
Switching of Redox Signaling by Prdx6 Expression Decides Cellular Fate by Hormetic Phenomena Involving Nrf2 and Reactive Oxygen Species




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Generation of Primary hLECs and SRA-hLECs
2.2. Generation and Validation of LECs Isolated from Lenses of Prdx6+/+ and Prdx6−/− Mice
2.3. Real-Time Quantitative Reverse Transcriptase-Polymerase Chain Reaction (RT-qPCR)
2.4. Protein Expression Analysis
2.5. Quantitation of Intracellular ROS Level by H2-DCF-DA and CellROX® Deep Red Reagent
2.6. Measurement of H2O2 Generation in Lens Epithelial Cells
2.7. Cell Survival Assay (MTS Assay)
2.8. Lentiviral (LV) Infection
2.9. Eukaryotic Plasmids
2.10. Extraction of Nuclear and Cytosolic Fraction
2.11. Transcription Factor Nrf2 Activation Assay
2.12. Chromatin Immunoprecipitation (ChIP) Assay for DNA-Protein Binding In Vivo
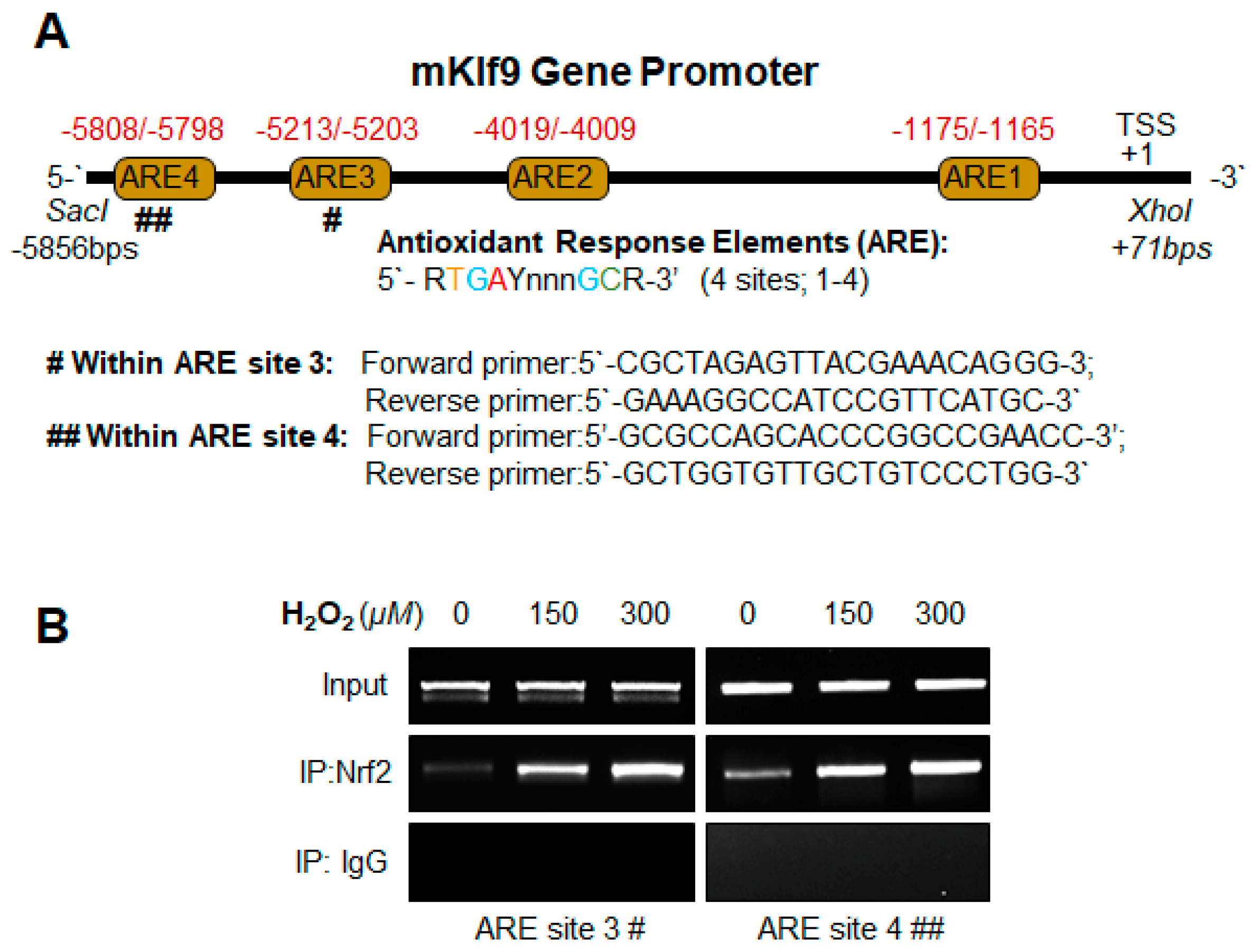
2.12.1. Mouse Klf9 Promoter ChIP Primers
- Klf9 ARE3:
- Forward primer: 5′-CGCTAGAGTTACGAAACAGGG-3′;
- Reverse primer: 5′-GAAAGGCCATCCGTTCATGC-3′
- Klf9 ARE4:
- Forward primer: 5′-GCGCCAGCACCCGGCCGAACC-3′;
- Reverse primer: 5′-GCTGGTGTTGCTGTCCCTGG-3′
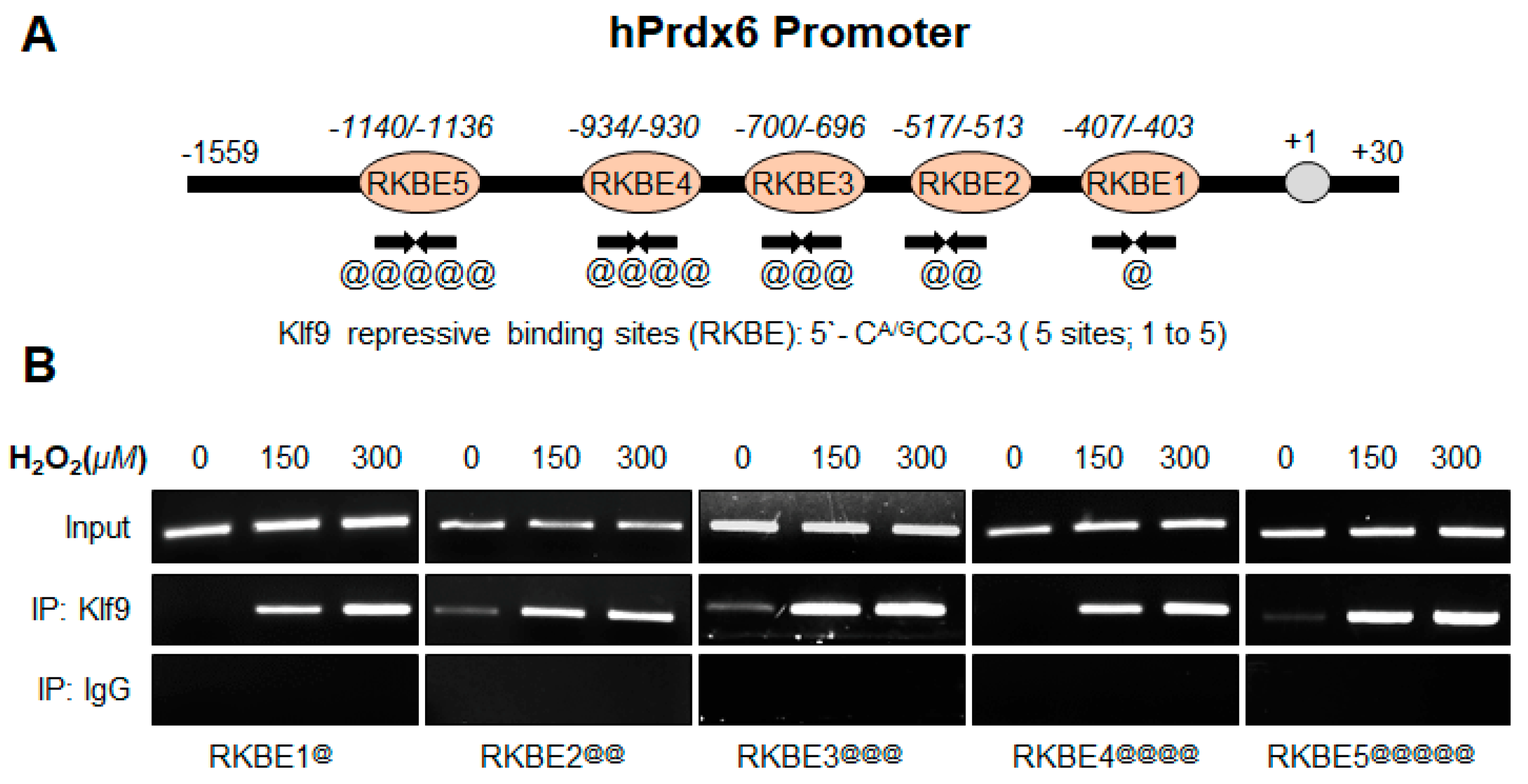
2.12.2. Human Prdx6 Promoter ChIP Primers
- Prdx6 RKBE1:
- Forward primer: 5′-GCTGTGCGAAGCCGCCGCA-3′;
- Reverse primer: 5′-GAAGCTTGAGGATGCGCCA-3
- Prdx6 RKBE2:
- Forward primer: 5′-GTAGTCGAGCAGTCACTCCA-3′;
- Reverse primer: 5′-GAAGGAAGAGGAACGCGGCAG-3
- Prdx6 RKBE3:
- Forward primer: 5′-GGTTCATAACAAACAGAAAGG-3′;
- Reverse primer: 5′-AGCCCAGCTACGATGAACTG-3
- Prdx6 RKBE4:
- Forward primer: 5′-GTCTGTCACCGGTTTCCCTT-3′;
- Reverse primer: 5′-GAGACCTACTGTGTGCAGGT-3
- Prdx6 RKBE5:
- Forward primer: 5′-CAGAGCACCTACCGTGAGCT-3′;
- Reverse primer: 5′-GAAGGGAAACCGGTGACAGA-3
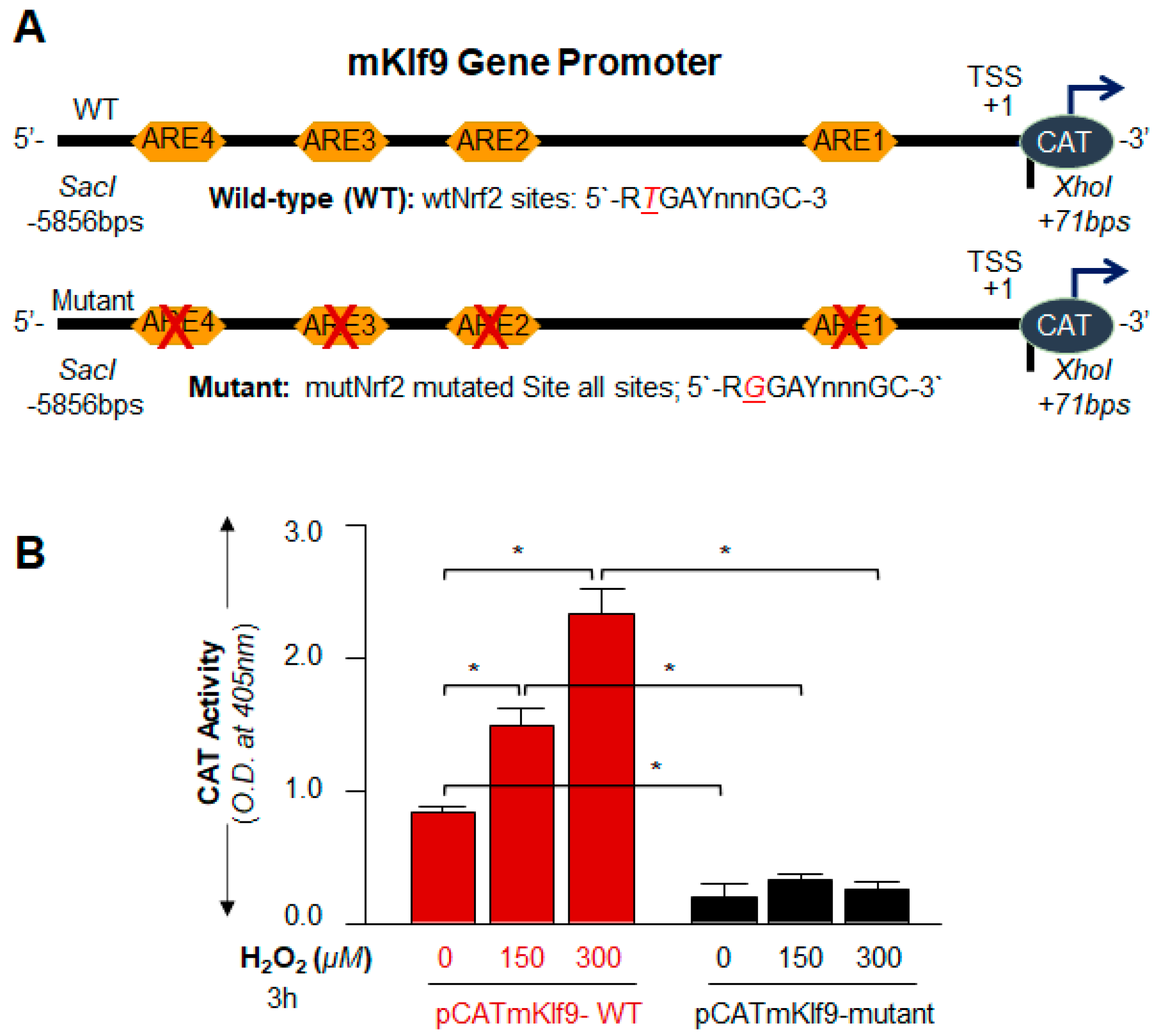
2.13. Preparation of Mouse Klf9 Promoter-Linked to Chloramphenicol Acetyltransferase (CAT) Reporter Plasmid
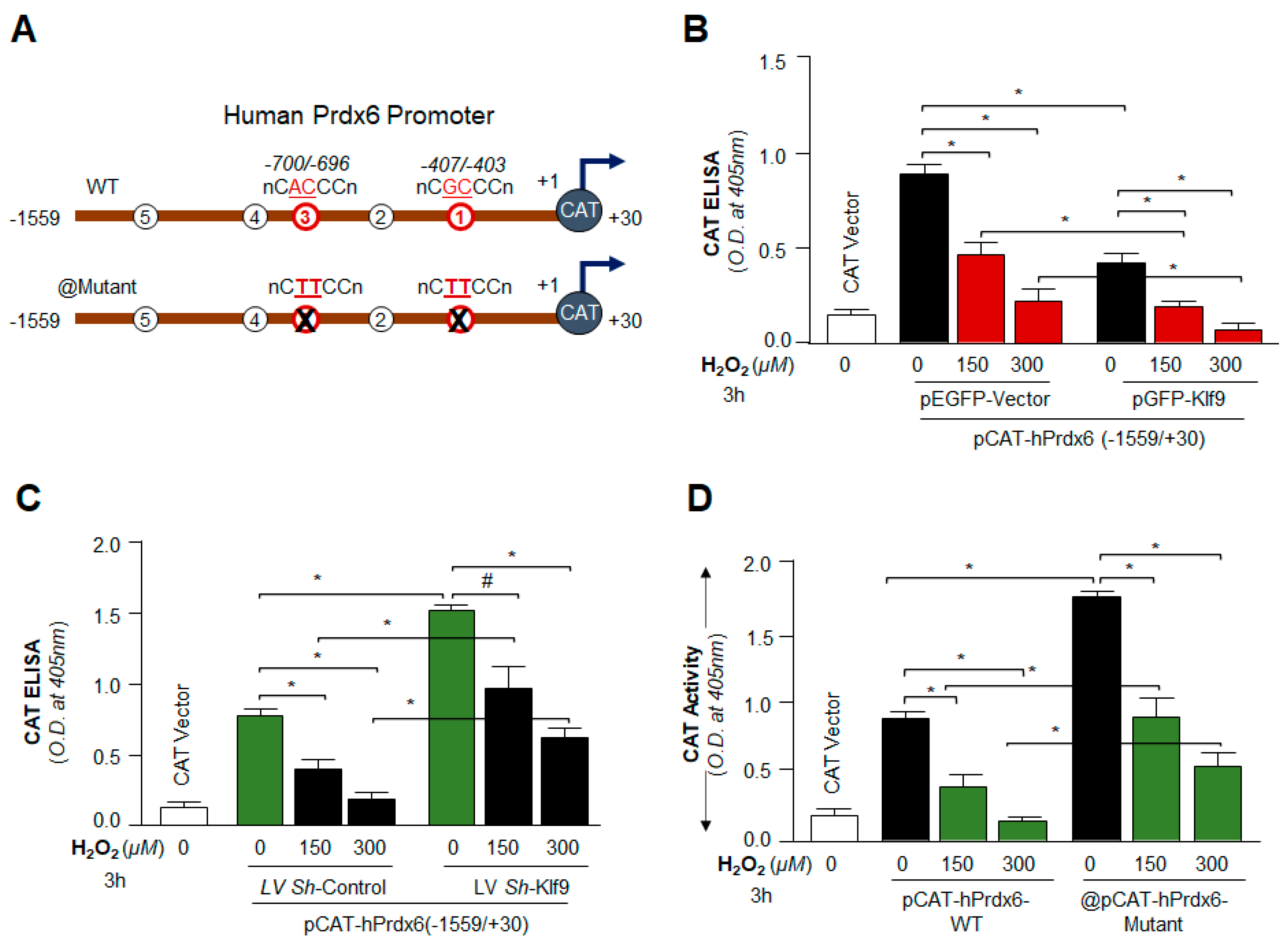
2.14. Preparation of Human Prdx6gene Promoter-Fused to Chloramphenicol Acetyltransferase (CAT) Reporter Plasmid Vector
2.15. Site-Directed Mutagenesis (SDM)
2.15.1. Nrf2/ARE SDM Primer
- Klf9 promoter, ARE site1 mutant (ARE1-mut; A to C, −1176 to −1164):
- Forward primer: 5′-CTCTAAAGCAGAGTCCGGAATCGGGAACC-3′
- Reverse primer: 5′-GGTTCCCGATTCCGGACTCTGCTTTAGAG-3′
- Klf9 promoter, ARE site2 mutant (ARE2-mut; T to G, −4020 to −4008):
- Forward primer: 5′-CAGATGAGGCACTGTTCGGAGAGAGCAAATCTTAC-3′
- Reverse primer: 5′-GTAAGATTTGCTCTCTCCGAACAGTGCCTCATCTG-3′
- Klf9 promoter, ARE site3 mutant (ARE3-mut; T to G, −5213 to −5203):
- Forward primer: 5′-CTGTCCTCAAAGGAACCTGCCTCCTC-3′
- Reverse primer: 5′-GAGGAGGCAGGTTCCTTTGAGGACAG-3′
- Klf9 promoter, ARE site4 mutant (ARE4-mut; A to C, −5808 to −5798):
- Forward primer: 5′-CGATTCCTGCAAAGTCCTCTCCACTCGCAC-3′
- Reverse primer: 5′-GTGCGAGTGGAGAGGACTTTGCAGGAATCG-3′
2.15.2. Klf9/RKBE SDM Primer
- Prdx6 promoter, RKBE1 mutant (RKBE1-mut; GC to TT, −407 to −403):
- Forward primer: 5′-CCCTAAAGCGCGTACTTCCTGCAGAGTCAAACC-3′
- Reverse primer: 5′-GGTTTGACTCTGCAGGAAGTACGCGCTTTAGGG-3
- Prdx6 promoter, RKBE3 mutant (RKBE3-mut; AC to TT, −700 to −696):
- Forward primer: 5′-CTCTGACATAAGGTCTTCCATACTTCTGGGTC-3′
- Reverse primer: 5′-GACCCAGAAGTATGGAAGACCTTATGTCAGAG-3
2.16. Statistical Analysis
3. Results
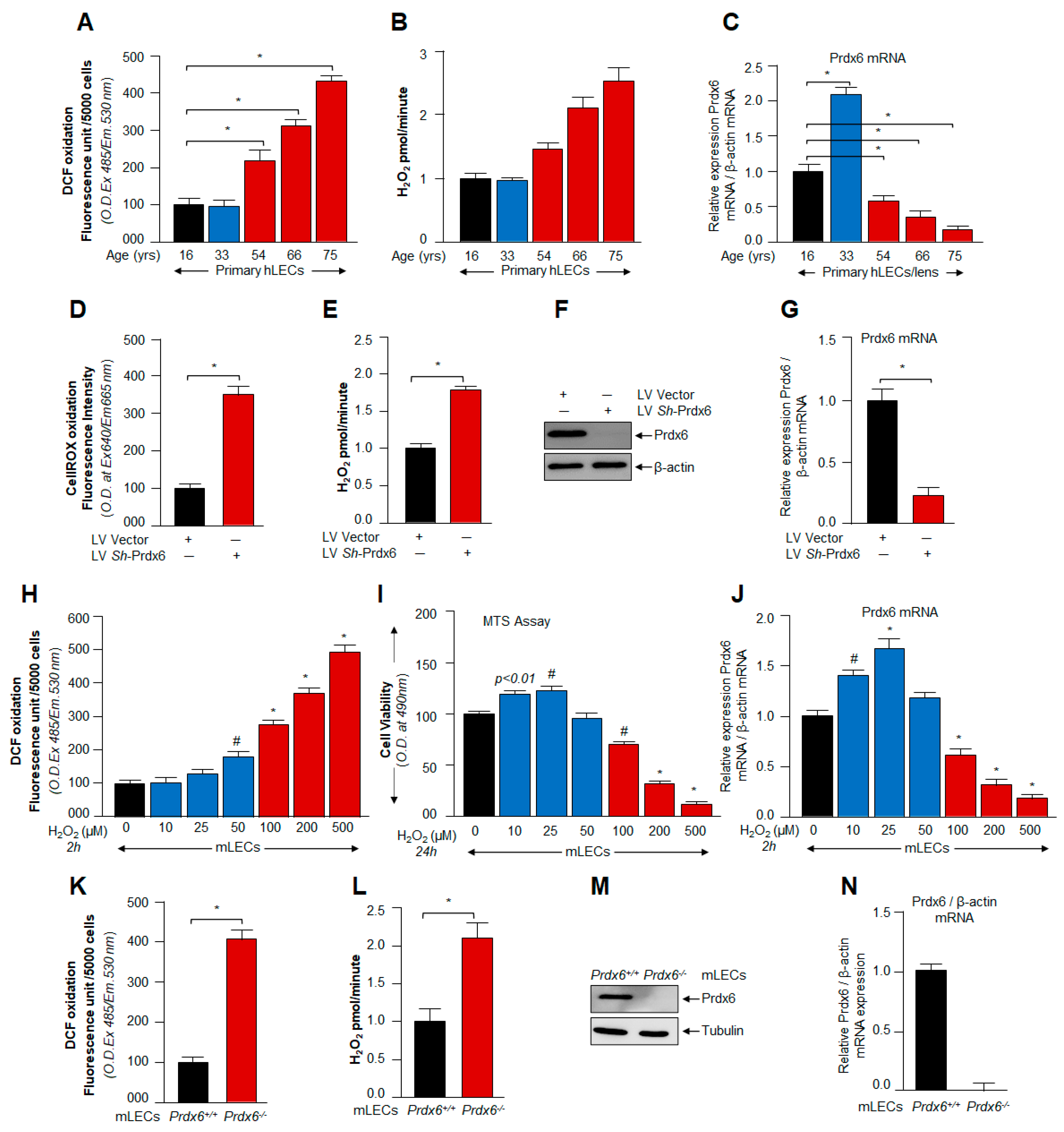
3.1. ROS/H2O2-Dependent Biological Response and Cellular Fate Was Linked to Cellular Level of Prdx6
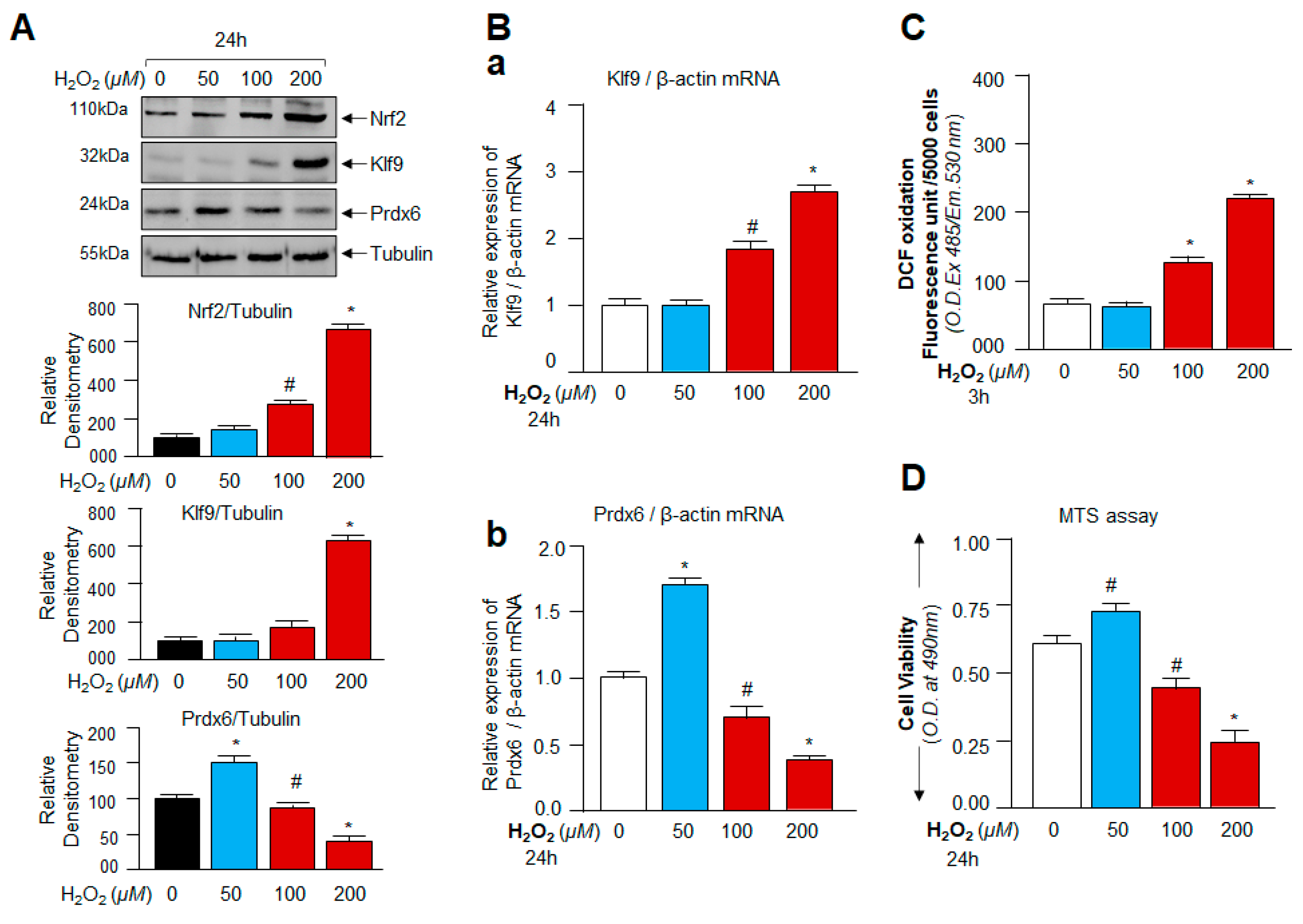
3.2. Oxidative Stress-Dependent Nrf2-Klf9 Expression Controlled Prdx6 Abundance-Mediated ROS Homeostasis and Cell Fate
3.3. Cellular Prdx6 Suppression and ROS Accumulation, and Cell Death Was Connected to Expression and Nuclear Accumulation of Nrf2 and Klf9
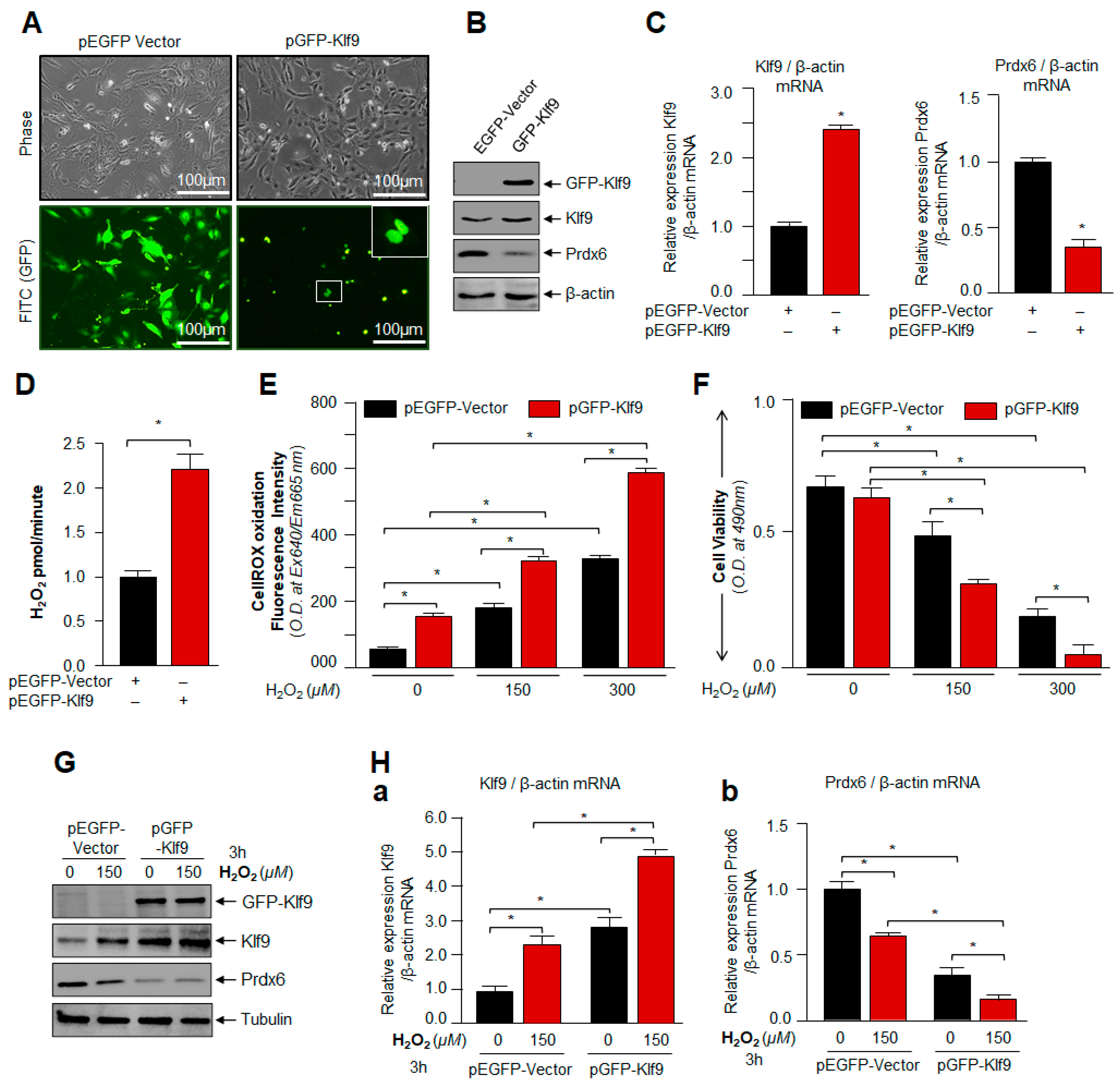
3.4. Overexpression of Klf9 Resulted in Increased ROS Production and Reduced Cells Viability with a Significant Reduction in Prdx6 Expression in hLECs during Oxidative Stress
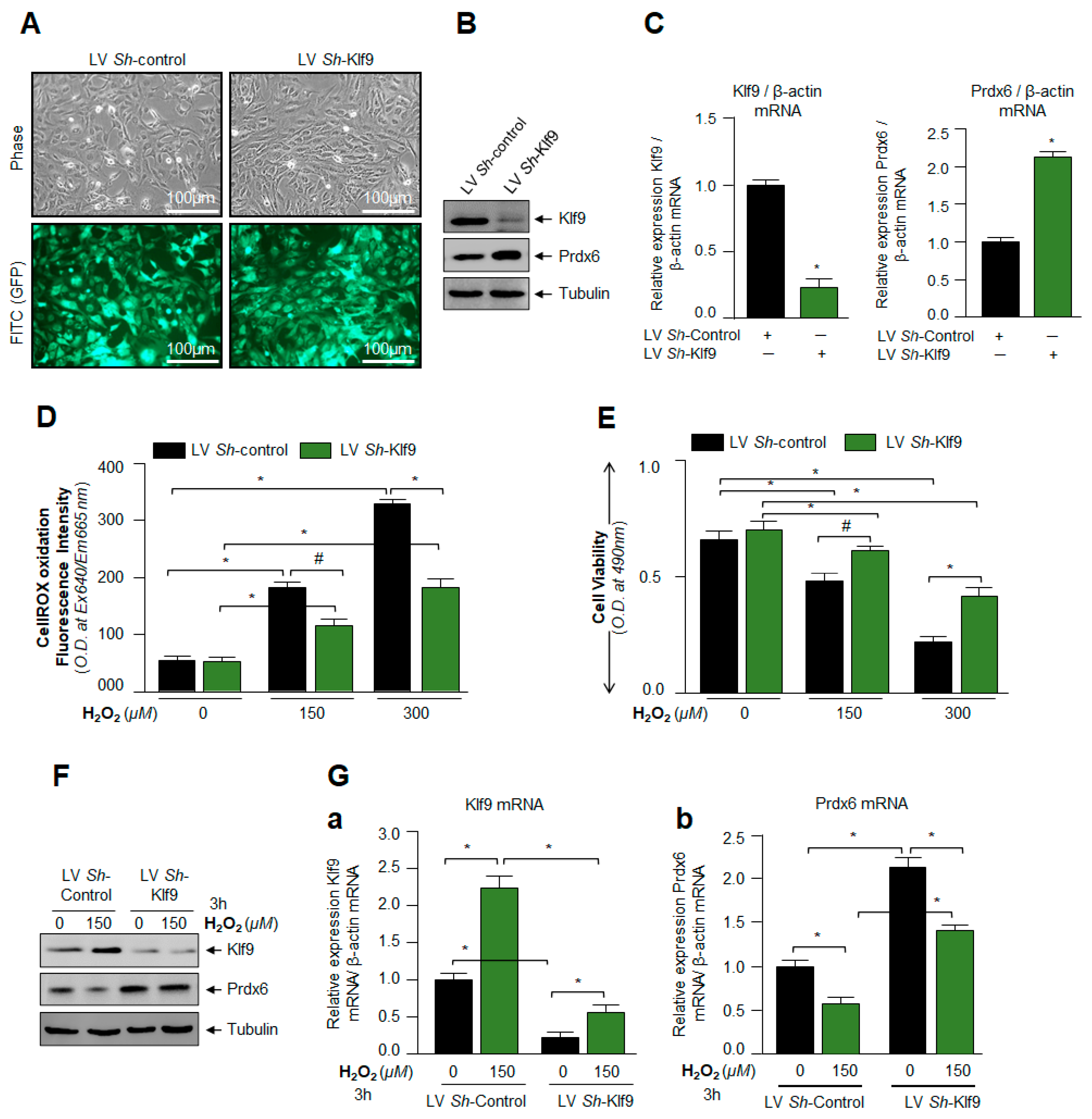
3.5. Klf9 Depletion Caused Reduction in ROS Level and Abated Cell Death by Increasing Prdx6 Expression
3.6. Klf9 Gene Bore Nrf2 Binding Sequences, ARE, and Nrf2/ARE Binding Increased with Increase in Oxidative Stress
3.7. Nrf2 Upregulated Klf9 Transcription through ARE Binding and the Transcriptional Activity Was Oxidative Load-Dependent
3.8. In Vivo DNA Binding Assay Disclosed That Oxidative Stress Level Stimulated Klf9 Binding to Its New Target Gene, Prdx6 in Dose-Dependent Fashion
3.9. Loss and Gain Experiments Disclosed That the Prdx6 Transcription Was Dependent on Cellular Availability of Klf9 during Oxidative Stress
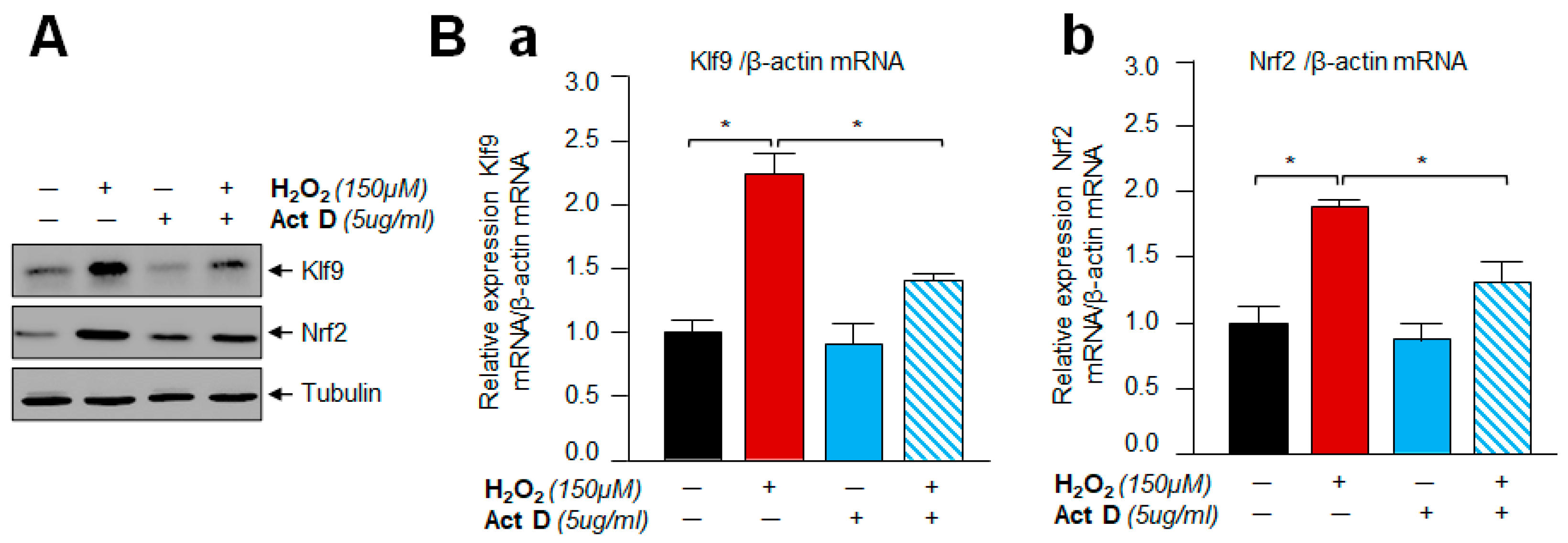
3.10. A Transcription Inhibitor, Actinomycin D Treatment Demonstrated That Klf9 Induction by Oxidative Stress Did Not Occur Post-Transcriptionally
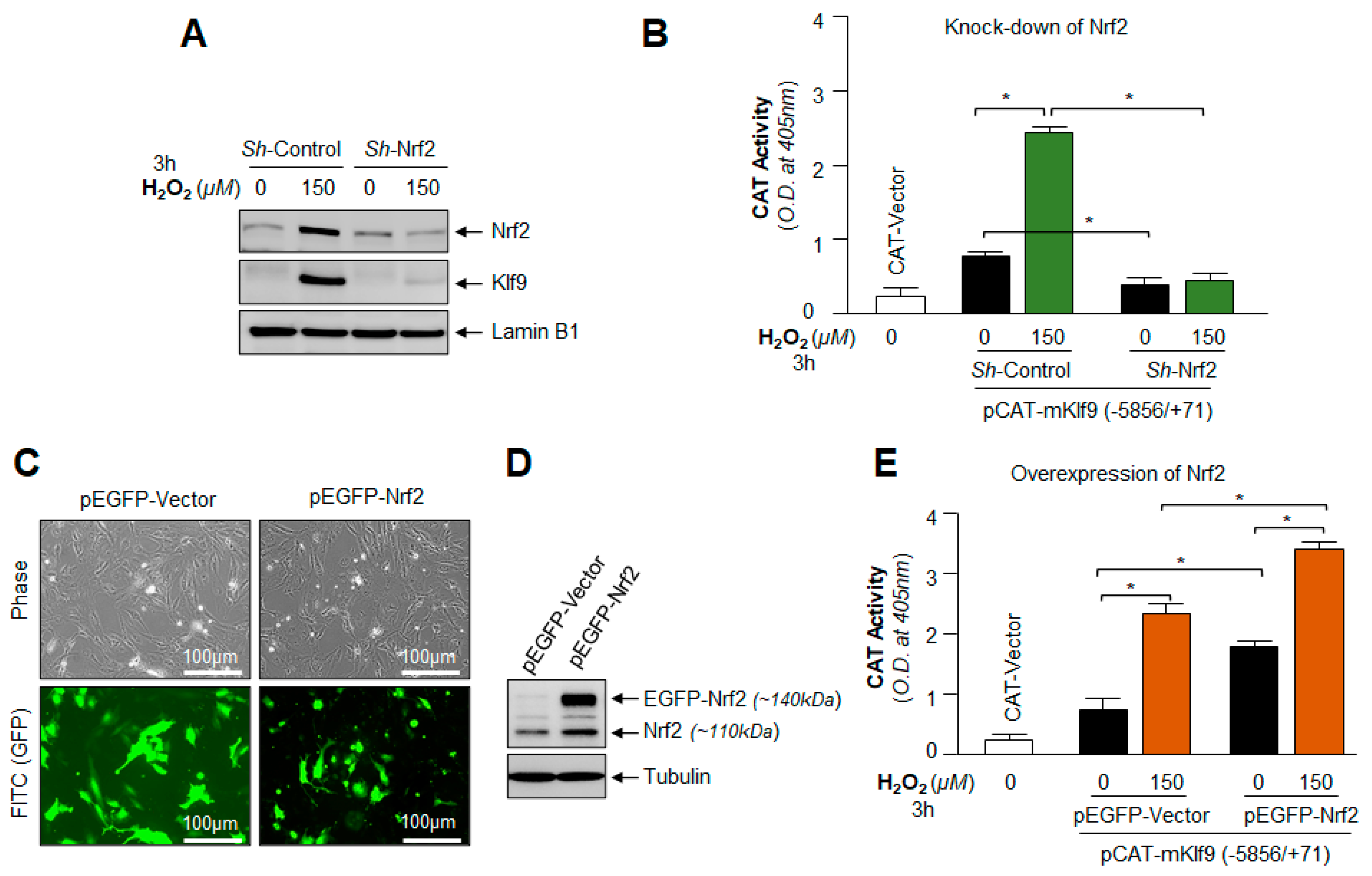
3.11. Under- and Over-Expression Experimentation Disclosed That Klf9 Expression Was Transcriptionally Regulated by Nrf2
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martins, I.; Galluzzi, L.; Kroemer, G. Hormesis, cell death and aging. Aging 2011, 3, 821–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, E.J. Toxicology rewrites its history and rethinks its future: Giving equal focus to both harmful and beneficial effects. Environ. Toxicol. Chem. 2011, 30, 2658–2673. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Blain, R.B. The hormesis database: The occurrence of hormetic dose responses in the toxicological literature. Regul. Toxicol. Pharmacol. 2011, 61, 73–81. [Google Scholar] [CrossRef]
- Ludovico, P.; Burhans, W.C. Reactive oxygen species, ageing and the hormesis police. FEMS Yeast Res. 2014, 14, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhunchha, B.; Fatma, N.; Kubo, E.; Singh, D.P. Aberrant sumoylation signaling evoked by reactive oxygen species impairs protective function of Prdx6 by destabilization and repression of its transcription. FEBS J. 2014, 281, 3357–3381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhunchha, B.; Singh, P.; Stamer, W.D.; Singh, D.P. Prdx6 retards senescence and restores trabecular meshwork cell health by regulating reactive oxygen species. Cell Death Discov. 2017, 3, 17060. [Google Scholar] [CrossRef] [Green Version]
- Kubo, E.; Hasanova, N.; Tanaka, Y.; Fatma, N.; Takamura, Y.; Singh, D.P.; Akagi, Y. Protein expression profiling of lens epithelial cells from Prdx6-depleted mice and their vulnerability to UV radiation exposure. Am. J. Physiol. Physiol. 2010, 298, C342–C354. [Google Scholar] [CrossRef]
- Singh, D.P.; Bhargavan, B.; Chhunchha, B.; Kubo, E.; Kumar, A.; Fatma, N. Transcriptional Protein Sp1 Regulates LEDGF Transcription by Directly Interacting with Its Cis-Elements in GC-Rich Region of TATA-Less Gene Promoter. PLoS ONE 2012, 7, e37012. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.P.; Chhunchha, B.; Fatma, N.; Kubo, E.; Singh, S.P.; Singh, D.P. Delivery of a protein transduction domain-mediated Prdx6 protein ameliorates oxidative stress-induced injury in human and mouse neuronal cells. Am. J. Physiol. Cell Physiol. 2016, 310, C1–C16. [Google Scholar] [CrossRef] [Green Version]
- Fatma, N.; Singh, P.; Chhunchha, B.; Kubo, E.; Shinohara, T.; Bhargavan, B.; Singh, D.P. Deficiency of Prdx6 in lens epithelial cells induces ER stress response-mediated impaired homeostasis and apoptosis. Am. J. Physiol. Physiol. 2011, 301, C954–C967. [Google Scholar] [CrossRef]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Sulforaphane-Induced Klf9/Prdx6 Axis Acts as a Molecular Switch to Control Redox Signaling and Determines Fate of Cells. Cells 2019, 8, 1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, E.; Chhunchha, B.; Singh, P.; Sasaki, H.; Singh, D.P. Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci. Rep. 2017, 7, 14130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi-Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 Amplifies Oxidative Stress via Induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, V.; Bates, T.E.; Mancuso, C.; Cornelius, C.; Ventimiglia, B.; Cambria, M.T.; Di Renzo, L.; De Lorenzo, A.; Dinkova-Kostova, A.T. Curcumin and the cellular stress response in free radical-related diseases. Mol. Nutr. Food Res. 2008, 52, 1062–1073. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.; Iavicoli, I.; di Paola, R.; Koverech, A.; Cuzzocrea, S.; Rizzarelli, E.; Calabrese, E.J. Cellular stress responses, hormetic phytochemicals and vitagenes in aging and longevity. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 753–783. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, V.; Cornelius, C.; Cuzzocrea, S.; Iavicoli, I.; Rizzarelli, E.; Calabrese, E.J. Hormesis, cellular stress response and vitagenes as critical determinants in aging and longevity. Mol. Asp. Med. 2011, 32, 279–304. [Google Scholar] [CrossRef]
- Hekimi, S.; Lapointe, J.; Wen, Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011, 21, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Labunskyy, V.; Gladyshev, V.N. Role of Reactive Oxygen Species-Mediated Signaling in Aging. Antioxid. Redox Signal. 2013, 19, 1362–1372. [Google Scholar] [CrossRef] [Green Version]
- Maryanovich, M.; Gross, A. A ROS rheostat for cell fate regulation. Trends Cell Biol. 2013, 23, 129–134. [Google Scholar] [CrossRef]
- Stancill, J.S.; Corbett, J.A. The Role of Thioredoxin/Peroxiredoxin in the beta-Cell Defense Against Oxidative Damage. Front. Endocrinol. 2021, 12, 718235. [Google Scholar] [CrossRef]
- Fatma, N.; Kubo, E.; Sharma, P.; Beier, D.R.; Singh, D.P. Impaired homeostasis and phenotypic abnormalities in Prdx6-/-mice lens epithelial cells by reactive oxygen species: Increased expression and activation of TGFbeta. Cell Death Differ. 2005, 12, 734–750. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zhang, X.; Zheng, M.L.; Li, Z.; Zhao, X.; Lai, W.; Shen, H.; Lv, J.; Yang, G.; Wang, Q.; et al. Peroxiredoxin 6 Is a Crucial Factor in the Initial Step of Mitochondrial Clearance and Is Upstream of the PINK1-Parkin Pathway. Antioxid. Redox Signal. 2016, 24, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Sorokina, E.M.; Feinstein, S.I.; Zhou, S.; Fisher, A.B. Intracellular targeting of peroxiredoxin 6 to lysosomal organelles requires MAPK activity and binding to 14-3-3epsilon. Am. J. Physiol. Cell Physiol. 2011, 300, C1430–C1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhunchha, B.; Kubo, E.; Fatma, N.; Singh, D.P. Sumoylation-deficient Prdx6 gains protective function by amplifying enzymatic activity and stability and escapes oxidative stress-induced aberrant Sumoylation. Cell Death Dis. 2017, 8, e2525. [Google Scholar] [CrossRef] [Green Version]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1–Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar] [CrossRef] [Green Version]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB Protein Is an Adaptor That Bridges Nrf2 to a Cul3-Based E3 Ligase: Oxidative Stress Sensing by a Cul3-Keap1 Ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent Activation of Nrf2 Contributes to Redox Homeostasis and Cell Survival following Endoplasmic Reticulum Stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, M.; Xiong, Y. BTB Protein Keap1 Targets Antioxidant Transcription Factor Nrf2 for Ubiquitination by the Cullin 3-Roc1 Ligase. Mol. Cell. Biol. 2005, 25, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase To Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Lo, S.-C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 Is a Redox-Regulated Substrate Adaptor Protein for a Cul3-Dependent Ubiquitin Ligase Complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [Green Version]
- Buckley, B.J.; Marshall, Z.M.; Whorton, A. Nitric oxide stimulates Nrf2 nuclear translocation in vascular endothelium. Biochem. Biophys. Res. Commun. 2003, 307, 973–979. [Google Scholar] [CrossRef]
- Ashino, T.; Yamanaka, R.; Yamamoto, M.; Shimokawa, H.; Sekikawa, K.; Iwakura, Y.; Shioda, S.; Numazawa, S.; Yoshida, T. Negative feedback regulation of lipopolysaccharide-induced inducible nitric oxide synthase gene expression by heme oxygenase-1 induction in macrophages. Mol. Immunol. 2008, 45, 2106–2115. [Google Scholar] [CrossRef] [PubMed]
- Fourquet, S.; Guerois, R.; Biard, D.; Toledano, M.B. Activation of NRF2 by Nitrosative Agents and H2O2 Involves KEAP1 Disulfide Formation. J. Biol. Chem. 2010, 285, 8463–8471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription Factor Nrf2 Coordinately Regulates a Group of Oxidative Stress-inducible Genes in Macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef] [Green Version]
- Wilson, L.A.; Gemin, A.; Espiritu, R.; Singh, G. ets-1 is transcriptionally up-regulated by H2O2 via an antioxidant response element. FASEB J. 2005, 19, 2085–2087. [Google Scholar] [CrossRef]
- Song, M.Y.; Lee, D.Y.; Chun, K.S.; Kim, E.H. The Role of NRF2/KEAP1 Signaling Pathway in Cancer Metabolism. Int. J. Mol. Sci. 2021, 22, 4376. [Google Scholar] [CrossRef]
- Rushmore, T.H.; Pickett, C.B. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J. Biol. Chem. 1990, 265, 14648–14653. [Google Scholar] [CrossRef]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Thangasamy, A.; Rogge, J.; Krishnegowda, N.K.; Freeman, J.W.; Ammanamanchi, S. Novel Function of Transcription Factor Nrf2 as an Inhibitor of RON Tyrosine Kinase Receptor-mediated Cancer Cell Invasion. J. Biol. Chem. 2011, 286, 32115–32122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Davies, K.J.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88 Pt B, 314–336. [Google Scholar] [CrossRef] [Green Version]
- Namani, A.; Li, Y.; Wang, X.J.; Tang, X. Modulation of NRF2 signaling pathway by nuclear receptors: Implications for cancer. Biochim. Biophys. Acta 2014, 1843, 1875–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Hayden, A.; Douglas, J.; Sommerlad, M.; Andrews, L.; Gould, K.; Hussain, S.; Thomas, G.J.; Packham, G.; Crabb, S.J. The Nrf2 transcription factor contributes to resistance to cisplatin in bladder cancer. Urol. Oncol. Semin. Orig. Investig. 2014, 32, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Simmen, F.A.; Su, Y.; Xiao, R.; Zeng, Z.; Simmen, R.C. The Krüppel-like factor 9 (KLF9) network in HEC-1-A endometrial carcinoma cells suggests the carcinogenic potential of dys-regulated KLF9 expression. Reprod. Biol. Endocrinol. 2008, 6, 41. [Google Scholar] [CrossRef] [Green Version]
- McConnell, B.B.; Ghaleb, A.M.; Nandan, M.O.; Yang, V.W. The diverse functions of Krüppel-like factors 4 and 5 in epithelial biology and pathobiology. BioEssays 2007, 29, 549–557. [Google Scholar] [CrossRef] [Green Version]
- Bonett, R.M.; Hu, F.; Bagamasbad, P.; Denver, R.J. Stressor and glucocorticoid-dependent induction of the immediate early gene kruppel-like factor 9: Implications for neural development and plasticity. Endocrinology 2009, 150, 1757–1765. [Google Scholar] [CrossRef]
- Good, K.L.; Tangye, S.G. Decreased expression of Krüppel-like factors in memory B cells induces the rapid response typical of secondary antibody responses. Proc. Natl. Acad. Sci. USA 2007, 104, 13420–13425. [Google Scholar] [CrossRef] [Green Version]
- Mannava, S.; Zhuang, D.; Nair, J.R.; Bansal, R.; Wawrzyniak, J.A.; Zucker, S.N.; Fink, E.E.; Moparthy, K.C.; Hu, Q.; Liu, S.; et al. KLF9 is a novel transcriptional regulator of bortezomib- and LBH589-induced apoptosis in multiple myeloma cells. Blood 2012, 119, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Suske, G.; Bruford, E.; Philipsen, S. Mammalian SP/KLF transcription factors: Bring in the family. Genomics 2005, 85, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Imataka, H.; Sogawa, K.; Yasumoto, K.; Kikuchi, Y.; Sasano, K.; Kobayashi, A.; Hayami, M.; Fujii-Kuriyama, Y. Two regulatory proteins that bind to the basic transcription element (BTE), a GC box sequence in the promoter region of the rat P-4501A1 gene. EMBO J. 1992, 11, 3663–3671. [Google Scholar] [CrossRef]
- Bagati, A.; Moparthy, S.; Fink, E.E.; Bianchi-Smiraglia, A.; Yun, D.H.; Kolesnikova, M.; Udartseva, O.O.; Wolff, D.W.; Roll, M.V.; Lipchick, B.C.; et al. KLF9-dependent ROS regulate melanoma progression in stage-specific manner. Oncogene 2019, 38, 3585–3597. [Google Scholar] [CrossRef]
- Arnér, E.S. Focus on mammalian thioredoxin reductases—Important selenoproteins with versatile functions. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2009, 1790, 495–526. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; He, B.; Hao, G.; Liu, Z.; Tang, J.; Fu, Q.; Jiang, C. KLF9 aggravates ischemic injury in cardiomyocytes through augmenting oxidative stress. Life Sci. 2019, 233, 116641. [Google Scholar] [CrossRef]
- Rattan, S.I.S.; Fernandes, R.A.; Demirovic, D.; Dymek, B.; Lima, C.F. Heat Stress and Hormetin-Induced Hormesis in Human Cells: Effects on Aging, Wound Healing, Angiogenesis, and Differentiation. Dose-Response 2009, 7, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Soriano-Arroquia, A.; Gostage, J.; Xia, Q.; Bardell, D.; McCormick, R.; McCloskey, E.; Bellantuono, I.; Clegg, P.; McDonagh, B.; Goljanek-Whysall, K. miR-24 and its target gene Prdx6 regulate viability and senescence of myogenic progenitors during aging. Aging Cell 2021, 20, e13475. [Google Scholar] [CrossRef]
- Kubo, E.; Fatma, N.; Akagi, Y.; Beier, D.R.; Singh, S.P.; Singh, D.P. TAT-mediated PRDX6 protein transduction protects against eye lens epithelial cell death and delays lens opacity. Am. J. Physiol. Physiol. 2008, 294, C842–C855. [Google Scholar] [CrossRef] [Green Version]
- McAvoy, J.W.; Chamberlain, C.G.; de Longh, R.U.; Hales, A.M.; Lovicu, F.J. Lens development. Eye 1999, 13, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Piatigorsky, J.; Rothschild, S.S. Loss during development of the ability of chick embryonic lens cells to elongate in culture: Inverse relationship between cell division and elongation. Dev. Biol. 1972, 28, 382–389. [Google Scholar] [CrossRef]
- Singh, D.P.; Kubo, E.; Takamura, Y.; Shinohara, T.; Kumar, A.; Chylack, L.T.; Fatma, N. DNA Binding Domains and Nuclear Localization Signal of LEDGF: Contribution of two Helix-Turn-Helix (HTH)-like Domains and a Stretch of 58 Amino Acids of the N-terminal to the Trans-activation Potential of LEDGF. J. Mol. Biol. 2006, 355, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Ohguro, N.; Kikuchi, T.; Sueno, T.; Reddy, V.N.; Yuge, K.; Chylack, L.T.; Shinohara, T. Lens Epithelium-Derived Growth Factor: Effects on Growth and Survival of Lens Epithelial Cells, Keratinocytes, and Fibroblasts. Biochem. Biophys. Res. Commun. 2000, 267, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Ibaraki, N.; Chen, S.-C.; Lin, L.-R.; Okamoto, H.; Pipas, J.M.; Reddy, V.N. Human Lens Epithelial Cell Line. Exp. Eye Res. 1998, 67, 577–585. [Google Scholar] [CrossRef]
- Chhunchha, B.; Fatma, N.; Bhargavan, B.; Kubo, E.; Kumar, A.; Singh, D.P. Specificity protein, Sp1-mediated increased expression of Prdx6 as a curcumin-induced antioxidant defense in lens epithelial cells against oxidative stress. Cell Death Dis. 2011, 2, e234. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Pakala, S.B.; Ohshiro, K.; Li, D.-Q.; Kumar, R. SUMOylation and SUMO-interacting Motif (SIM) of Metastasis Tumor Antigen 1 (MTA1) Synergistically Regulate Its Transcriptional Repressor Function. J. Biol. Chem. 2011, 286, 43793–43808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Clock Protein Bmal1 and Nrf2 Cooperatively Control Aging or Oxidative Response and Redox Homeostasis by Regulating Rhythmic Expression of Prdx6. Cells 2020, 9, 1861. [Google Scholar] [CrossRef]
- Fatma, N.; Kubo, E.; Takamura, Y.; Ishihara, K.; Garcia, C.; Beebe, D.C.; Singh, D.P. Loss of NF-kappaB control and repression of Prdx6 gene transcription by reactive oxygen species-driven SMAD3-mediated transforming growth factor beta signaling. J. Biol. Chem. 2009, 284, 22758–22772. [Google Scholar] [CrossRef] [Green Version]
- Fatma, N.; Kubo, E.; Toris, C.B.; Stamer, W.D.; Camras, C.B.; Singh, D.P. PRDX6 attenuates oxidative stress- and TGFbeta-induced abnormalities of human trabecular meshwork cells. Free Radic. Res. 2009, 43, 783–795. [Google Scholar] [CrossRef] [Green Version]
- Kubo, E.; Miyazawa, T.; Fatma, N.; Akagi, Y.; Singh, D.P. Development- and age-associated expression pattern of peroxiredoxin 6, and its regulation in murine ocular lens. Mech. Ageing Dev. 2006, 127, 249–256. [Google Scholar] [CrossRef]
- Kubo, E.; Singh, D.P.; Fatma, N.; Akagi, Y. TAT-mediated peroxiredoxin 5 and 6 protein transduction protects against high-glucose-induced cytotoxicity in retinal pericytes. Life Sci. 2009, 84, 857–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhunchha, B.; Kubo, E.; Singh, P.; Singh, D.P. Sumoylation-deficient Prdx6 repairs aberrant Sumoylation-mediated Sp1 dysregulation-dependent Prdx6 repression and cell injury in aging and oxidative stress. Aging 2018, 10, 2284–2315. [Google Scholar] [CrossRef] [PubMed]
- Karakuzu, O.; Cruz, M.R.; Liu, Y.; Garsin, D.A. Amplex Red Assay for Measuring Hydrogen Peroxide Production from Caenorhabditis elegans. Bio-Protocol 2019, 9, e3409. [Google Scholar] [CrossRef] [PubMed]
- Cory, A.H.; Owen, T.C.; Barltrop, J.A.; Cory, J.G. Use of an Aqueous Soluble Tetrazolium/Formazan Assay for Cell Growth Assays in Culture. Cancer Commun. 1991, 3, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Fatma, N.; Singh, D.P.; Shinohara, T.; Chylack, L.T. Transcriptional Regulation of the Antioxidant Protein 2 Gene, a Thiol-specific Antioxidant, by Lens Epithelium-derived Growth Factor to Protect Cells from Oxidative Stress. J. Biol. Chem. 2001, 276, 48899–48907. [Google Scholar] [CrossRef] [Green Version]
- Semchyshyn, H.M.; Valishkevych, B.V. Hormetic Effect of H2O2 in Saccharomyces cerevisiae: Involvement of TOR and Glutathione Reductase. Dose Response 2016, 14, 1559325816636130. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Albrecht, S.C.; Barata, A.G.; Großhans, J.; Teleman, A.; Dick, T.P. In Vivo Mapping of Hydrogen Peroxide and Oxidized Glutathione Reveals Chemical and Regional Specificity of Redox Homeostasis. Cell Metab. 2011, 14, 819–829. [Google Scholar] [CrossRef] [Green Version]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. H2O2, a Necessary Evil for Cell Signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J. Use and abuse of exogenous H2O2 in studies of signal transduction. Free Radic. Biol. Med. 2007, 42, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Costantini, D.; Monaghan, P.; Metcalfe, N. Early life experience primes resistance to oxidative stress. J. Exp. Biol. 2012, 215, 2820–2826. [Google Scholar] [CrossRef] [Green Version]
- Thorpe, G.W.; Reodica, M.; Davies, M.J.; Heeren, G.; Jarolim, S.; Pillay, B.; Breitenbach, M.; Higgins, V.J.; Dawes, I.W. Superoxide radicals have a protective role during H2O2 stress. Mol. Biol. Cell 2013, 24, 2876–2884. [Google Scholar] [CrossRef] [PubMed]
- Mane, N.R.; Gajare, K.A.; Deshmukh, A.A. Mild heat stress induces hormetic effects in protecting the primary culture of mouse prefrontal cerebrocortical neurons from neuropathological alterations. IBRO Rep. 2018, 5, 110–115. [Google Scholar] [CrossRef]
- Bartling, B.; Friedrich, I.; Silber, R.-E.; Simm, A. Ischemic preconditioning is not cardioprotective in senescent human myocardium. Ann. Thorac. Surg. 2003, 76, 105–111. [Google Scholar] [CrossRef]
- Rodriguez, M.; Snoek, L.B.; Riksen, J.A.; Bevers, R.P.; Kammenga, J.E. Genetic variation for stress-response hormesis in C. elegans lifespan. Exp. Gerontol. 2012, 47, 581–587. [Google Scholar] [CrossRef]
- Sodagam, L.; Lewinska, A.; Kwasniewicz, E.; Kokhanovska, S.; Wnuk, M.; Siems, K.; Rattan, S.I.S. Phytochemicals Rosmarinic Acid, Ampelopsin, and Amorfrutin-A Can Modulate Age-Related Phenotype of Serially Passaged Human Skin Fibroblasts in vitro. Front. Genet. 2019, 10, 81. [Google Scholar] [CrossRef]
- Poljsak, B.; Šuput, D.; Milisav, I. Achieving the Balance between ROS and Antioxidants: When to Use the Synthetic Antioxidants. Oxidative Med. Cell. Longev. 2013, 2013, 956792. [Google Scholar] [CrossRef]
- Pinazo-Durán, M.D.; Gallego-Pinazo, R.; Garcia-Medina, J.J.; Zanón-Moreno, V.; Nucci, C.; Dolz-Marco, R.; Martinez-Castillo, S.; Galbis-Estrada, C.; Marco-Ramirez, C.; Lopez-Galvez, M.I.; et al. Oxidative stress and its downstream signaling in aging eyes. Clin. Interv. Aging 2014, 9, 637–652. [Google Scholar] [CrossRef] [Green Version]
- Brennan, L.A.; Kantorow, M. Mitochondrial function and redox control in the aging eye: Role of MsrA and other repair systems in cataract and macular degenerations. Exp. Eye Res. 2009, 88, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Babizhayev, M.A.; Vishnyakova, K.S.; Yegorov, Y.E. Oxidative Damage Impact on Aging and Age-Related Diseases: Drug Targeting of Telomere Attrition and Dynamic Telomerase Activity Flirting with Imidazole-Containing Dipeptides. Recent Pat. Drug Deliv. Formul. 2014, 8, 163–192. [Google Scholar] [CrossRef]
- Pacifici, F.; Della-Morte, D.; Piermarini, F.; Arriga, R.; Scioli, M.G.; Capuani, B.; Pastore, D.; Coppola, A.; Rea, S.; Donadel, G.; et al. Prdx6 Plays a Main Role in the Crosstalk between Aging and Metabolic Sarcopenia. Antioxidants 2020, 9, 329. [Google Scholar] [CrossRef]
- Fernández-Ortiz, M.; Sayed, R.K.; Fernández-Martínez, J.; Cionfrini, A.; Aranda-Martínez, P.; Escames, G.; de Haro, T.; Acuña-Castroviejo, D. Melatonin/Nrf2/NLRP3 Connection in Mouse Heart Mitochondria during Aging. Antioxidants 2020, 9, 1187. [Google Scholar] [CrossRef]
- Chhunchha, B.; Fatma, N.; Kubo, E.; Rai, P.; Singh, S.P.; Singh, D.P. Curcumin abates hypoxia-induced oxidative stress based-ER stress-mediated cell death in mouse hippocampal cells (HT22) by controlling Prdx6 and NF-kappaB regulation. Am. J. Physiol. Cell Physiol. 2013, 304, C636–C655. [Google Scholar] [CrossRef] [Green Version]
- Rybka, J.; Kędziora-Kornatowska, K.; Banaś-Leżańska, P.; Majsterek, I.; Carvalho, L.A.; Cattaneo, A.; Anacker, C.; Kędziora, J. Interplay between the pro-oxidant and antioxidant systems and proinflammatory cytokine levels, in relation to iron metabolism and the erythron in depression. Free Radic. Biol. Med. 2013, 63, 187–194. [Google Scholar] [CrossRef]
- Shibata, S.; Shibata, N.; Shibata, T.; Sasaki, H.; Singh, D.P.; Kubo, E. The role of Prdx6 in the protection of cells of the crystalline lens from oxidative stress induced by UV exposure. Jpn. J. Ophthalmol. 2016, 60, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, B.; Hecht, H.J.; Flohe, L. Peroxiredoxins. Biol. Chem. 2002, 383, 347–364. [Google Scholar] [CrossRef]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef]
- Fatma, N.; Kubo, E.; Sen, M.; Agarwal, N.; Thoreson, W.B.; Camras, C.B.; Singh, D.P. Peroxiredoxin 6 delivery attenuates TNF-alpha-and glutamate-induced retinal ganglion cell death by limiting ROS levels and maintaining Ca2+ homeostasis. Brain Res. 2008, 1233, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Tomalin, L.E.; Day, A.M.; Underwood, Z.E.; Smith, G.R.; Pezze, P.D.; Rallis, C.; Patel, W.; Dickinson, B.C.; Bähler, J.; Brewer, T.F.; et al. Increasing extracellular H2O2 produces a bi-phasic response in intracellular H2O2, with peroxiredoxin hyperoxidation only triggered once the cellular H2O2-buffering capacity is overwhelmed. Free Radic. Biol. Med. 2016, 95, 333–348. [Google Scholar] [CrossRef] [Green Version]
- Fink, E.E.; Moparthy, S.; Bagati, A.; Bianchi-Smiraglia, A.; Lipchick, B.C.; Wolff, D.W.; Roll, M.V.; Wang, J.; Liu, S.; Bakin, A.V.; et al. XBP1-KLF9 Axis Acts as a Molecular Rheostat to Control the Transition from Adaptive to Cytotoxic Unfolded Protein Response. Cell Rep. 2018, 25, 212–223.e4. [Google Scholar] [CrossRef] [Green Version]
- Fink, E.E.; Mannava, S.; Bagati, A.; Bianchi-Smiraglia, A.; Nair, J.R.; Moparthy, K.; Lipchick, B.C.; Drokov, M.; Utley, A.; Ross, J.; et al. Mitochondrial thioredoxin reductase regulates major cytotoxicity pathways of proteasome inhibitors in multiple myeloma cells. Leukemia 2016, 30, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Linnane, A.W.; Kios, M.; Vitetta, L. Coenzyme Q10—Its role as a prooxidant in the formation of superoxide anion/hydrogen peroxide and the regulation of the metabolome. Mitochondrion 2007, 7, S51–S61. [Google Scholar] [CrossRef]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Tang, L.-Y.; Fu, W.-W.; Yuan, J.; Sheng, Y.-Y.; Yang, Q.-P. Low-concentration hydrogen peroxide can upregulate keratinocyte intracellular calcium and PAR-2 expression in a human keratinocyte–melanocyte co-culture system. Arch. Dermatol. Res. 2016, 308, 723–731. [Google Scholar] [CrossRef]
- Adler, S.; Stahl, R.A.; Baker, P.J.; Chen, Y.P.; Pritzl, P.M.; Couser, W.G. Biphasic effect of oxygen radicals on prostaglandin production by rat mesangial cells. Am. J. Physiol. Physiol. 1987, 252, F743–F749. [Google Scholar] [CrossRef]
- Mu, P.; Liu, Q.; Zheng, R. Biphasic regulation of H2O2 on angiogenesis implicated NADPH oxidase. Cell Biol. Int. 2010, 34, 1013–1020. [Google Scholar] [CrossRef]
- Wu, Y.; Feinstein, S.I.; Manevich, Y.; Chowdhury, I.; Pak, J.H.; Kazi, A.; Dodia, C.; Speicher, D.W.; Fisher, A.B. Mitogen-activated protein kinase-mediated phosphorylation of peroxiredoxin 6 regulates its phospholipase A2 activity. Biochem. J. 2009, 419, 669–679. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chhunchha, B.; Kubo, E.; Singh, D.P. Switching of Redox Signaling by Prdx6 Expression Decides Cellular Fate by Hormetic Phenomena Involving Nrf2 and Reactive Oxygen Species. Cells 2022, 11, 1266. https://doi.org/10.3390/cells11081266
Chhunchha B, Kubo E, Singh DP. Switching of Redox Signaling by Prdx6 Expression Decides Cellular Fate by Hormetic Phenomena Involving Nrf2 and Reactive Oxygen Species. Cells. 2022; 11(8):1266. https://doi.org/10.3390/cells11081266
Chicago/Turabian StyleChhunchha, Bhavana, Eri Kubo, and Dhirendra P. Singh. 2022. "Switching of Redox Signaling by Prdx6 Expression Decides Cellular Fate by Hormetic Phenomena Involving Nrf2 and Reactive Oxygen Species" Cells 11, no. 8: 1266. https://doi.org/10.3390/cells11081266
APA StyleChhunchha, B., Kubo, E., & Singh, D. P. (2022). Switching of Redox Signaling by Prdx6 Expression Decides Cellular Fate by Hormetic Phenomena Involving Nrf2 and Reactive Oxygen Species. Cells, 11(8), 1266. https://doi.org/10.3390/cells11081266