
Steady-State Levels of Miro1 Linked to Phosphorylation at Serine 156 and Mitochondrial Respiration in Dopaminergic Neurons


Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation Neural Precursor Cells (NPCs) and Differentiation in Midbrain Dopaminergic Neurons (hDaNs)
2.2. RT-qPCR
2.3. Immunofluorescence Staining
2.4. SDS-PAGE and Western Blotting
2.5. Imaging Analysis of Mitochondrial Morphology and Membrane Potential
2.6. Imaging Analysis of Mitochondrial Movement
2.7. Imaging Analysis of Mitochondrial Turnover Using MitoTimer Construct
2.8. Flow Cytometry
2.9. Respiratory Analyses
2.10. Miro1 Knockdown in Undifferentiated SH-SY5Y
2.11. Miro1 Knockdown in Differentiated SH-SY5Y
2.12. Statistical Analysis
3. Results
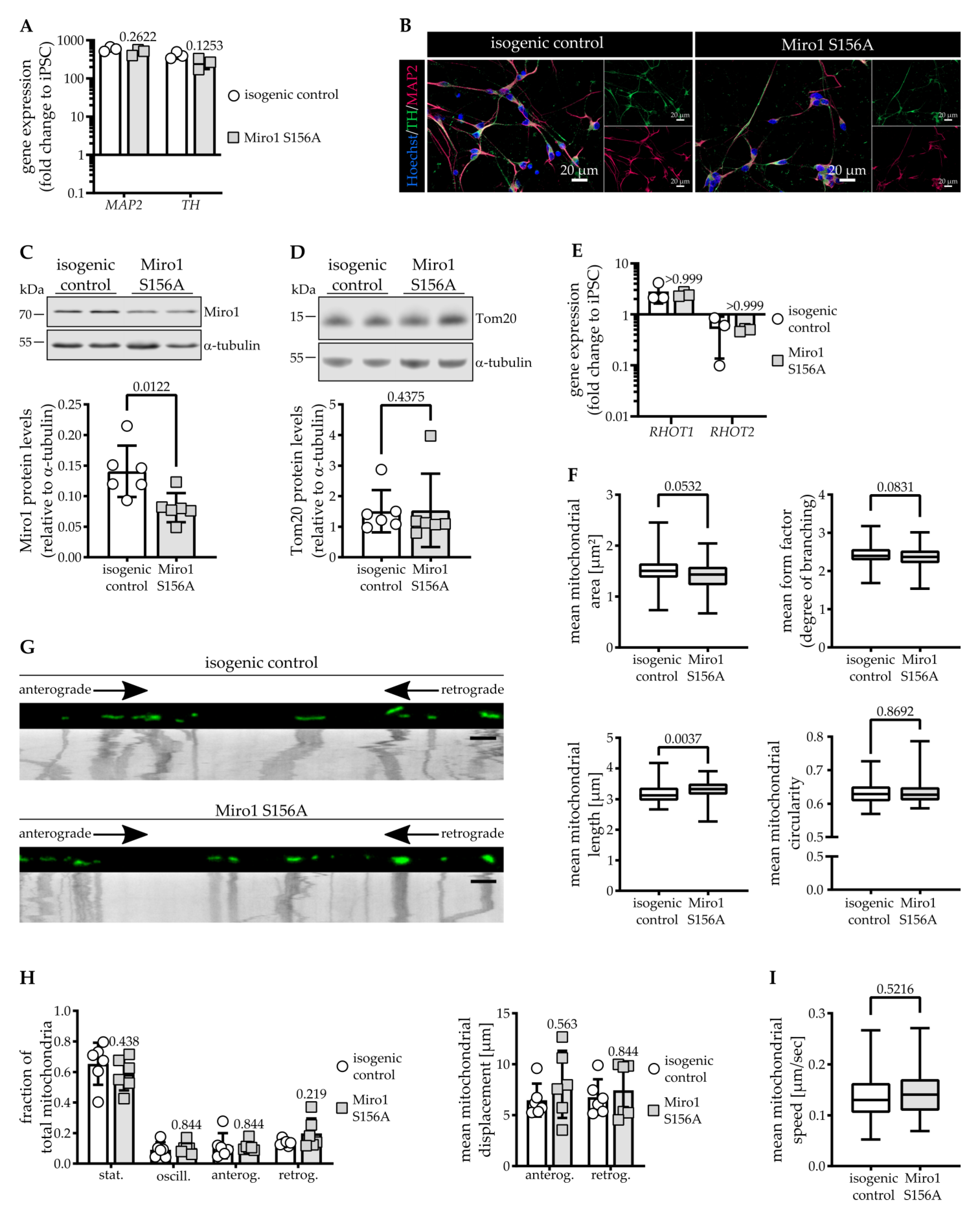
3.1. Miro1 S156A Protein Levels Are Reduced in Dopaminergic Neurons
3.2. Miro1 Degradation during CCCP-Induced Mitophagy Is Imparied by S156A Mutation
3.3. Miro1 S156A Reduces Mitochondrial Respiration in hDaNs
3.4. The S156A Mutation Reduces Miro1 Protein Levels Affecting OXPHOS in Postmitotic Differentiated Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fransson, A.; Ruusala, A.; Aspenstrom, P. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J. Biol. Chem. 2003, 278, 6495–6502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fransson, S.; Ruusala, A.; Aspenstrom, P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem. Biophys. Res. Commun. 2006, 344, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Klosowiak, J.L.; Focia, P.J.; Chakravarthy, S.; Landahl, E.C.; Freymann, D.M.; Rice, S.E. Structural coupling of the EF hand and C-terminal GTPase domains in the mitochondrial protein Miro. EMBO Rep. 2013, 14, 968–974. [Google Scholar] [CrossRef] [PubMed]
- MacAskill, A.F.; Brickley, K.; Stephenson, F.A.; Kittler, J.T. GTPase dependent recruitment of Grif-1 by Miro1 regulates mitochondrial trafficking in hippocampal neurons. Mol. Cell Neurosci. 2009, 40, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.J.; Louie, K.; Wellington, A.; Macleod, G.T.; Hu, F.; Panchumarthi, S.; Zinsmaier, K.E. Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J. Neurosci. 2009, 29, 5443–5455. [Google Scholar] [CrossRef]
- Lopez-Domenech, G.; Higgs, N.F.; Vaccaro, V.; Ros, H.; Arancibia-Carcamo, I.L.; MacAskill, A.F.; Kittler, J.T. Loss of Dendritic Complexity Precedes Neurodegeneration in a Mouse Model with Disrupted Mitochondrial Distribution in Mature Dendrites. Cell Rep. 2016, 17, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.; Oh, S.S.; Weaver, D.; Lewandowska, A.; Maxfield, D.; Schuler, M.H.; Smith, N.K.; Macfarlane, J.; Saunders, G.; Palmer, C.A.; et al. Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease. Proc. Natl. Acad. Sci. USA 2014, 111, E3631–E3640. [Google Scholar] [CrossRef] [Green Version]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef]
- Chang, K.T.; Niescier, R.F.; Min, K.T. Mitochondrial matrix Ca2+ as an intrinsic signal regulating mitochondrial motility in axons. Proc. Natl. Acad. Sci. USA 2011, 108, 15456–15461. [Google Scholar] [CrossRef] [Green Version]
- Macaskill, A.F.; Rinholm, J.E.; Twelvetrees, A.E.; Arancibia-Carcamo, I.L.; Muir, J.; Fransson, A.; Aspenstrom, P.; Attwell, D.; Kittler, J.T. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 2009, 61, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Niescier, R.F.; Hong, K.; Park, D.; Min, K.T. MCU Interacts with Miro1 to Modulate Mitochondrial Functions in Neurons. J. Neurosci. 2018, 38, 4666–4677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saotome, M.; Safiulina, D.; Szabadkai, G.; Das, S.; Fransson, A.; Aspenstrom, P.; Rizzuto, R.; Hajnóczky, G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. USA 2008, 105, 20728–20733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Schwarz, T.L. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell 2009, 136, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weihofen, A.; Thomas, K.J.; Ostaszewski, B.L.; Cookson, M.R.; Selkoe, D.J. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 2009, 48, 2045–2052. [Google Scholar] [CrossRef] [Green Version]
- Birsa, N.; Norkett, R.; Wauer, T.; Mevissen, T.E.; Wu, H.C.; Foltynie, T.; Bhatia, K.; Hirst, W.D.; Komander, D.; Plun-Favreau, H.; et al. Lysine 27 ubiquitination of the mitochondrial transport protein Miro is dependent on serine 65 of the Parkin ubiquitin ligase. J. Biol. Chem. 2014, 289, 14569–14582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exner, N.; Lutz, A.K.; Haass, C.; Winklhofer, K.F. Mitochondrial dysfunction in Parkinson’s disease: Molecular mechanisms and pathophysiological consequences. EMBO J. 2012, 31, 3038–3062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Valente Enza, M.; Abou-Sleiman Patrick, M.; Caputo, V.; Muqit Miratul, M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio Anna, R.; Healy Daniel, G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010, 6, 1090–1106. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Sawada, T.; Lee, S.; Yu, W.; Silverio, G.; Alapatt, P.; Millan, I.; Shen, A.; Saxton, W.; Kanao, T.; et al. Parkinson’s disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 2012, 8, e1002537. [Google Scholar] [CrossRef]
- Kazlauskaite, A.; Kelly, V.; Johnson, C.; Baillie, C.; Hastie, C.J.; Peggie, M.; Macartney, T.; Woodroof, H.I.; Alessi, D.R.; Pedrioli, P.G.; et al. Phosphorylation of Parkin at Serine65 is essential for activation: Elaboration of a Miro1 substrate-based assay of Parkin E3 ligase activity. Open Biol. 2014, 4, 130213. [Google Scholar] [CrossRef] [Green Version]
- Shlevkov, E.; Kramer, T.; Schapansky, J.; LaVoie, M.J.; Schwarz, T.L. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc. Natl. Acad. Sci. USA 2016, 113, E6097–E6106. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.H.; Li, L.; Vanhauwaert, R.; Nguyen, K.T.; Davis, M.D.; Bu, G.; Wszolek, Z.K.; Wang, X. Miro1 Marks Parkinson’s Disease Subset and Miro1 Reducer Rescues Neuron Loss in Parkinson’s Models. Cell Metab. 2019, 30, 1131.e1137–1140.e1137. [Google Scholar] [CrossRef]
- Schwarz, L.; Casadei, N.; Fitzgerald, J.C. Generation of R272Q, S156A and K572R RHOT1/Miro1 point mutations in iPSCs from a healthy individual using FACS-assisted CRISPR/Cas9 genome editing. Stem Cell Res. 2021, 55, 102469. [Google Scholar] [CrossRef]
- Reinhardt, P.; Glatza, M.; Hemmer, K.; Tsytsyura, Y.; Thiel, C.S.; Hoing, S.; Moritz, S.; Parga, J.A.; Wagner, L.; Bruder, J.M.; et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS ONE 2013, 8, e59252. [Google Scholar] [CrossRef] [Green Version]
- Bus, C.; Zizmare, L.; Feldkaemper, M.; Geisler, S.; Zarani, M.; Schaedler, A.; Klose, F.; Admard, J.; Mageean, C.J.; Arena, G.; et al. Human Dopaminergic Neurons Lacking PINK1 Exhibit Disrupted Dopamine Metabolism Related to Vitamin B6 Co-Factors. iScience 2020, 23, 101797. [Google Scholar] [CrossRef]
- Merrill, R.A.; Flippo, K.H.; Strack, S. Measuring Mitochondrial Shape with ImageJ. In Techniques to Investigate Mitochondrial Function in Neurons; Neuromethods; Springer: Berlin, Germany, 2017; pp. 31–48. [Google Scholar]
- Kocsis, E.; Trus, B.L.; Steer, C.J.; Bisher, M.E.; Steven, A.C. Image averaging of flexible fibrous macromolecules: The clathrin triskelion has an elastic proximal segment. J. Struct. Biol. 1991, 107, 6–14. [Google Scholar] [CrossRef]
- Tinevez, J.Y.; Perry, N.; Schindelin, J.; Hoopes, G.M.; Reynolds, G.D.; Laplantine, E.; Bednarek, S.Y.; Shorte, S.L.; Eliceiri, K.W. TrackMate: An open and extensible platform for single-particle tracking. Methods 2017, 115, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, G.; Thornton, C.; Stotland, A.; Lui, D.; Sin, J.; Ramil, J.; Magee, N.; Andres, A.; Quarato, G.; Carreira, R.S.; et al. MitoTimer: A novel tool for monitoring mitochondrial turnover. Autophagy 2013, 9, 1852–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrone, L.; Bus, C.; Schondorf, D.; Fitzgerald, J.C.; Kubler, M.; Schmid, B.; Reinhardt, P.; Reinhardt, L.; Deleidi, M.; Levin, T.; et al. Generation of iPSCs carrying a common LRRK2 risk allele for in vitro modeling of idiopathic Parkinson’s disease. PLoS ONE 2018, 13, e0192497. [Google Scholar] [CrossRef] [Green Version]
- Babic, M.; Russo, G.J.; Wellington, A.J.; Sangston, R.M.; Gonzalez, M.; Zinsmaier, K.E. Miro’s N-terminal GTPase domain is required for transport of mitochondria into axons and dendrites. J. Neurosci. 2015, 35, 5754–5771. [Google Scholar] [CrossRef] [Green Version]
- Frederick, R.L.; McCaffery, J.M.; Cunningham, K.W.; Okamoto, K.; Shaw, J.M. Yeast Miro GTPase, Gem1p, regulates mitochondrial morphology via a novel pathway. J. Cell Biol. 2004, 167, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.; Hess, S.; Chan, D.C. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 2011, 20, 1726–1737. [Google Scholar] [CrossRef]
- Lopez-Domenech, G.; Howden, J.H.; Covill-Cooke, C.; Morfill, C.; Patel, J.V.; Burli, R.; Crowther, D.; Birsa, N.; Brandon, N.J.; Kittler, J.T. Loss of neuronal Miro1 disrupts mitophagy and induces hyperactivation of the integrated stress response. EMBO J. 2021, 40, e100715. [Google Scholar] [CrossRef]
- Alshaabi, H.; Shannon, N.; Gravelle, R.; Milczarek, S.; Messier, T.; Cunniff, B. Miro1-mediated mitochondrial positioning supports subcellular redox status. Redox Biol. 2021, 38, 101818. [Google Scholar] [CrossRef]
- Shaltouki, A.; Hsieh, C.H.; Kim, M.J.; Wang, X. Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson’s models. Acta Neuropathol. 2018, 136, 607–620. [Google Scholar] [CrossRef] [PubMed]
- López-Doménech, G.; Covill-Cooke, C.; Howden, J.H.; Birsa, N.; Morfill, C.; Brandon, N.J.; Kittler, J.T. Miro ubiquitination is critical for efficient damage-induced PINK1/Parkin-mediated mitophagy. bioRxiv 2018, 414664. [Google Scholar] [CrossRef]
- Schuler, M.H.; Lewandowska, A.; Caprio, G.D.; Skillern, W.; Upadhyayula, S.; Kirchhausen, T.; Shaw, J.M.; Cunniff, B. Miro1-mediated mitochondrial positioning shapes intracellular energy gradients required for cell migration. Mol. Biol. Cell 2017, 28, 2159–2169. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, S.; Wu, Z.; Klinkenberg, M.; Sun, Y.; Auburger, G.; Guo, S.; Lu, B. PINK1 and Parkin control localized translation of respiratory chain component mRNAs on mitochondria outer membrane. Cell Metab. 2015, 21, 95–108. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| GAPDH | CGAGATCCCTCCAAAATCAAG | GCAGAGATGATGACCCTTTTG |
| NESTIN | GGCAGCGTTGGAACAGAGGTTGGA | CTCTAAACTGGAGTGGTCAGGGCT |
| SOX2 | TTCACATGTCCCAGCACTACCAGA | TCACATGTGTGAGAGGGGCAGTGTGC |
| PAX6 | GTGTCCAACGGATGTGTGAG | CTAGCCAGGTTGCGAAGAAC |
| TH | TGTCTGAGGAGCCTGAGATTCG | GCTTGTCCTTGGCGTCACTG |
| MAP2 | CCGTGTGGACCATGGGGCTG | GTCGTCGGGGTGATGCCACG |
| RHOT1 | TGTCACCCCAGAGAGAGTTC | GCCTGCTGTCTTTGTCTGTT |
| RHOT2 | ATTGAGACCTGCGTGGAGTG | AAGCGTTGAGCTCTTCGTCA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwarz, L.; Fitzgerald, J.C. Steady-State Levels of Miro1 Linked to Phosphorylation at Serine 156 and Mitochondrial Respiration in Dopaminergic Neurons. Cells 2022, 11, 1269. https://doi.org/10.3390/cells11081269
Schwarz L, Fitzgerald JC. Steady-State Levels of Miro1 Linked to Phosphorylation at Serine 156 and Mitochondrial Respiration in Dopaminergic Neurons. Cells. 2022; 11(8):1269. https://doi.org/10.3390/cells11081269
Chicago/Turabian StyleSchwarz, Lisa, and Julia C. Fitzgerald. 2022. "Steady-State Levels of Miro1 Linked to Phosphorylation at Serine 156 and Mitochondrial Respiration in Dopaminergic Neurons" Cells 11, no. 8: 1269. https://doi.org/10.3390/cells11081269