
N-VEGF, the Autoregulatory Arm of VEGF-A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Plasmid Construction
2.3. Lentiviral Production
2.4. NIH3T3 Infection
2.5. Nuclear and Cytoplasmic Fractionation
2.6. SDS-PAGE and Western Blotting
2.7. Immunofluorescence Staining and Microscopy
2.8. Real-Time RT-PCR
2.9. RNA-Seq
2.10. CRISPR/Cas9-Mediated Deletion of N-VEGF
2.11. Flow Cytometry
2.12. Hypoxia Treatment
2.13. ApoTox-GloTM Triplex Assay
2.14. Cobalt Chloride and Tunicamycin Treatments
2.15. Statistical Analysis
3. Results
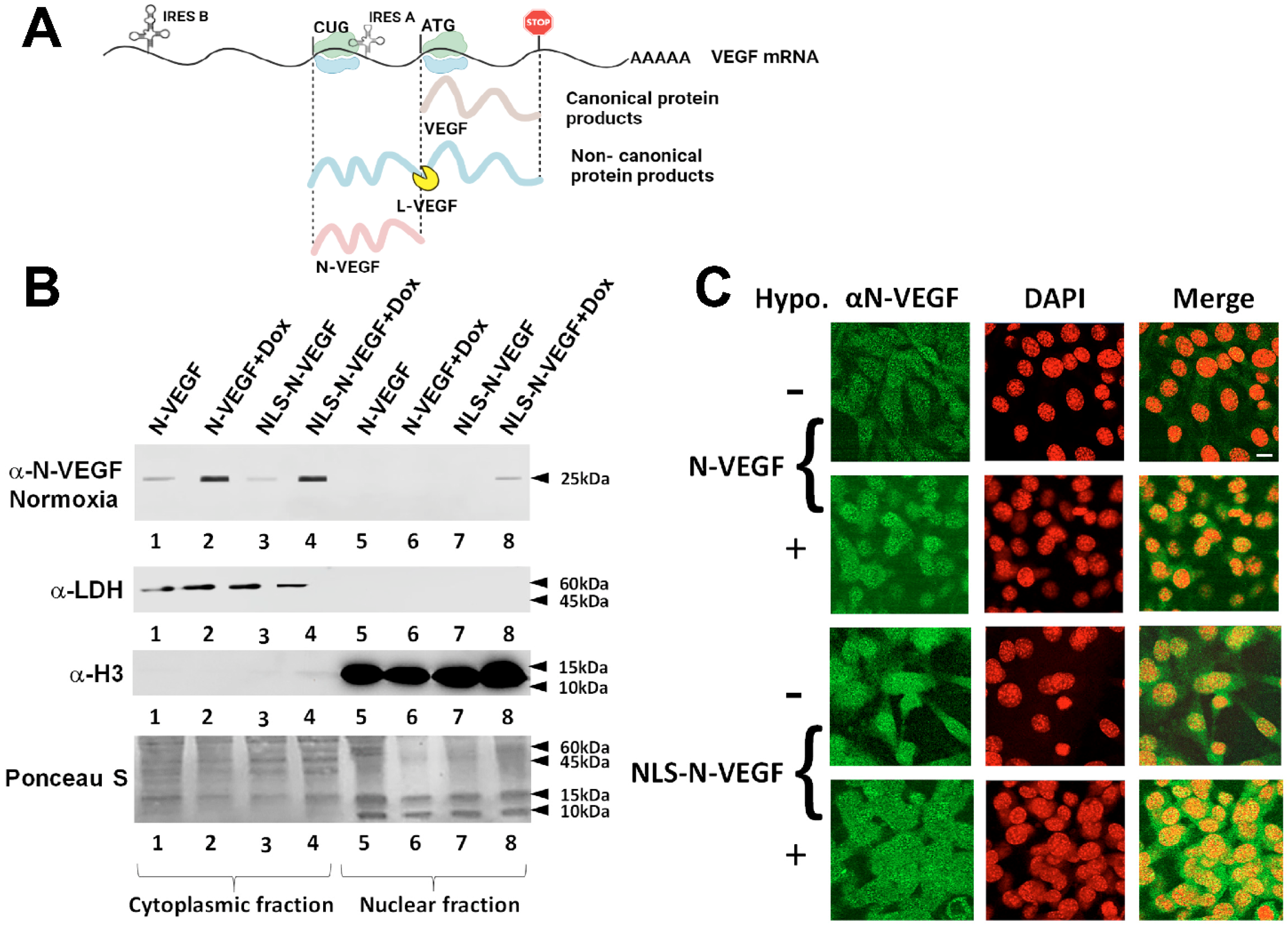
3.1. Expression and Hypoxia-Independent Mobilization of N-VEGF
3.2. Hypoxia-Independent Nuclear Mobilization of N-VEGF Induces Part of the Hypoxic Transcriptional Program
3.3. Genetic Deletion of N-VEGF Using CRISPR-Cas9
3.4. Impact of N-VEGF Deletion on Hypoxia-Induced Genes
3.5. N-VEGF Deletion Leads to Increased Hypoxia-Mediated Cell Death
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arcondeguy, T.; Lacazette, E.; Millevoi, S.; Prats, H.; Touriol, C. VEGF-A mRNA processing, stability and translation: A paradigm for intricate regulation of gene expression at the post-transcriptional level. Nucleic Acids Res. 2013, 41, 7997–8010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meiron, M.; Anunu, R.; Scheinman, E.J.; Hashmueli, S.; Levi, B.Z. New isoforms of VEGF are translated from alternative initiation CUG codons located in its 5’UTR. Biochem. Biophys. Res. Commun. 2001, 282, 1053–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tee, M.K.; Jaffe, R.B. A precursor form of vascular endothelial growth factor arises by initiation from an upstream in-frame CUG codon. Biochem. J. 2001, 359, 219–226. [Google Scholar] [CrossRef]
- Huez, I.; Bornes, S.; Bresson, D.; Creancier, L.; Prats, H. New vascular endothelial growth factor isoform generated by internal ribosome entry site-driven CUG translation initiation. Mol. Endocrinol. 2001, 15, 2197–2210. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum-Dekel, Y.; Fuchs, A.; Yakirevich, E.; Azriel, A.; Mazareb, S.; Resnick, M.B. Nuclear localization of long-VEGF is associated with hypoxia and tumor angiogenesis. Biochem. Biophys. Res. Commun. 2005, 332, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.; Azriel, A.; Cohen, T.; Meraro, D.; Hashmueli, S.; Bech-Otschir, D. Interaction between interferon consensus sequence-binding protein and COP9/signalosome subunit CSN2 (Trip15). A possible link between interferon regulatory factor signaling and the COP9/signalosome. J. Biol. Chem. 2000, 275, 39081–39089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Labun, K.; Montague, T.G.; Krause, M.; Torres-Cleuren, Y.N.; Tjeldnes, H.; Valen, E. CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019, 47, 171–174. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, E.; Nakayama, M. VCre/VloxP and SCre/SloxP: New site-specific recombination systems for genome engineering. Nucleic Acids Res. 2011, 39, e49. [Google Scholar] [CrossRef]
- Shih, S.C.; Claffey, K.P. Hypoxia-mediated regulation of gene expression in mammalian cells. Int. J. Exp. Pathol. 1998, 79, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Gossen, M.; Freundlieb, S.; Bender, G.; Muller, G.; Hillen, W.; Bujard, H. Transcriptional activation by tetracyclines in mammalian cells. Science 1995, 268, 1766–1769. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.J.; Bui, T.; Gruber, M.; Gordan, J.D.; DeBerardinis, R.J.; Covello, K.L. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007, 21, 1037–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bono, H.; Hirota, K. Meta-Analysis of Hypoxic Transcriptomes from Public Databases. Biomedicines 2020, 8, 10. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1301–1303. [Google Scholar] [CrossRef]
- Wilhelm, M.; Kukekov, N.V.; Schmit, T.L.; Biagas, K.V.; Sproul, A.A.; Gire, S. Sh3rf2/POSHER protein promotes cell survival by ring-mediated proteasomal degradation of the c-Jun N-terminal kinase scaffold POSH (Plenty of SH3s) protein. J. Biol. Chem. 2012, 287, 2247–2256. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.H.; Joo, H.; Bae, J.; Hyeon, S.J.; Her, S.; Ko, E. Brain injury induces HIF-1alpha-dependent transcriptional activation of LRRK2 that exacerbates brain damage. Cell Death Dis. 2018, 9, 1125. [Google Scholar] [CrossRef]
- Abu-Ghazaleh, R.; Kabir, J.; Jia, H.; Lobo, M.; Zachary, I. Src mediates stimulation by vascular endothelial growth factor of the phosphorylation of focal adhesion kinase at tyrosine 861, and migration and anti-apoptosis in endothelial cells. Biochem. J. 2001, 360, 255–264. [Google Scholar] [CrossRef]
- Varela-Guruceaga, M.; Belaidi, E.; Lebeau, L.; Aka, E.; Andriantsitohaina, R.; Giorgetti-Peraldi, S. Intermittent Hypoxia Mediates Caveolae Disassembly That Parallels Insulin Resistance Development. Front. Physiol. 2020, 11, 565486. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Xiao, E.; Huang, M. MEK/ERK pathway is positively involved in hypoxia-induced vasculogenic mimicry formation in hepatocellular carcinoma which is regulated negatively by protein kinase A. Med. Oncol. 2015, 32, 408. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Kumari, S.; Ali, Z.; Pasha, M.Q. Acute hypobaric hypoxia augments ROCK2 protein level and activity. Exp. Lung Res. 2017, 43, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Koritzinsky, M.; Magagnin, M.G.; van den Beucken, T.; Seigneuric, R.; Savelkouls, K.; Dostie, J. Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. Embo J. 2006, 25, 1114–1125. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Thompson, M.A.; Edmonds, M.D.; Liang, S.; McClintock-Treep, S.; Wang, X.; Li, S. miR-31 and miR-17-5p levels change during transformation of follicular lymphoma. Hum. Pathol. 2016, 50, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Chao, L.M.; Sun, W.; Chen, H.; Liu, B.Y.; Li, P.F.; Zhao, D.W. MicroRNA-31 inhibits osteosarcoma cell proliferation, migration and invasion by targeting PIK3C2A. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7205–7213. [Google Scholar] [CrossRef]
- Yamazato, Y.; Shiozaki, A.; Ichikawa, D.; Kosuga, T.; Shoda, K.; Arita, T. Aquaporin 1 suppresses apoptosis and affects prognosis in esophageal squamous cell carcinoma. Oncotarget 2018, 9, 29957–29974. [Google Scholar] [CrossRef]
- Bai, J.A.; Xu, G.F.; Yan, L.J.; Zeng, W.W.; Ji, Q.Q.; Wu, J.D. SGK1 inhibits cellular apoptosis and promotes proliferation via the MEK/ERK/p53 pathway in colitis. World J. Gastroenterol. 2015, 21, 6180–6193. [Google Scholar] [CrossRef]
- Han, Z.; Jiang, G.; Zhang, Y.; Xu, J.; Chen, C.; Zhang, L. Effects of RNA interference-mediated NRP-1 silencing on the proliferation and apoptosis of breast cancer cells. Mol. Med. Rep. 2015, 12, 513–519. [Google Scholar] [CrossRef]
- Sandner, P.; Wolf, K.; Bergmair, U.; Gess, B.; Kurtz, A. Hypoxia and cobalt stimulate vascular endothelial growth factor receptor gene expression in rats. Pflugers Arch. 1997, 433, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, H.; Xie, M.; Hu, M.; Ge, S.; Yang, D. Abundant expression of Dec1/stra13/sharp2 in colon carcinoma: Its antagonizing role in serum deprivation-induced apoptosis and selective inhibition of procaspase activation. Biochem. J. 2002, 367, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Bull, V.H.; Thiede, B. Proteome analysis of tunicamycin-induced ER stress. Electrophoresis 2012, 33, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.G.; Hill, R.P. Hypoxia, DNA Repair and Genetic Instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef]
- Bastide, A.; Karaa, Z.; Bornes, S.; Hieblot, C.; Lacazette, E.; Prats, H. An upstream open reading frame within an IRES controls expression of a specific VEGF-A isoform. Nucleic Acids Res. 2008, 36, 2434–2445. [Google Scholar] [CrossRef] [Green Version]
- Bornes, S.; Prado-Lourenco, L.; Bastide, A.; Zanibellato, C.; Iacovoni, J.S.; Lacazette, E. Translational induction of VEGF internal ribosome entry site elements during the early response to ischemic stress. Circ. Res. 2007, 100, 305–308. [Google Scholar] [CrossRef] [Green Version]
- Cawley, N.X.; Li, Z.; Loh, Y.P. 60 YEARS OF POMC: Biosynthesis, trafficking, and secretion of pro-opiomelanocortin-derived peptides. J. Mol. Endocrinol. 2016, 56, 77–97. [Google Scholar] [CrossRef] [Green Version]
- Henrique, D.; Schweisguth, F. Mechanisms of Notch signaling: A simple logic deployed in time and space. Development 2019, 146, dev172148. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katsman, M.; Azriel, A.; Horev, G.; Reizel, Y.; Levi, B.-Z. N-VEGF, the Autoregulatory Arm of VEGF-A. Cells 2022, 11, 1289. https://doi.org/10.3390/cells11081289
Katsman M, Azriel A, Horev G, Reizel Y, Levi B-Z. N-VEGF, the Autoregulatory Arm of VEGF-A. Cells. 2022; 11(8):1289. https://doi.org/10.3390/cells11081289
Chicago/Turabian StyleKatsman, Marina, Aviva Azriel, Guy Horev, Yitzhak Reizel, and Ben-Zion Levi. 2022. "N-VEGF, the Autoregulatory Arm of VEGF-A" Cells 11, no. 8: 1289. https://doi.org/10.3390/cells11081289