kanneganti, T.-D. Concerted Activation of the AIM2 and NLRP3 Inflammasomes Orchestrates Host Protection against Aspergillus Infection. Cell Host Microbe 2015, 17, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Man, S.M.; Karki, R.; Briard, B.; Burton, A.; Gingras, S.; Pelletier, S.; Kanneganti, T.-D. Differential Roles of Caspase-1 and Caspase-11 in Infection and Inflammation. Sci. Rep. 2017, 7, 45126. [Google Scholar] [CrossRef] [Green Version]
- Aymonnier, K.; Ng, J.; Fredenburgh, L.E.; Zambrano-Vera, K.; Münzer, P.; Gutch, S.; Fukui, S.; Desjardins, M.; Subramaniam, M.; Baron, R.; et al. Inflammasome Activation in Neutrophils of Patients with Severe COVID-19. Blood Adv. 2022, 6, 2001–2013. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.B.; Cooper, M.A.; Graf, T.; Hornung, V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuya, M.Y.; Asano, T.; Sumichika, Y.; Sato, S.; Kobayashi, H.; Watanabe, H.; Suzuki, E.; Kozuru, H.; Yatsuhashi, H.; Koga, T.; et al. Tofacitinib Inhibits Granulocyte–Macrophage Colony-Stimulating Factor-Induced NLRP3 Inflammasome Activation in Human Neutrophils. Arthritis Res. Ther. 2018, 20, 196. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.R.; Gu, G.J.; Jung, D.; Kim, Y.W.; Na, J.; Woo, J.S.; Cho, J.Y.; Youn, H.; Seok, S.H. GM-CSF Induces Inflammatory Macrophages by Regulating Glycolysis and Lipid Metabolism. J. Immunol. 2016, 197, 4101–4109. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Groß, C.J.; Sotomayor, F.V.; Stacey, K.J.; Tschopp, J.; Sweet, M.J.; Schroder, K. The Neutrophil NLRC4 Inflammasome Selectively Promotes IL-1β Maturation without Pyroptosis during Acute Salmonella Challenge. Cell Rep. 2014, 8, 570–582. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.W.; Lawlor, K.E.; von Pein, J.B.; Boucher, D.; Gerlic, M.; Croker, B.A.; Bezbradica, J.S.; Vince, J.E.; Schroder, K. Cutting Edge: Blockade of Inhibitor of Apoptosis Proteins Sensitizes Neutrophils to TNF- but Not Lipopolysaccharide-Mediated Cell Death and IL-1β Secretion. J. Immunol. 2018, 200, 3341–3346. [Google Scholar] [CrossRef]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 Self-Cleavage Is an Intrinsic Mechanism to Terminate Inflammasome Activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018, 48, 35–44.e6. [Google Scholar] [CrossRef] [Green Version]
- Heilig, R.; Dick, M.S.; Sborgi, L.; Meunier, E.; Hiller, S.; Broz, P. The Gasdermin-D Pore Acts as a Conduit for IL-1β Secretion in Mice. Eur. J. Immunol. 2018, 48, 584–592. [Google Scholar] [CrossRef] [Green Version]
- Karmakar, M.; Minns, M.; Greenberg, E.N.; Diaz-Aponte, J.; Pestonjamasp, K.; Johnson, J.L.; Rathkey, J.K.; Abbott, D.W.; Wang, K.; Shao, F.; et al. N-GSDMD Trafficking to Neutrophil Organelles Facilitates IL-1β Release Independently of Plasma Membrane Pores and Pyroptosis. Nat. Commun. 2020, 11, 2212. [Google Scholar] [CrossRef]
- Carty, M.; Kearney, J.; Shanahan, K.A.; Hams, E.; Sugisawa, R.; Connolly, D.; Doran, C.G.; Muñoz-Wolf, N.; Gürtler, C.; Fitzgerald, K.A.; et al. Cell Survival and Cytokine Release after Inflammasome Activation Is Regulated by the Toll-IL-1R Protein SARM. Immunity 2019, 50, 1412–1424.e6. [Google Scholar] [CrossRef]
- Maianski, N.A.; Geissler, J.; Srinivasula, S.M.; Alnemri, E.S.; Roos, D.; Kuijpers, T.W. Functional Characterization of Mitochondria in Neutrophils: A Role Restricted to Apoptosis. Cell Death Differ. 2004, 11, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhao, J.; Mu, D.; Wang, Z.; Liu, Q.; Zhang, Y.; Yang, D. Pyroptosis Mediates Neutrophil Extracellular Trap Formation during Bacterial Infection in Zebrafish. J. Immunol. 2021, 206, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, H.M.; Mueller, J.L.; Broide, D.H.; Wanderer, A.A.; Kolodner, R.D. Mutation of a New Gene Encoding a Putative Pyrin-like Protein Causes Familial Cold Autoinflammatory Syndrome and Muckle–Wells Syndrome. Nat. Genet. 2001, 29, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuemmerle-Deschner, J.B. CAPS—Pathogenesis, Presentation and Treatment of an Autoinflammatory Disease. Semin. Immunopathol. 2015, 37, 377–385. [Google Scholar] [CrossRef]
- Brydges, S.D.; Mueller, J.L.; McGeough, M.D.; Pena, C.A.; Misaghi, A.; Gandhi, C.; Putnam, C.D.; Boyle, D.L.; Firestein, G.S.; Horner, A.A.; et al. Inflammasome-Mediated Disease Animal Models Reveal Roles for Innate but Not Adaptive Immunity. Immunity 2009, 30, 875–887. [Google Scholar] [CrossRef] [Green Version]
- Dalbeth, N.; Merriman, T.R.; Stamp, L.K. Gout. Lancet 2016, 388, 2039–2052. [Google Scholar] [CrossRef]
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Baßler, K.; Schlickeiser, S.; Zhang, B.; Krämer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, 1419–1440.e23. [Google Scholar] [CrossRef]
- Courjon, J.; Dufies, O.; Robert, A.; Bailly, L.; Torre, C.; Chirio, D.; Contenti, J.; Vitale, S.; Loubatier, C.; Doye, A.; et al. Heterogeneous NLRP3 Inflammasome Signature in Circulating Myeloid Cells as a Biomarker of COVID-19 Severity. Blood Adv. 2021, 5, 1523–1534. [Google Scholar] [CrossRef]
- Silvin, A.; Chapuis, N.; Dunsmore, G.; Goubet, A.-G.; Dubuisson, A.; Derosa, L.; Almire, C.; Hénon, C.; Kosmider, O.; Droin, N.; et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell 2020, 182, 1401–1418.e18. [Google Scholar] [CrossRef]
- Lima, T.S.; Gov, L.; Lodoen, M.B. Evasion of Human Neutrophil-Mediated Host Defense during Toxoplasma Gondii Infection. mBio 2018, 9, e02027-17. [Google Scholar] [CrossRef] [Green Version]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of Interleukin-1β Induces Gastric Inflammation and Cancer and Mobilizes Myeloid-Derived Suppressor Cells in Mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy Drugs Induce Pyroptosis through Caspase-3 Cleavage of a Gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by Caspase-3 during Apoptosis Mediates Progression to Secondary Necrotic/Pyroptotic Cell Death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoni, K.; Pericat, D.; Pinilla, M.; Bagayoko, S.; Hessel, A.; Leon-Icaza, S.-A.; Bellard, E.; Mazères, S.; Doz-Deblauwe, E.; Winter, N.; et al. Caspase-1-Driven Neutrophil Pyroptosis Promotes an Incomplete NETosis upon Pseudomonas Aeruginosa Infection. bioRxiv 2022. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Pathway | Steps | Macrophages | Neutrophils | |
|---|---|---|---|---|
| Canonical pathway | Priming | Known sensors | TLRs, TNF-R, IL-1R, NOD1/2, CLRs | TLRs, TNF-R, IL-1R, NOD2, CLRs |
| Known activated pathways | NF-κB, MAPK, ASK, CASP8/FADD and AP-1 | NF-κB | ||
| Activation | K+ efflux induced by pore-forming toxins and extracellular ATP - Particles and crystals (strong activation) - Bacterial infection - Dectin-1/Syk activation induced by fungal infection and fungal PAMPs | K+ efflux induced by pore-forming toxins and extracellular ATP - Particles and crystals (weak activation) - Bacterial infection - Syk activation induced by serum amyloid A | ||
| NLRP3-dependent cytokine secretion (on a per cell basis) | +++ | + | ||
| Cell death | GSDMD-dependent pyroptosis | No pyroptosis | ||
| Non-canonical pathway | Caspase-11 activation | LPS binding to caspase-11 - Bacterial intracellular LPS exposure through GBPs and IRGB10 cooperation in type I IFN dependant-manner | LPS binding to caspase-11 - Caspase-11 activation dependent to type I IFN signalling during fungal infection | |
| GSDMD cleavage | Caspase-11-dependent activation | Caspase-11-dependent activation | ||
| NLRP3 activation | K+ efflux through GSDMD pores | K+ efflux through GSDMD pores | ||
| NLRP3-dependent cytokine secretion (on a per cell basis) | +++ | + | ||
| Cell death | GSDMD-dependent pyroptosis | Extrusion of chromatin through NETosis process with GSDMD-dependent mechanism | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paget, C.; Doz-Deblauwe, E.; Winter, N.; Briard, B. Specific NLRP3 Inflammasome Assembling and Regulation in Neutrophils: Relevance in Inflammatory and Infectious Diseases. Cells 2022, 11, 1188. https://doi.org/10.3390/cells11071188
Paget C, Doz-Deblauwe E, Winter N, Briard B. Specific NLRP3 Inflammasome Assembling and Regulation in Neutrophils: Relevance in Inflammatory and Infectious Diseases. Cells. 2022; 11(7):1188. https://doi.org/10.3390/cells11071188
Chicago/Turabian StylePaget, Christophe, Emilie Doz-Deblauwe, Nathalie Winter, and Benoit Briard. 2022. "Specific NLRP3 Inflammasome Assembling and Regulation in Neutrophils: Relevance in Inflammatory and Infectious Diseases" Cells 11, no. 7: 1188. https://doi.org/10.3390/cells11071188
APA StylePaget, C., Doz-Deblauwe, E., Winter, N., & Briard, B. (2022). Specific NLRP3 Inflammasome Assembling and Regulation in Neutrophils: Relevance in Inflammatory and Infectious Diseases. Cells, 11(7), 1188. https://doi.org/10.3390/cells11071188