
Proteomic Analysis of the Role of the Adenylyl Cyclase–cAMP Pathway in Red Blood Cell Mechanical Responses


Abstract
:1. Introduction
2. Materials and Methods
2.1. Blood Sampling and Drug Treatments
2.2. Application of Physiological Shear Stress to Blood
2.3. RBC Deformability Measurement
2.4. RBC Membrane Isolation and Protein Preparation
2.5. Proteomics Experimental Design
2.6. In Gel Digestion, nLC-MS/MS, and Data Acquisition
2.7. Validation Experiments
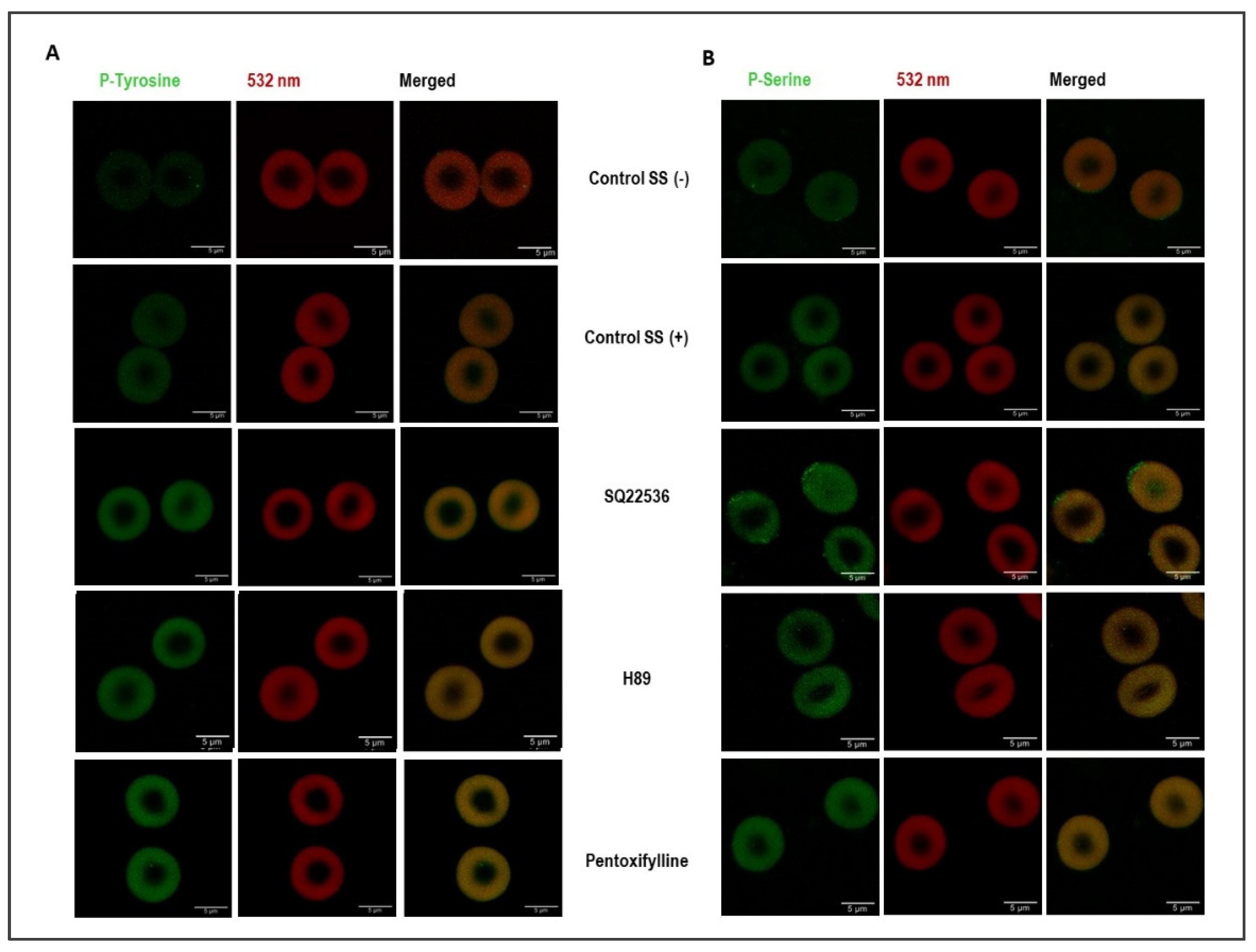
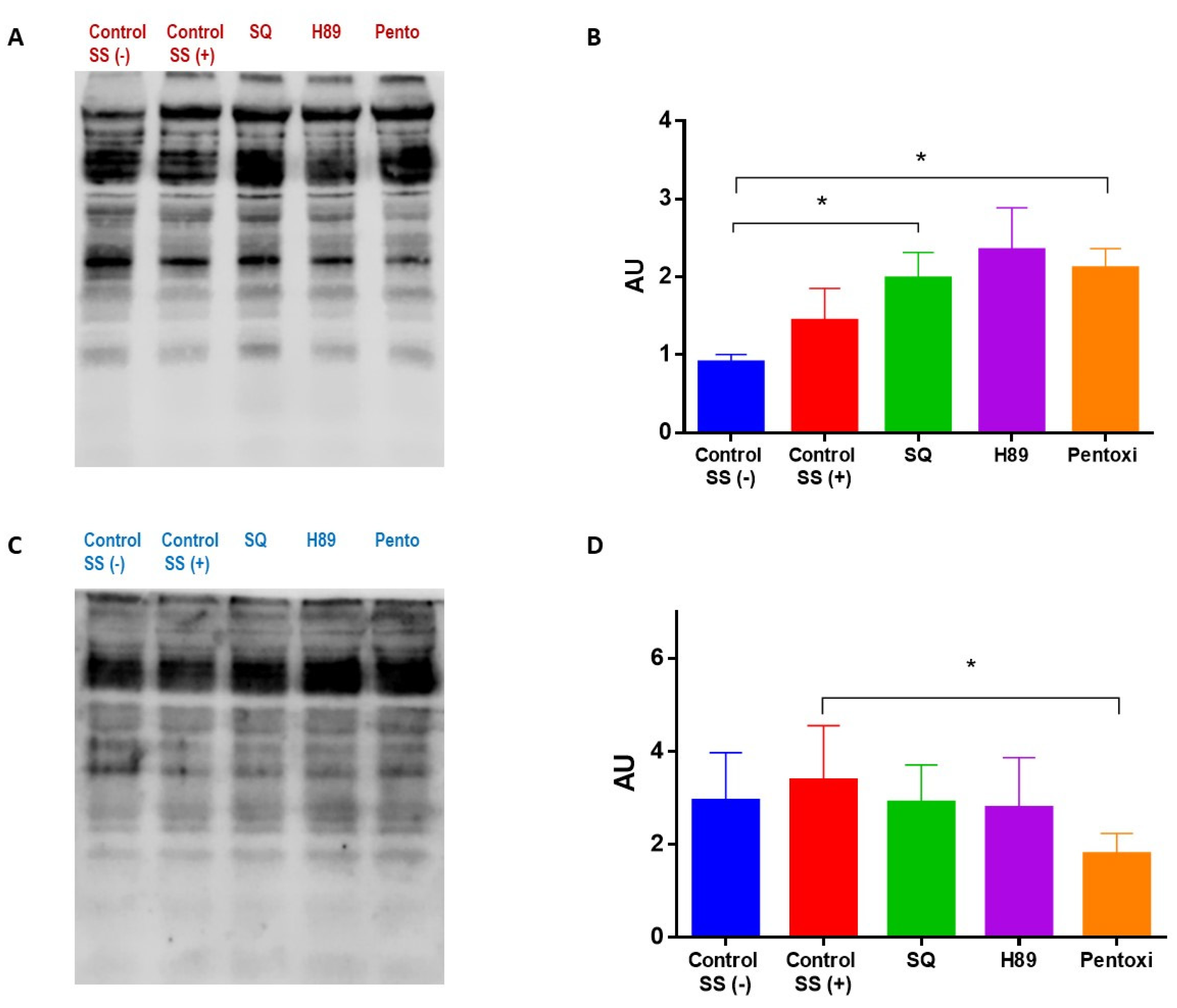
2.8. Protein Phosphorylation Studies
2.9. Statistical Analysis
3. Results
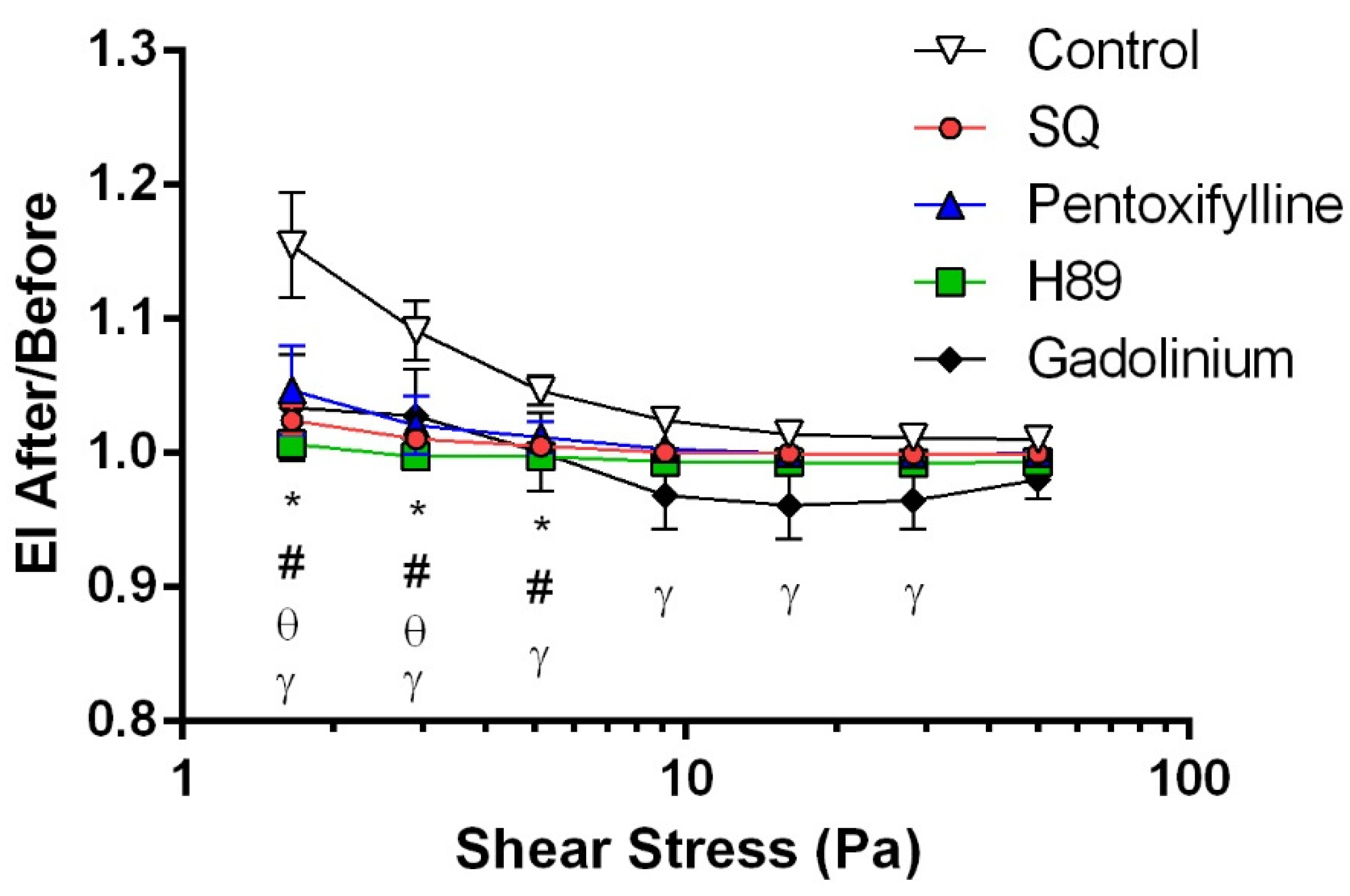
3.1. cAMP/PKA Signaling Pathway and Piezo1 Contribute to Shear-Induced RBC Deformability
3.2. Shear Stress and cAMP/PKA Signaling Do Not Alter Protein Composition in RBC Membranes
3.3. Shear Stress and cAMP/PKA Signaling Induce Changes in Phosphorylation
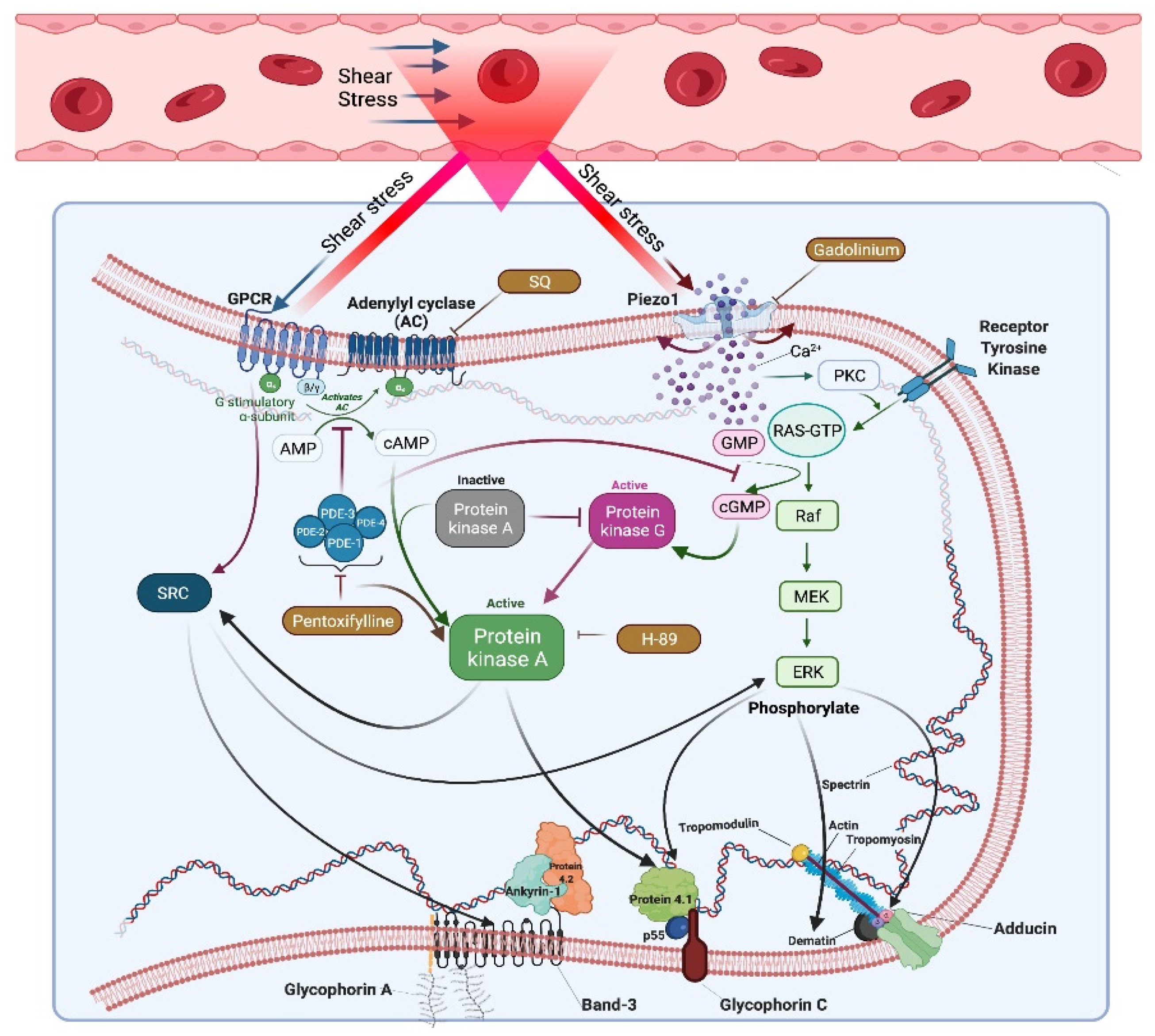
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Danielczok, J.G.; Terriac, E.; Hertz, L.; Petkova-Kirova, P.; Lautenschläger, F.; Laschke, M.W.; Kaestner, L. Red Blood Cell Passage of Small Capillaries Is Associated with Transient Ca2+-mediated Adaptations. Front. Physiol. 2017, 8, 979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alapan, Y.; Little, J.A.; Gurkan, U.A. Heterogeneous Red Blood Cell Adhesion and Deformability in Sickle Cell Disease. Sci. Rep. 2014, 4, 7173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.D.; Ghali, H.S.; Zhao, Z.; Thomas, L.I.; Friedman, E.A. Association of reduced red blood cell deformability and diabetic nephropathy. Kidney Int. 2005, 67, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, K. Red blood cell abnormalities and hypertension. Hypertens. Res. 2020, 43, 72–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, M.R.; Mohandas, N.; Shohet, S.B. Osmotic Gradient Ektacytometry: Comprehensive Characterization of Red Cell Volume and Surface Maintenance. Blood 1983, 61, 899–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chasis, J.A.; Mohandas, N. Erythrocyte membrane deformability and stability: Two distinct membrane properties that are independently regulated by skeletal protein associations. J. Cell Biol. 1986, 103, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diez-Silva, M.; Dao, M.; Han, J.; Lim, C.T.; Suresh, S. Shape and Biomechanical Characteristics of Human Red Blood Cells in Health and Disease. MRS Bull. 2010, 35, 382–388. [Google Scholar] [CrossRef]
- Gallagher, P.G. Disorders of red cell volume regulation. Curr. Opin. Hematol. 2013, 20, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Baskurt, O.K.; Temiz, A.; Meiselman, H.J. Effect of superoxide anions on red blood cell rheologic properties. Free Radic. Biol. Med. 1998, 24, 102–110. [Google Scholar] [CrossRef]
- Fischer, D.J.; Torrence, N.J.; Sprung, R.J.; Spence, D.M. Determination of erythrocyte deformability and its correlation to cellular ATP release using microbore tubing with diameters that approximate resistance vessels in vivo. Analyst 2003, 128, 1163–1168. [Google Scholar] [CrossRef]
- Kuhn, V.; Diederich, L.; Keller, T.C.S.; Kramer, C.M.; Lückstädt, W.; Panknin, C.; Suvorava, T.; Isakson, B.E.; Kelm, M.; Cortese-Krott, M.M. Red Blood Cell Function and Dysfunction: Redox Regulation, Nitric Oxide Metabolism, Anemia. Antioxid. Redox Signal. 2017, 26, 718–742. [Google Scholar] [CrossRef] [PubMed]
- Huisjes, R.; Bogdanova, A.; van Solinge, W.W.; Schiffelers, R.M.; Kaestner, L.; van Wijk, R. Squeezing for Life—Properties of Red Blood Cell Deformability. Front. Physiol. 2018, 9, 656. [Google Scholar] [CrossRef]
- Nigra, A.D.; Casale, C.H.; Santander, V.S. Human erythrocytes: Cytoskeleton and its origin. Cell Mol. Life Sci. 2020, 77, 1681–1694. [Google Scholar] [CrossRef]
- Cahalan, S.M.; Lukacs, V.; Ranade, S.S.; Chien, S.; Bandell, M.; Patapoutian, A. Piezo1 links mechanical forces to red blood cell volume. eLife 2015, 4, e07370. [Google Scholar] [CrossRef]
- Cinar, E.; Zhou, S.; DeCourcey, J.; Wang, Y.; Waugh, R.E.; Wan, J. Piezo1 regulates mechanotransductive release of ATP from human RBCs. Proc. Natl. Acad. Sci. USA 2015, 112, 11783–11788. [Google Scholar] [CrossRef] [Green Version]
- Buerck, J.P.; Burke, D.K.; Schmidtke, D.W.; Snyder, T.A.; Papavassiliou, D.V.; O’Rear, E.A. Production of erythrocyte microparticles in a sub-hemolytic environment. J. Artif. Organs 2021, 24, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Ristenpart, W.D.; Stone, H.A. Dynamics of shear-induced ATP release from red blood cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16432–16437. [Google Scholar] [CrossRef] [Green Version]
- Doss, J.F.; Corcoran, D.L.; Jima, D.D.; Telen, M.J.; Dave, S.S.; Chi, J.T. A comprehensive joint analysis of the long and short RNA transcriptomes of human erythrocytes. BMC Genom. 2015, 16, 952. [Google Scholar] [CrossRef] [Green Version]
- Gurkan, U.A. Biophysical and rheological biomarkers of red blood cell physiology and pathophysiology. Curr. Opin. Hematol. 2021, 28, 138–149. [Google Scholar] [CrossRef]
- Noomuna, P.; Risinger, M.; Zhou, S.; Seu, K.; Man, Y.; An, R.; Sheik, D.A.; Wan, J.; Little, J.A.; Gurkan, U.A.; et al. Inhibition of Band 3 tyrosine phosphorylation: A new mechanism for treatment of sickle cell disease. Br. J. Haematol. 2020, 190, 599–609. [Google Scholar] [CrossRef]
- Strader, M.B.; Jana, S.; Meng, F.; Heaven, M.R.; Shet, A.S.; Thein, S.L.; Alayash, A.I. Post-translational modification as a response to cellular stress induced by hemoglobin oxidation in sickle cell disease. Sci. Rep. 2020, 10, 14218. [Google Scholar] [CrossRef] [PubMed]
- Adderley, J.D.; John von Freyend, S.; Jackson, S.A.; Bird, M.J.; Burns, A.L.; Anar, B.; Metcalf, T.; Semblat, J.P.; Billker, O.; Wilson, D.W.; et al. Analysis of erythrocyte signalling pathways during Plasmodium falciparum infection identifies targets for host-directed antimalarial intervention. Nat. Commun. 2020, 11, 4015. [Google Scholar] [CrossRef] [PubMed]
- Zuccala, E.S.; Satchwell, T.J.; Angrisano, F.; Tan, Y.H.; Wilson, M.C.; Heesom, K.J.; Baum, J. Quantitative phospho-proteomics reveals the Plasmodium merozoite triggers pre-invasion host kinase modification of the red cell cytoskeleton. Sci. Rep. 2016, 6, 19766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantaleo, A.; Ferru, E.; Pau, M.C.; Khadjavi, A.; Mandili, G.; Matte, A.; Spano, A.; De Franceschi, L.; Pippia, P.; Turrini, F. Band 3 Erythrocyte Membrane Protein Acts as Redox Stress Sensor Leading to Its Phosphorylation by p (72) Syk. Oxid. Med. Cell. Longev. 2016, 2016, 6051093. [Google Scholar] [CrossRef] [Green Version]
- Azouzi, S.; Romana, M.; Arashiki, N.; Takakuwa, Y.; El Nemer, W.; Peyrard, T.; Colin, Y.; Amireault, P.; Le Van Kim, C. Band 3 phosphorylation induces irreversible alterations of stored red blood cells. Am. J. Hematol. 2018, 93, E110–E112. [Google Scholar] [CrossRef]
- Boivin, P. Role of the phosphorylation of red blood cell membrane proteins. Biochem. J. 1988, 256, 689–695. [Google Scholar] [CrossRef] [Green Version]
- Longo, V.; Marrocco, C.; Zolla, L.; Rinalducci, S. Label-free quantitation of phosphopeptide changes in erythrocyte membranes: Towards molecular mechanisms underlying deformability alterations in stored red blood cells. Haematologica 2014, 99, e122–e125. [Google Scholar] [CrossRef] [Green Version]
- Muravyov, A.V.; Tikhomirova, I.A. Role molecular signaling pathways in changes of red blood cell deformability. Clin. Hemorheol. Microcirc. 2013, 53, 45–59. [Google Scholar] [CrossRef]
- Muravyov, A.V.; Tikhomirova, I.A.; Maimistova, A.A.; Bulaeva, S.V. Extra- and intracellular signaling pathways under red blood cell aggregation and deformability changes. Clin. Hemorheol. Microcirc. 2009, 43, 223–232. [Google Scholar] [CrossRef]
- Semenov, A.N.; Shirshin, E.A.; Muravyov, A.V.; Priezzhev, A.V. The Effects of Different Signaling Pathways in Adenylyl Cyclase Stimulation on Red Blood Cells Deformability. Front. Physiol. 2019, 10, 923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugurel, E.; Connes, P.; Yavas, G.; Eglenen, B.; Turkay, M.; Aksu, A.C.; Renoux, C.; Joly, P.; Gauthier, A.; Hot, A.; et al. Differential effects of adenylyl cyclase-protein kinase A cascade on shear-induced changes of sickle cell deformability. Clin. Hemorheol. Microcirc. 2019, 73, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Horga, J.F.; Gisbert, J.; De Agustín, J.C.; Hernández, M.; Zapater, P. A Beta-2-Adrenergic Receptor Activates Adenylate Cyclase in Human Erythrocyte Membranes at Physiological Calcium Plasma Concentrations. Blood Cells Mol. Dis. 2000, 26, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Oonishi, T.; Sakashita, K.; Uyesaka, N. Regulation of red blood cell filterability by Ca2+ influx and cAMP-mediated signaling pathways. Am. J. Physiol.-Cell Physiol. 1997, 273, C1828–C1834. [Google Scholar] [CrossRef]
- Moura, P.A.-O.; Lizarralde Iragorri, M.A.; Français, O.; Le Pioufle, B.; Dobbe, J.G.G.; Streekstra, G.J.; El Nemer, W.A.-O.X.; Toye, A.A.-O.; Satchwell, T.A.-O.X. Reticulocyte and red blood cell deformation triggers specific phosphorylation events. Blood Adv. 2019, 3, 2653–2663. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, T.G.; Stefanadis, C. Vascular wall shear stress: Basic principles and methods. Hell. J. Cardiol. 2005, 46, 9–15. [Google Scholar]
- Meram, E.; Yilmaz, B.D.; Bas, C.; Atac, N.; Yalcin, O.; Meiselman, H.J.; Baskurt, O.K. Shear stress-induced improvement of red blood cell deformability. Biorheology 2013, 50, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Pfitzner, J. Poiseuille and his law. Anaesthesia 1976, 31, 273–275. [Google Scholar] [CrossRef]
- Sutera, S.P.; Skalak, R. The History of Poiseuille’s Law. Annu. Rev. Fluid Mech. 1993, 25, 1–20. [Google Scholar] [CrossRef]
- Hardeman, M.R.; Goedhart, P.T.; Dobbe, J.G.G.; Lettinga, K.P. Laser-assisted optical rotational cell analyser (L.O.R.C.A.); I. A new instrument for measurement of various structural hemorheological parameters. Clin. Hemorheol. Microcirc. 1994, 14, 605–618. [Google Scholar] [CrossRef]
- Baskurt, O.K.; Hardeman, M.R.; Uyuklu, M.; Ulker, P.; Cengiz, M.; Nemeth, N.; Shin, S.; Alexy, T.; Meiselman, H.J. Parameterization of red blood cell elongation index--shear stress curves obtained by ektacytometry. Scand. J. Clin. Lab. Investig. 2009, 69, 777–788. [Google Scholar] [CrossRef]
- Baskurt, O.K.; Meiselman, H.J. Data reduction methods for ektacytometry in clinical hemorheology. Clin. Hemorheol. Microcirc. 2013, 54, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, S.R.; Hughes, K.M.; Kakhniashvili, D.G.; Neelam, S. The isolation of reticulocyte-free human red blood cells. Exp. Biol. Med. 2007, 232, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Pesciotta, E.N.; Sriswasdi, S.; Tang, H.Y.; Mason, P.J.; Bessler, M.; Speicher, D.W. A label-free proteome analysis strategy for identifying quantitative changes in erythrocyte membranes induced by red cell disorders. J. Proteom. 2012, 76, 194–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Yan, R.; Li, W.; Xu, K. Super-Resolution Microscopy Reveals the Native Ultrastructure of the Erythrocyte Cytoskeleton. Cell Rep. 2018, 22, 1151–1158. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, N.; Sogor, V.; Kiss, F.; Ulker, P. Interspecies diversity of erythrocyte mechanical stability at various combinations in magnitude and duration of shear stress, and osmolality. Clin. Hemorheol. Microcirc. 2016, 63, 381–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muravyov, A.V.; Tikhomirova, I.A.; Maimistova, A.A.; Bulaeva, S.V.; Zamishlayev, A.V.; Batalova, E.A. Crosstalk between adenylyl cyclase signaling pathway and Ca2+ regulatory mechanism under red blood cell microrheological changes. Clin. Hemorheol. Microcirc. 2010, 45, 337–345. [Google Scholar] [CrossRef]
- Sprague, R.; Bowles, E.; Stumpf, M.; Ricketts, G.; Freidman, A.; Hou, W.H.; Stephenson, A.; Lonigro, A. Rabbit erythrocytes possess adenylyl cyclase type II that is activated by the heterotrimeric G proteins Gs and Gi. Pharm. Rep. 2005, 57, 222–228. [Google Scholar]
- Dyrda, A.; Cytlak, U.; Ciuraszkiewicz, A.; Lipinska, A.; Cueff, A.; Bouyer, G.; Egée, S.; Bennekou, P.; Lew, V.L.; Thomas, S.L.Y. Local Membrane Deformations Activate Ca2+-Dependent K+ and Anionic Currents in Intact Human Red Blood Cells. PLoS ONE 2010, 5, e9447. [Google Scholar] [CrossRef] [Green Version]
- Thuet, K.M.; Bowles, E.A.; Ellsworth, M.I.; Sprague, R.S.; Stephenson, A.H. The Rho kinase inhibitor Y-27632 increases erythrocyte deformability and low oxygen tension-induced ATP release. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1891–H1896. [Google Scholar] [CrossRef] [Green Version]
- Tiftik, R.N.; Başkurt, O.K.; Kul, S.; Büyükafşar, K. The functional significance of the rho/rho-kinase pathway in human erythrocytes. Turk. J. Haematol. 2014, 31, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Bor-Kucukatay, M.; Wenby, R.B.; Meiselman, H.J.; Baskurt, O.K. Effects of nitric oxide on red blood cell deformability. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1577–H1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grau, M.; Jerke, M.; Nader, E.; Schenk, A.; Renoux, C.; Collins, B.; Dietz, T.; Bizjak, D.A.; Joly, P.; Bloch, W.; et al. Effect of acute exercise on RBC deformability and RBC nitric oxide synthase signalling pathway in young sickle cell anaemia patients. Sci. Rep. 2019, 9, 11813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grau, M.; Pauly, S.; Ali, J.; Walpurgis, K.; Thevis, M.; Bloch, W.; Suhr, F. RBC-NOS-Dependent S-Nitrosylation of Cytoskeletal Proteins Improves RBC Deformability. PLoS ONE 2013, 8, e56759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baskurt, O.K.; Meiselman, H.J. Blood rheology and hemodynamics. Semin. Thromb. Hemost. 2003, 29, 435–450. [Google Scholar] [CrossRef] [Green Version]
- Chien, S. Red cell deformability and its relevance to blood flow. Annu. Rev. Physiol. 1987, 49, 177–192. [Google Scholar] [CrossRef]
- Ugurel, E.; Kisakurek, Z.B.; Aksu, Y.; Goksel, E.; Cilek, N.; Yalcin, O. Calcium/protein kinase C signaling mechanisms in shear-induced mechanical responses of red blood cells. Microvasc. Res. 2021, 135, 104124. [Google Scholar] [CrossRef]
- Li, J.; Hou, B.; Tumova, S.; Muraki, K.; Bruns, A.; Ludlow, M.J.; Sedo, A.; Hyman, A.J.; McKeown, L.; Young, R.S.; et al. Piezo1 integration of vascular architecture with physiological force. Nature 2014, 515, 279–282. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, S.A.; Li, W.; Ma, Y.; Shen, J.; Wang, Y.; Shen, Y.; Chen, J.; Ji, Y.; Xie, Y.; et al. Piezo1-Mediated Mechanotransduction Promotes Cardiac Hypertrophy by Impairing Calcium Homeostasis to Activate Calpain/Calcineurin Signaling. Hypertension 2021, 78, 647–660. [Google Scholar] [CrossRef]
- Gavi, S.; Shumay, E.; Wang, H.Y.; Malbon, C.C. G-protein-coupled receptors and tyrosine kinases: Crossroads in cell signaling and regulation. Trends Endocrinol. Metab. 2006, 17, 48–54. [Google Scholar] [CrossRef]
- McGarrigle, D.; Huang, X.Y. GPCRs signaling directly through Src-family kinases. Sci. STKE 2007, 392, pe35. [Google Scholar] [CrossRef]
- Wang, W.; Qiao, Y.; Li, Z. New Insights into Modes of GPCR Activation. Trends Pharmacol. Sci. 2018, 39, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Bordin, L.; Ion-Popa, F.; Brunati, A.M.; Clari, G.; Low, P.S. Effector-induced Syk-mediated phosphorylation in human erythrocytes. Biochim. Biophys. Acta 2005, 1745, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Brunati, A.M.; Bordin, L.; Clari, G.; James, P.; Quadroni, M.; Baritono, E.; Pinna, L.A.; Donella-Deana, A. Sequential phosphorylation of protein band 3 by Syk and Lyn tyrosine kinases in intact human erythrocytes: Identification of primary and secondary phosphorylation sites. Blood 2000, 96, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Sadek, M.S.; Cachorro, E.; El-Armouche, A.; Kämmerer, S. Therapeutic Implications for PDE2 and cGMP/cAMP Mediated Crosstalk in Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 7462. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Movsesian, M.A. cAMP and cGMP signaling cross-talk: Role of phosphodiesterases and implications for cardiac pathophysiology. Circ. Res. 2007, 100, 1569–1578. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.Y.; Greenstein, J.L.; Winslow, R.L. Roles of phosphodiesterases in the regulation of the cardiac cyclic nucleotide cross-talk signaling network. J. Mol. Cell. Cardiol. 2016, 91, 215–227. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Xi, X.; Gu, M.; Feil, R.; Ye, R.D.; Eigenthaler, M.; Hofmann, F.; Du, X. A stimulatory role for cGMP-dependent protein kinase in platelet activation. Cell 2003, 112, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Zennadi, R.; Whalen, E.J.; Soderblom, E.J.; Alexander, S.C.; Thompson, J.W.; Dubois, L.G.; Moseley, M.A.; Telen, M.J. Erythrocyte plasma membrane-bound ERK1/2 activation promotes ICAM-4-mediated sickle red cell adhesion to endothelium. Blood 2012, 119, 1217–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.H. Dimerization of cell surface receptors in signal transduction. Cell 1995, 80, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.H.; McAvoy, T.; Rakhilin, S.V.; Nishi, A.; Greengard, P.; Nairn, A.C. Protein kinase A activates protein phosphatase 2A by phosphorylation of the B56delta subunit. Proc. Natl. Acad. Sci. USA 2007, 104, 2979–2984. [Google Scholar] [CrossRef] [Green Version]
- Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011148. [Google Scholar] [CrossRef] [PubMed]
- Nassar, A.; Mahony, M.; Morshedi, M.; Lin, M.H.; Srisombut, C.; Oehninger, S. Modulation of sperm tail protein tyrosine phosphorylation by pentoxifylline and its correlation with hyperactivated motility. Fertil. Steril. 1999, 71, 919–923. [Google Scholar] [CrossRef]
- Yunes, R.; Fernández, P.; Doncel, G.F.; Acosta, A.A. Cyclic nucleotide phosphodiesterase inhibition increases tyrosine phosphorylation and hyper motility in normal and pathological human spermatozoa. Biocell 2005, 29, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Obara, Y.; Labudda, K.; Dillon, T.J.; Stork, P.J. PKA phosphorylation of Src mediates Rap1 activation in NGF and cAMP signaling in PC12 cells. J. Cell Sci. 2004, 117, 6085–6094. [Google Scholar] [CrossRef] [Green Version]
- Baker, M.A.; Hetherington, L.; Aitken, R.J. Identification of SRC as a key PKA-stimulated tyrosine kinase involved in the capacitation-associated hyperactivation of murine spermatozoa. J. Cell Sci. 2006, 119, 3182–3192. [Google Scholar] [CrossRef] [Green Version]
- Sapio, L.; Di Maiolo, F.; Illiano, M.; Esposito, A.; Chiosi, E.; Spina, A.; Naviglio, S. Targeting protein kinase A in cancer therapy: An update. EXCLI J. 2014, 13, 843–855. [Google Scholar]
- Tortora, G.; Ciardiello, F. Antisense Targeting Protein Kinase A Type I as a Drug for Integrated Strategies of Cancer Therapy. Ann. N. Y. Acad. Sci. 2003, 1002, 236–243. [Google Scholar] [CrossRef]
- Lee, L.C.Y.; Maurice, D.H.; Baillie, G.S. Targeting protein–protein interactions within the cyclic AMP signaling system as a therapeutic strategy for cardiovascular disease. Future Med. Chem. 2013, 5, 451–464. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Viña, D.; Seoane, N.; Vasquez, E.C.; Campos-Toimil, M. cAMP Compartmentalization in Cerebrovascular Endothelial Cells: New Therapeutic Opportunities in Alzheimer’s Disease. Cells 2021, 10, 1951. [Google Scholar] [CrossRef] [PubMed]
- Berthouze, M.; Laurent, A.-C.; Breckler, M.; Lezoualc’h, F. New Perspectives in cAMP-Signaling Modulation. Curr. Heart Fail. Rep. 2011, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Lezoualc’h, F.; Fazal, L.; Laudette, M.; Conte, C. Cyclic AMP Sensor EPAC Proteins and Their Role in Cardiovascular Function and Disease. Circ. Res. 2016, 118, 881–897. [Google Scholar] [CrossRef]
- Thorens, B.; Dériaz, N.; Bosco, D.; DeVos, A.; Pipeleers, D.; Schuit, F.; Meda, P.; Porret, A. Protein Kinase A-dependent Phosphorylation of GLUT2 in Pancreatic β Cells. J. Biol. Chem. 1996, 271, 8075–8081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikuta, T.; Gutsaeva, D.; Parkerson, J.; Yerigenahally, S.; Head, C.A. Hydroxyurea Induces Fetal Hemoglobin Expression by Activating cAMP Signaling Pathways In a cAMP- and cGMP-Dependent Manner; New Hypothesis to Account for a Role of Non-Erythroid Cells In Fetal Hemoglobin Induction. Blood 2010, 116, 1622. [Google Scholar] [CrossRef]
- Ikuta, T.; Kuroyanagi, Y.; Odo, N.; Liu, S. A common signaling pathway is activated in erythroid cells expressing high levels of fetal hemoglobin: A potential role for cAMP-elevating agents in β-globin disorders. J. Blood Med. 2013, 4, 149–159. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, S.; Silva-Herdade, A.S.; Saldanha, C. Modulation of erythrocyte deformability by PKC activity. Clin. Hemorheol. Microcirc. 2008, 39, 363–373. [Google Scholar] [CrossRef]
- Ling, E.; Danilov, Y.N.; Cohen, C.M. Modulation of red cell band 4.1 function by cAMP-dependent kinase and protein kinase C phosphorylation. J. Biol. Chem. 1998, 263, 2209–2216. [Google Scholar] [CrossRef]
- Barbul, A.; Zipser, Y.; Nachles, A.; Korenstein, R. Deoxygenation and elevation of intracellular magnesium induce tyrosine phosphorylation of band 3 in human erythrocytes. FEBS Lett. 1999, 455, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Zipser, Y.; Piade, A.; Barbul, A.; Korenstein, R.; Kosower, N.S. Ca2+ promotes erythrocyte band 3 tyrosine phosphorylation via dissociation of phosphotyrosine phosphatase from band 3. Biochem. J. 2002, 368, 137–144. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UniProt No | Protein | Coverage (%) | UP 1 | Peptide | PSM 2 |
|---|---|---|---|---|---|
| P60709 | Actin, cytoplasmic 1 | 41.87 | 9 | 12 | 30 |
| P11171 | Protein 4.1 | 28.82 | 21 | 24 | 59 |
| P02549 | Spectrin alpha chain, erythrocytic 1 | 33.11 | 75 | 78 | 179 |
| H0YK48 | Tropomyosin alpha-1 chain | 65.32 | 4 | 24 | 60 |
| O43396 | Thioredoxin-like protein 1 | 30.10 | 7 | 8 | 24 |
| P28289 | Tropomodulin-1 | 60.72 | 23 | 24 | 71 |
| P17980 | 26S protease regulatory subunit 6A | 35.54 | 12 | 15 | 30 |
| Q14254 | Flotillin-2 | 54.91 | 21 | 24 | 57 |
| P60900 | Proteasome subunit alpha type-6 | 43.50 | 9 | 10 | 25 |
| P62879 | Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-2 | 37.65 | 4 | 11 | 36 |
| O75955 | Flotillin-1 | 31.15 | 12 | 12 | 26 |
| Q08495 | Dematin | 62.96 | 24 | 29 | 302 |
| P02549 | Spectrin alpha chain, erythrocytic 1 | 70.28 | 172 | 192 | 732 |
| P17980 | 26S protease regulatory subunit 6A | 53.99 | 17 | 21 | 49 |
| P02549 | Spectrin alpha chain, erythrocytic 1 | 44.11 | 99 | 111 | 307 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ugurel, E.; Goksel, E.; Cilek, N.; Kaga, E.; Yalcin, O. Proteomic Analysis of the Role of the Adenylyl Cyclase–cAMP Pathway in Red Blood Cell Mechanical Responses. Cells 2022, 11, 1250. https://doi.org/10.3390/cells11071250
Ugurel E, Goksel E, Cilek N, Kaga E, Yalcin O. Proteomic Analysis of the Role of the Adenylyl Cyclase–cAMP Pathway in Red Blood Cell Mechanical Responses. Cells. 2022; 11(7):1250. https://doi.org/10.3390/cells11071250
Chicago/Turabian StyleUgurel, Elif, Evrim Goksel, Neslihan Cilek, Elif Kaga, and Ozlem Yalcin. 2022. "Proteomic Analysis of the Role of the Adenylyl Cyclase–cAMP Pathway in Red Blood Cell Mechanical Responses" Cells 11, no. 7: 1250. https://doi.org/10.3390/cells11071250
APA StyleUgurel, E., Goksel, E., Cilek, N., Kaga, E., & Yalcin, O. (2022). Proteomic Analysis of the Role of the Adenylyl Cyclase–cAMP Pathway in Red Blood Cell Mechanical Responses. Cells, 11(7), 1250. https://doi.org/10.3390/cells11071250