
SMYD2 Inhibition Downregulates TMPRSS2 and Decreases SARS-CoV-2 Infection in Human Intestinal and Airway Epithelial Cells

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Biosafety
2.2. Cell Culture
2.3. LDH Cytotoxicity Assay
2.4. Cloning and Reconstitution of recSARS-CoV-2_d6-YFP_Spike Delta
2.5. Infection Experiments
2.6. Detection, Quantification and Analysis of Infected Cells
2.7. Plaque Assay
2.8. Western Blot Analysis
2.9. RNA Extraction and Quantitative Real-Time PCR
2.10. RNA Sequencing
2.11. Immunofluorescence Staining
3. Results
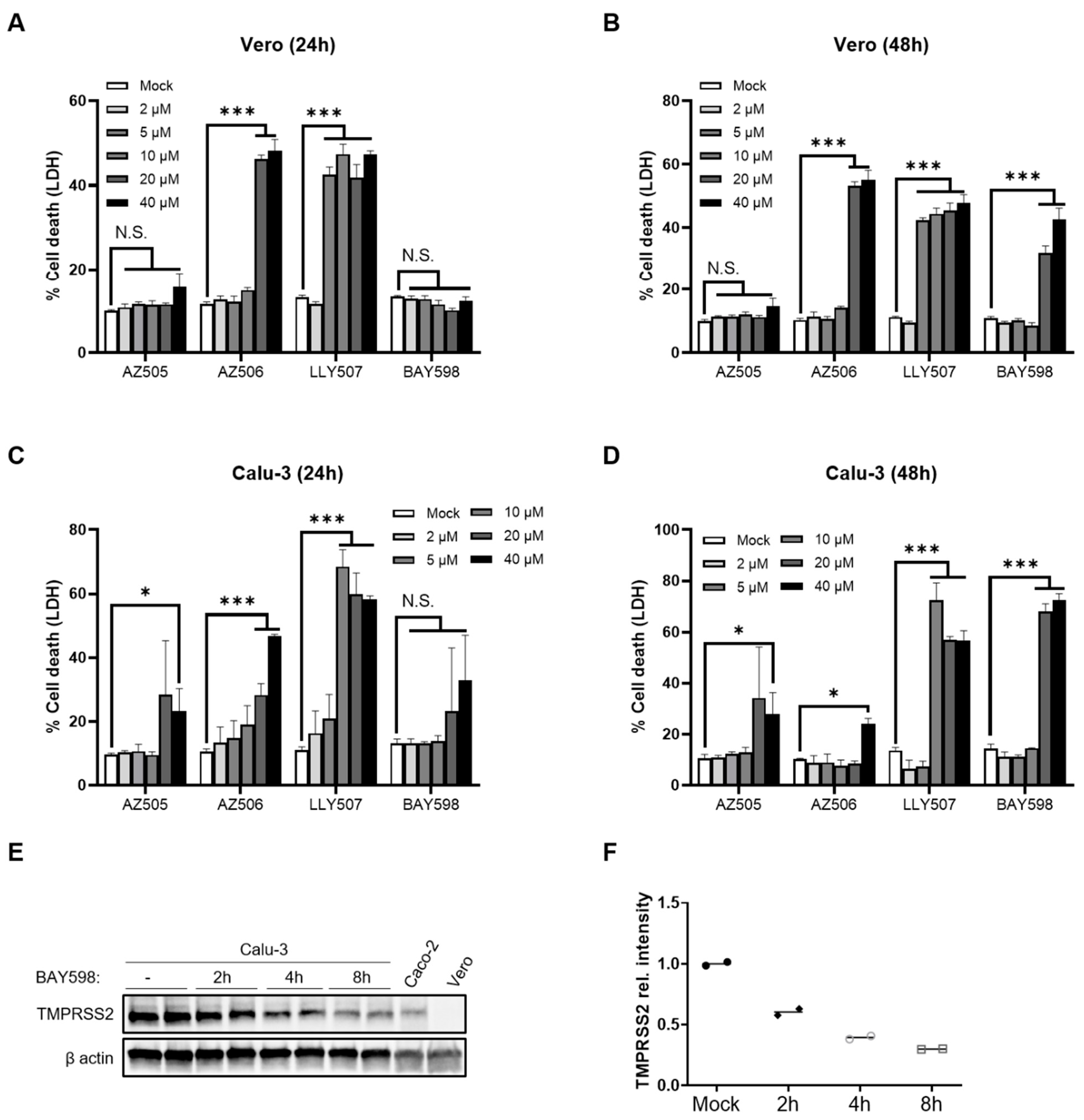
3.1. SMYD2 Deficiency or Inhibition Results in the Downregulation of TMPRSS2 in HT-29 Cells
3.2. Inhibition of SMYD2 Impedes SARS-CoV-2 Infection of Caco-2 Cells
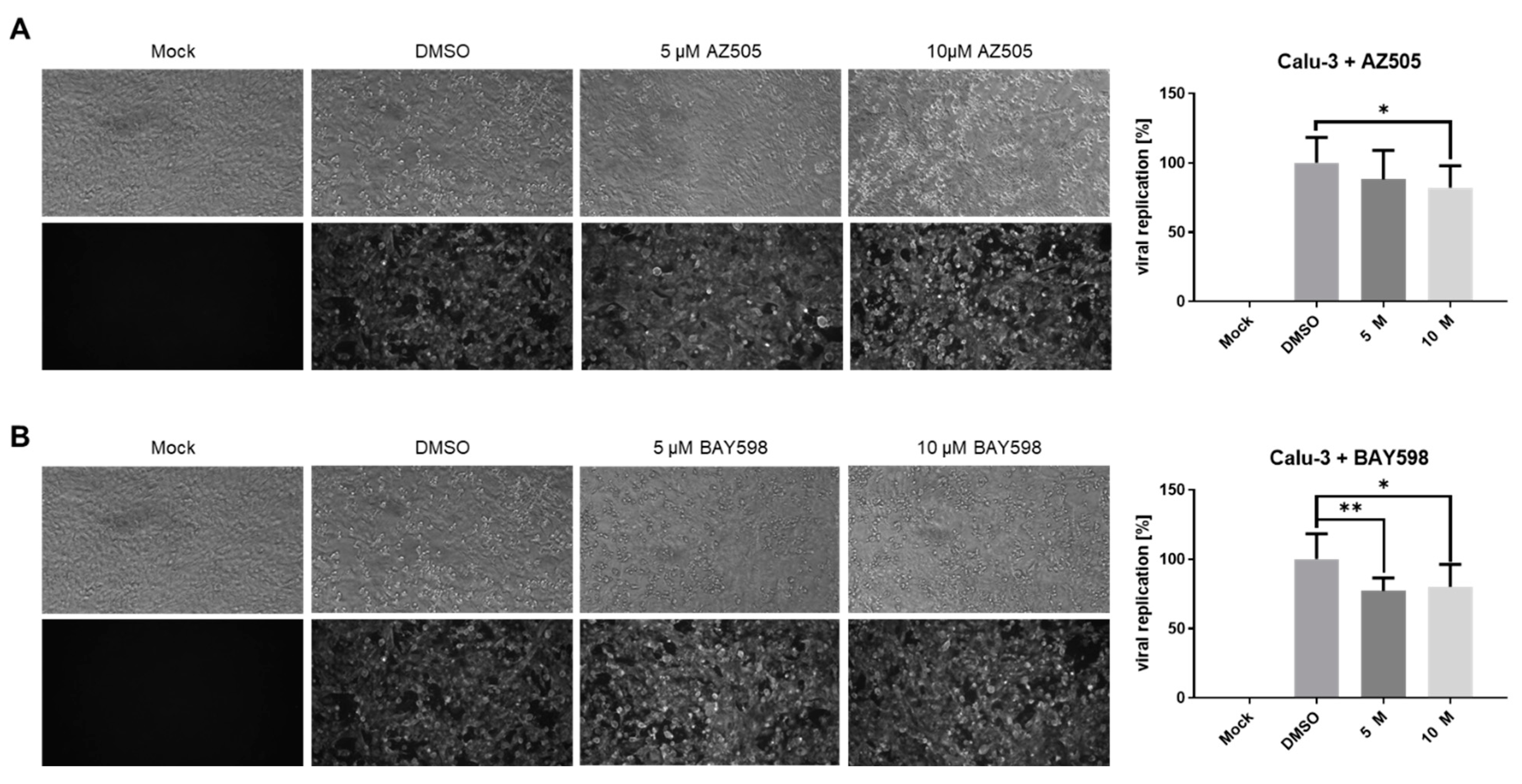
3.3. Inhibition of SMYD2 Only Slightly Affects SARS-CoV-2 Infection in Calu-3 Cells
3.4. BAY598 Similarly Inhibits Replication of Variants of Concern (VOC)
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Muller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.; Zaki, A.; Fouchier, R.A.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2021, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Jaimes, J.A.; Millet, J.K.; Whittaker, G.R. Proteolytic Cleavage of the SARS-CoV-2 Spike Protein and the Role of the Novel S1/S2 Site. iScience 2020, 23, 101212. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Gottlieb, R.L.; Vaca, C.E.; Paredes, R.; Mera, J.; Webb, B.J.; Perez, G.; Oguchi, G.; Ryan, P.; Nielsen, B.U.; Brown, M.; et al. Early Remdesivir to Prevent Progression to Severe Covid-19 in Outpatients. N. Engl. J. Med. 2022, 386, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martin-Quiros, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Drozdzal, S.; Rosik, J.; Lechowicz, K.; Machaj, F.; Szostak, B.; Przybycinski, J.; Lorzadeh, S.; Kotfis, K.; Ghavami, S.; Los, M.J. An update on drugs with therapeutic potential for SARS-CoV-2 (COVID-19) treatment. Drug Resist. Updates 2021, 59, 100794. [Google Scholar] [CrossRef]
- Mahase, E. Covid-19: Pfizer’s paxlovid is 89% effective in patients at risk of serious illness, company reports. BMJ 2021, 375, n2713. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 2021, 184, 1171–1187.e20. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2021, 602, 657–663. [Google Scholar] [CrossRef]
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.G.; et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature 2021, 602, 654–656. [Google Scholar] [CrossRef]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, S.; Wu, B.; Yang, Q.; Chen, A.; Li, Y.; Zhang, Y.; Pan, T.; Zhang, H.; He, X. SARS-CoV-2 Omicron strain exhibits potent capabilities for immune evasion and viral entrance. Signal Transduct. Target. Ther. 2021, 6, 430. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Heinlein, C.; Hackman, R.C.; Nelson, P.S. Phenotypic analysis of mice lacking the Tmprss2-encoded protease. Mol. Cell Biol. 2006, 26, 965–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Farha, M.; Lambert, J.P.; Al-Madhoun, A.S.; Elisma, F.; Skerjanc, I.S.; Figeys, D. The tale of two domains: Proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol. Cell Proteom. 2008, 7, 560–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benayoun, B.A.; Veitia, R.A. A post-translational modification code for transcription factors: Sorting through a sea of signals. Trends Cell Biol. 2009, 19, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, R.; Saloura, V.; Nakamura, Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat Rev. Cancer 2015, 15, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Boriack-Sjodin, P.A.; Swinger, K.K. Protein Methyltransferases: A Distinct, Diverse, and Dynamic Family of Enzymes. Biochemistry 2016, 55, 1557–1569. [Google Scholar] [CrossRef]
- Cho, H.S.; Hayami, S.; Toyokawa, G.; Maejima, K.; Yamane, Y.; Suzuki, T.; Dohmae, N.; Kogure, M.; Kang, D.; Neal, D.E.; et al. RB1 methylation by SMYD2 enhances cell cycle progression through an increase of RB1 phosphorylation. Neoplasia 2012, 14, 476–486. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, S.; Ichikawa, D.; Hirajima, S.; Nagata, H.; Nishimura, Y.; Kawaguchi, T.; Miyamae, M.; Okajima, W.; Ohashi, T.; Konishi, H.; et al. Overexpression of SMYD2 contributes to malignant outcome in gastric cancer. Br. J. Cancer 2015, 112, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, S.; Imoto, I.; Tsuda, H.; Kozaki, K.I.; Muramatsu, T.; Shimada, Y.; Aiko, S.; Yoshizumi, Y.; Ichikawa, D.; Otsuji, E.; et al. Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis 2009, 30, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, L.H.; Andrade, R.V.; Felipe, M.S.; Motoyama, A.B.; Pittella Silva, F. SMYD2 is highly expressed in pediatric acute lymphoblastic leukemia and constitutes a bad prognostic factor. Leuk. Res. 2014, 38, 496–502. [Google Scholar] [CrossRef]
- Spellmon, N.; Holcomb, J.; Trescott, L.; Sirinupong, N.; Yang, Z. Structure and function of SET and MYND domain-containing proteins. Int. J. Mol. Sci. 2015, 16, 1406–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tracy, C.; Warren, J.S.; Szulik, M.; Wang, L.; Garcia, J.; Makaju, A.; Russell, K.; Miller, M.; Franklin, S. The Smyd Family of Methyltransferases: Role in Cardiac and Skeletal Muscle Physiology and Pathology. Curr. Opin. Physiol. 2018, 1, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Eggert, E.; Hillig, R.C.; Koehr, S.; Stockigt, D.; Weiske, J.; Barak, N.; Mowat, J.; Brumby, T.; Christ, C.D.; Ter Laak, A.; et al. Discovery and Characterization of a Highly Potent and Selective Aminopyrazoline-Based in Vivo Probe (BAY-598) for the Protein Lysine Methyltransferase SMYD2. J. Med. Chem. 2016, 59, 4578–4600. [Google Scholar] [CrossRef]
- Ferguson, A.D.; Larsen, N.A.; Howard, T.; Pollard, H.; Green, I.; Grande, C.; Cheung, T.; Garcia-Arenas, R.; Cowen, S.; Wu, J.; et al. Structural basis of substrate methylation and inhibition of SMYD2. Structure 2011, 19, 1262–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.; Allali-Hassani, A.; Antonysamy, S.; Chang, S.; Chen, L.H.; Curtis, C.; Emtage, S.; Fan, L.; Gheyi, T.; Li, F.; et al. LLY-507, a Cell-active, Potent, and Selective Inhibitor of Protein-lysine Methyltransferase SMYD2. J. Biol. Chem. 2015, 290, 13641–13653. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.Q.; Thonn, V.; Patankar, J.V.; Thoma, O.M.; Waldner, M.; Zielinska, M.; Bao, L.L.; Gonzalez-Acera, M.; Wallmuller, S.; Engel, F.B.; et al. SMYD2 targets RIPK1 and restricts TNF-induced apoptosis and necroptosis to support colon tumor growth. Cell Death Dis. 2022, 13, 52. [Google Scholar] [CrossRef]
- Herrmann, A.; Jungnickl, D.; Cordsmeier, A.; Peter, A.S.; Uberla, K.; Ensser, A. Cloning of a Passage-Free SARS-CoV-2 Genome and Mutagenesis Using Red Recombination. Int. J. Mol. Sci. 2021, 22, 188. [Google Scholar] [CrossRef]
- Peter, A.S.; Roth, E.; Schulz, S.R.; Fraedrich, K.; Steinmetz, T.; Damm, D.; Hauke, M.; Richel, E.; Mueller-Schmucker, S.; Habenicht, K.; et al. A pair of non-competing neutralizing human monoclonal antibodies protecting from disease in a SARS-CoV-2 infection model. Eur. J. Immunol. 2021. [Google Scholar] [CrossRef]
- Cowen, S.D.; Russell, D.; Dakin, L.A.; Chen, H.; Larsen, N.A.; Godin, R.; Throner, S.; Zheng, X.; Molina, A.; Wu, J.; et al. Design, Synthesis, and Biological Activity of Substrate Competitive SMYD2 Inhibitors. J. Med. Chem. 2016, 59, 11079–11097. [Google Scholar] [CrossRef]
- Hahn, F.; Hage, S.; Herrmann, A.; Wangen, C.; Kicuntod, J.; Jungnickl, D.; Tillmanns, J.; Muller, R.; Fraedrich, K.; Uberla, K.; et al. Methodological Development of a Multi-Readout Assay for the Assessment of Antiviral Drugs against SARS-CoV-2. Pathogens 2021, 10, 1076. [Google Scholar] [CrossRef]
- Trogen, B.; Pirofski, L.A. Understanding vaccine hesitancy in COVID-19. Med 2021, 2, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Rzymski, P.; Camargo, C.A., Jr.; Fal, A.; Flisiak, R.; Gwenzi, W.; Kelishadi, R.; Leemans, A.; Nieto, J.J.; Ozen, A.; Perc, M.; et al. COVID-19 Vaccine Boosters: The Good, the Bad, and the Ugly. Vaccines 2021, 9, 1299. [Google Scholar] [CrossRef]
- Tartof, S.Y.; Slezak, J.M.; Puzniak, L.; Hong, V.; Frankland, T.B.; Ackerson, B.K.; Takhar, H.S.; Ogun, O.A.; Simmons, S.R.; Zamparo, J.M.; et al. Effectiveness of a third dose of BNT162b2 mRNA COVID-19 vaccine in a large US health system: A retrospective cohort study. Lancet Reg. Health Am. 2022, 100198. [Google Scholar] [CrossRef]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Smith, J.C.; Kruger, N.; Arora, P.; Sorensen, L.K.; Sogaard, O.S.; Hasselstrom, J.B.; Winkler, M.; Hempel, T.; et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine 2021, 65, 103255. [Google Scholar] [CrossRef]
- Hoffmann, M.; Schroeder, S.; Kleine-Weber, H.; Muller, M.A.; Drosten, C.; Pohlmann, S. Nafamostat Mesylate Blocks Activation of SARS-CoV-2: New Treatment Option for COVID-19. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [Green Version]
- Koch, J.; Uckeley, Z.M.; Doldan, P.; Stanifer, M.; Boulant, S.; Lozach, P.Y. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. EMBO J. 2021, 40, e107821. [Google Scholar] [CrossRef]
- Kishimoto, M.; Uemura, K.; Sanaki, T.; Sato, A.; Hall, W.W.; Kariwa, H.; Orba, Y.; Sawa, H.; Sasaki, M. TMPRSS11D and TMPRSS13 Activate the SARS-CoV-2 Spike Protein. Viruses 2021, 13, 384. [Google Scholar] [CrossRef]
- Abe, M.; Tahara, M.; Sakai, K.; Yamaguchi, H.; Kanou, K.; Shirato, K.; Kawase, M.; Noda, M.; Kimura, H.; Matsuyama, S.; et al. TMPRSS2 is an activating protease for respiratory parainfluenza viruses. J. Virol. 2013, 87, 11930–11935. [Google Scholar] [CrossRef] [Green Version]
- Bertram, S.; Dijkman, R.; Habjan, M.; Heurich, A.; Gierer, S.; Glowacka, I.; Welsch, K.; Winkler, M.; Schneider, H.; Hofmann-Winkler, H.; et al. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J. Virol. 2013, 87, 6150–6160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottcher, E.; Matrosovich, T.; Beyerle, M.; Klenk, H.D.; Garten, W.; Matrosovich, M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 2006, 80, 9896–9898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gierer, S.; Bertram, S.; Kaup, F.; Wrensch, F.; Heurich, A.; Kramer-Kuhl, A.; Welsch, K.; Winkler, M.; Meyer, B.; Drosten, C.; et al. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J. Virol. 2013, 87, 5502–5511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef] [Green Version]
- Shirato, K.; Kawase, M.; Matsuyama, S. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J. Virol. 2013, 87, 12552–12561. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.-Q.; Herrmann, A.; Thonn, V.; Cordsmeier, A.; Neurath, M.F.; Ensser, A.; Becker, C. SMYD2 Inhibition Downregulates TMPRSS2 and Decreases SARS-CoV-2 Infection in Human Intestinal and Airway Epithelial Cells. Cells 2022, 11, 1262. https://doi.org/10.3390/cells11081262
Yu Y-Q, Herrmann A, Thonn V, Cordsmeier A, Neurath MF, Ensser A, Becker C. SMYD2 Inhibition Downregulates TMPRSS2 and Decreases SARS-CoV-2 Infection in Human Intestinal and Airway Epithelial Cells. Cells. 2022; 11(8):1262. https://doi.org/10.3390/cells11081262
Chicago/Turabian StyleYu, Yu-Qiang, Alexandra Herrmann, Veronika Thonn, Arne Cordsmeier, Markus F. Neurath, Armin Ensser, and Christoph Becker. 2022. "SMYD2 Inhibition Downregulates TMPRSS2 and Decreases SARS-CoV-2 Infection in Human Intestinal and Airway Epithelial Cells" Cells 11, no. 8: 1262. https://doi.org/10.3390/cells11081262