
High-Fat Diet-Induced Dysregulation of Immune Cells Correlates with Macrophage Phenotypes and Chronic Inflammation in Adipose Tissue




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Induction of Obesity in Mice by HFD
2.3. Preparation of Single-Cell Suspensions
2.4. Measurement of Cytokines and Chemokines
2.5. Flow Cytometry Analysis
2.6. RT-qPCR Analysis
2.7. Immunoblot Analysis
2.8. Histology
2.9. Statistics
3. Results
3.1. Changes in Body Weight and Metabolic Parameters after HFD Induction
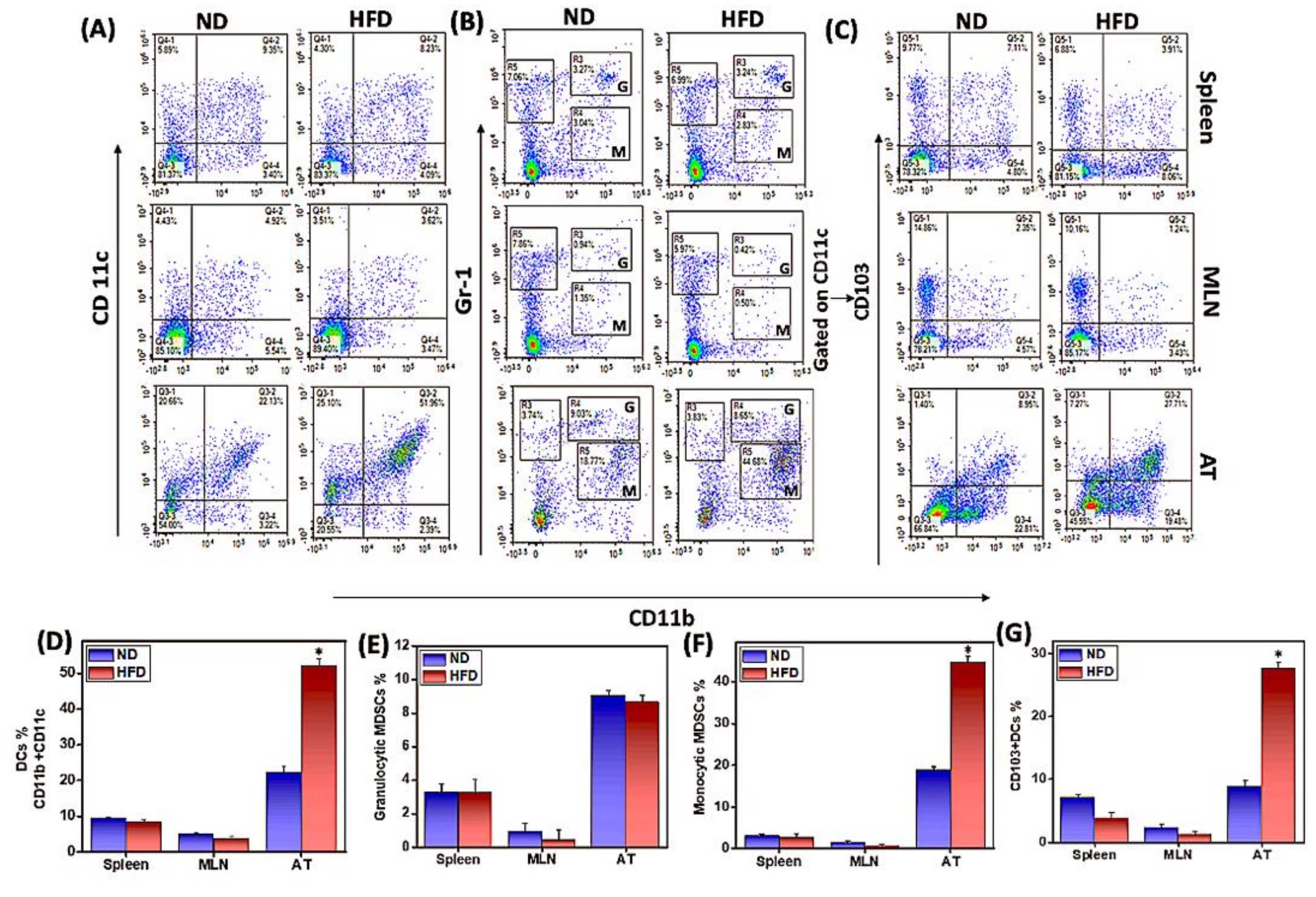
3.2. HFD Induces M1 Macrophages, CD103+ Dendritic Cells, and Monocytic Myeloid-Derived Suppressor Cells (MDSCs) in AT
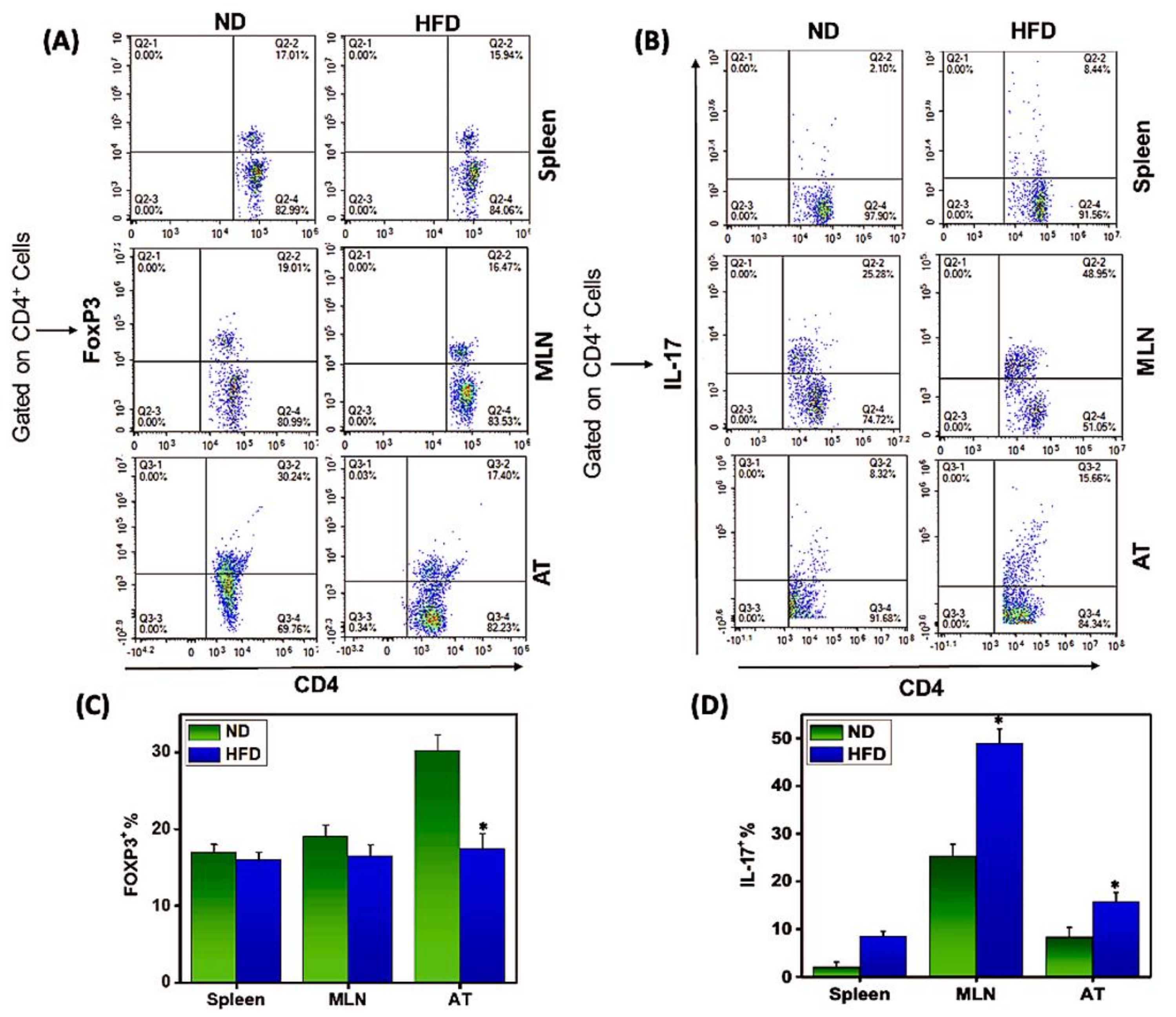
3.3. Th17/Tregs Dysregulation in AT Correlates with Inflammation
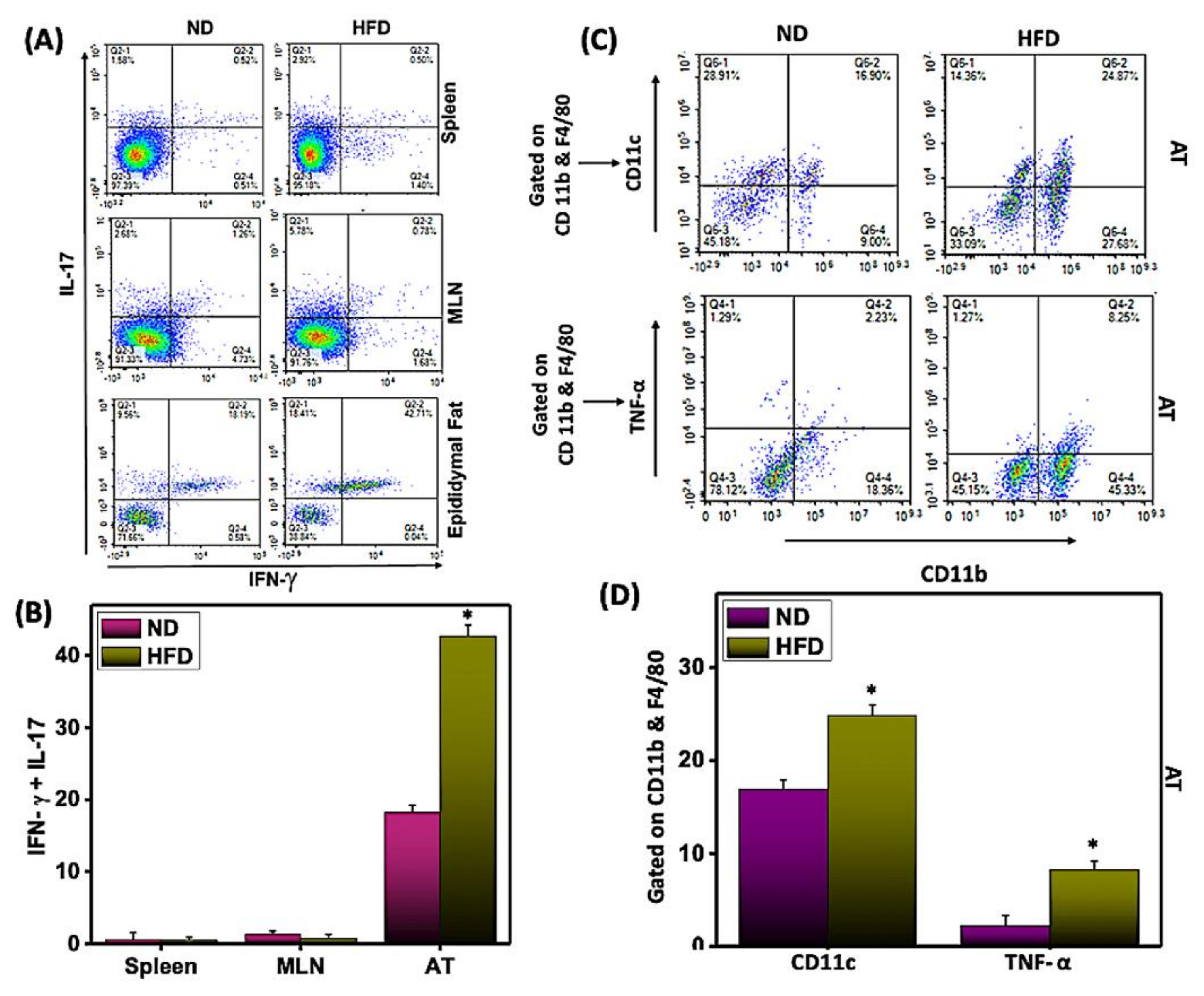
3.4. HFD-Induced Th17 Mediates Th1 and M1 Macrophage Response in AT
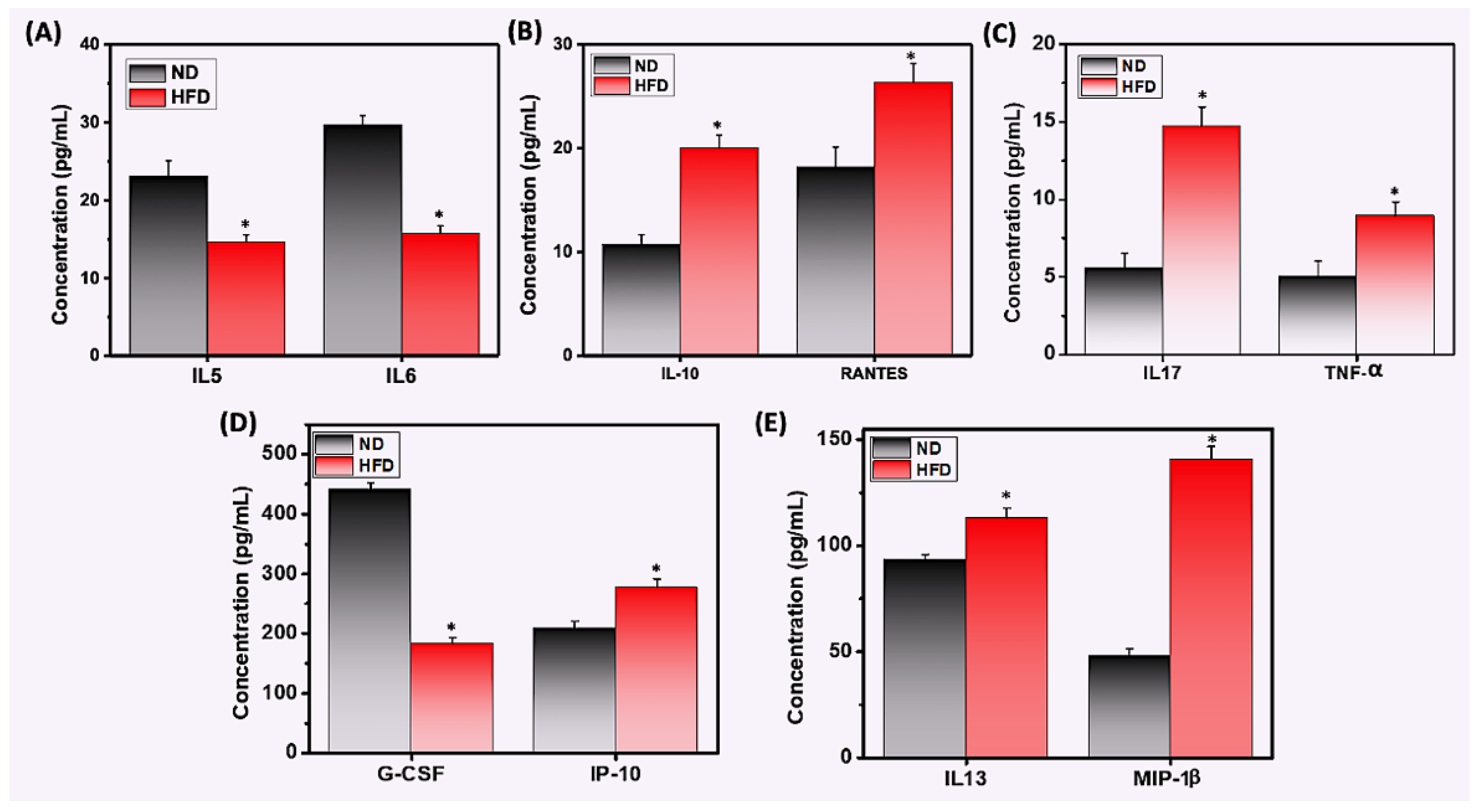
3.5. HFD Differentially Mediates Th1/Th17 Cytokine and Chemokine Response
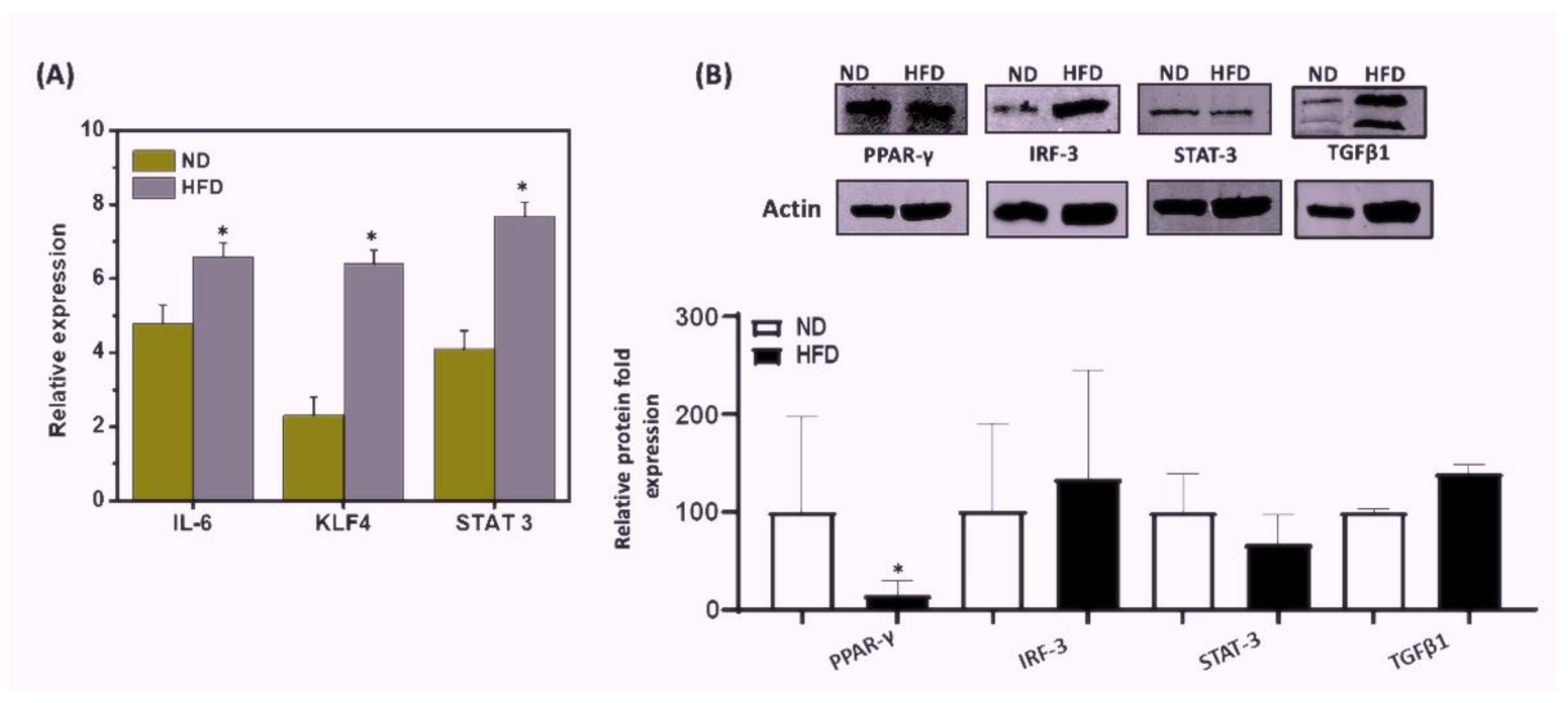
3.6. HFD Induced Differential Signaling Pathways to Sustain Chronic AT Inflammation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Flegal, K.M.; Carroll, M.D.; Ogden, C.L.; Curtin, L.R. Prevalence and trends in obesity among US adults, 1999–2008. JAMA 2010, 303, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, D.E.; Kaufman, K.D. Metabolic syndrome: A clinical and molecular perspective. Annu. Rev. Med. 2005, 56, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Malecka-Tendera, E.; Mazur, A. Childhood obesity: A pandemic of the twenty-first century. Int. J. Obes. 2006, 30, S1–S3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [Green Version]
- Lumeng, C.N.; Saltiel, A. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.K.; Gutierrez, D.A.; Hasty, A.H. Adipose tissue recruitment of leukocytes. Curr. Opin. Lipidol. 2010, 21, 172–177. [Google Scholar] [CrossRef]
- Winer, S.; Winer, D.A. The adaptive immune system as a fundamental regulator of adipose tissue inflammation and insulin resistance. Immunol. Cell Biol. 2012, 90, 755–762. [Google Scholar] [CrossRef]
- Surendar, J.; Frohberger, S.J.; Karunakaran, I.; Schmitt, V.; Stamminger, W.; Neumann, A.-L.; Wilhelm, C.; Hoerauf, A.; Hübner, M.P. Adiponectin Limits IFN-γ and IL-17 Producing CD4 T Cells in Obesity by Restraining Cell Intrinsic Glycolysis. Front. Immunol. 2019, 10, 2555. [Google Scholar] [CrossRef]
- Ahmed, M.; Gaffen, S.L. IL-17 in obesity and adipogenesis. Cytokine Growth Factor Rev. 2010, 21, 449–453. [Google Scholar] [CrossRef] [Green Version]
- Łuczyński, W.; Grubczak, K.; Moniuszko, M.; Głowińska-Olszewska, B.; Bossowski, A. Elevated levels of Th17 cells in children with central obesity. Scand. J. Clin. Lab. Investig. 2015, 75, 595–601. [Google Scholar] [CrossRef]
- Schindler, T.I.; Wagner, J.-J.; Goedicke-Fritz, S.; Rogosch, T.; Coccejus, V.; Laudenbach, V.; Nikolaizik, W.; Härtel, C.; Maier, R.F.; Kerzel, S.; et al. TH17 Cell Frequency in Peripheral Blood Is Elevated in Overweight Children without Chronic Inflammatory Diseases. Front. Immunol. 2017, 8, 1543. [Google Scholar] [CrossRef] [Green Version]
- Giles, D.A.; Moreno-Fernandez, M.; Divanovic, S.; Moreno-Fernandez, M.E. IL-17 Axis Driven Inflammation in Non-Alcoholic Fatty Liver Disease Progression. Curr. Drug Targets 2015, 16, 1315–1323. [Google Scholar] [CrossRef] [Green Version]
- Endo, Y.; Yokote, K.; Nakayama, T. The obesity-related pathology and Th17 cells. Cell Mol. Life Sci. 2016, 74, 1231–1245. [Google Scholar] [CrossRef]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef]
- Martinez-Sanchez, M.E.; Hiriart, M.; Alvarez-Buylla, E.R. The CD4+ T cell regulatory network mediates inflammatory responses during acute hyperinsulinemia: A simulation study. BMC Syst. Biol. 2017, 11, 64. [Google Scholar] [CrossRef]
- Fabbrini, E.; Cella, M.; Mccartney, S.A.; Fuchs, A.; Abumrad, N.A.; Pietka, T.A.; Chen, Z.; Finck, B.N.; Han, D.H.; Magkos, F.; et al. Association between specific adipose tissue CD4+ T-cell populations and insulin resistance in obese individuals. Gastroenterology 2013, 145, 366–374.e3. [Google Scholar] [CrossRef] [Green Version]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef]
- Zeng, C.; Shi, X.; Zhang, B.; Liu, H.; Zhang, L.; Ding, W.; Zhao, Y. The imbalance of Th17/Th1/Tregs in patients with type 2 diabetes: Relationship with metabolic factors and complications. J. Mol. Med. 2012, 90, 175–186. [Google Scholar] [CrossRef]
- Kiran, S.; Kumar, V.; Murphy, E.A.; Enos, R.T.; Singh, U.P. High Fat Diet-Induced CD8+ T Cells in Adipose Tissue Mediate Macrophages to Sustain Low-Grade Chronic Inflammation. Front. Immunol. 2021, 12, 680944. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Kanneganti, T.-D.; Dixit, V.D. Immunological complications of obesity. Nat. Immunol. 2012, 13, 707–712. [Google Scholar] [CrossRef]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative Activation of Macrophages: An Immunologic Functional Perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [Green Version]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.-I.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Ferrante, A.W., Jr. Macrophages, fat, and the emergence of immunometabolism. J. Clin. Investig. 2013, 123, 4992–4993. [Google Scholar] [CrossRef]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef]
- Toussirot, E.; Streit, G.; Nguyen, N.U.; Dumoulin, G.; Le Huédé, G.; Saas, P.; Wendling, D. Adipose tissue, serum adipokines, and ghrelin in patients with ankylosing spondylitis. Metabolism 2007, 56, 1383–1389. [Google Scholar] [CrossRef]
- Bertola, A.; Ciucci, T.; Rousseau, D.; Bourlier, V.; Duffaut, C.; Bonnafous, S.; Blin-Wakkach, C.; Anty, R.; Iannelli, A.; Gugenheim, J.; et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes 2012, 61, 2238–2247. [Google Scholar] [CrossRef] [Green Version]
- Singer, K.; Maley, N.; Mergian, T.; DelProposto, J.; Cho, K.W.; Zamarron, B.F.; Martinez-Santibanez, G.; Geletka, L.; Muir, L.; Wachowiak, P.; et al. Differences in Hematopoietic Stem Cells Contribute to Sexually Dimorphic Inflammatory Responses to High Fat Diet-induced Obesity. J. Biol. Chem. 2015, 290, 13250–13262. [Google Scholar] [CrossRef] [Green Version]
- Domínguez, P.M.; Ardavín, C. Differentiation and function of mouse monocyte-derived dendritic cells in steady state and inflammation. Immunol. Rev. 2010, 234, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic-Racic, M.; Yang, X.; Turner, M.S.; Mantell, B.S.; Stolz, D.B.; Sumpter, T.L.; Sipula, I.J.; Dedousis, N.; Scott, D.K.; Morel, P.A.; et al. Dendritic Cells Promote Macrophage Infiltration and Comprise a Substantial Proportion of Obesity-Associated Increases in CD11c+ Cells in Adipose Tissue and Liver. Diabetes 2012, 61, 2330–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crook, K.R.; Liu, P. Role of myeloid-derived suppressor cells in autoimmune disease. World J. Immunol. 2014, 4, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Kontaki, E.; Boumpas, D.T.; Tzardi, M.; Mouzas, I.A.; Papadakis, K.A.; Verginis, P. Aberrant function of myeloid-derived suppressor cells (MDSCs) in experimental colitis and in inflammatory bowel disease (IBD) immune responses. Autoimmunity 2017, 50, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Sha, H.; Yang, L.; Ji, Y.; Ostrand-Rosenberg, S.; Qi, L. Gr-1+ CD11b+ myeloid-derived suppressor cells suppress inflammation and promote insulin sensitivity in obesity. J. Biol. Chem. 2011, 286, 23591–23599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zúñiga, L.A.; Shen, W.-J.; Joyce-Shaikh, B.; Pyatnova, E.A.; Richards, A.G.; Thom, C.; Andrade, S.M.; Cua, D.J.; Kraemer, F.; Butcher, E.C. IL-17 Regulates Adipogenesis, Glucose Homeostasis, and Obesity. J. Immunol. 2010, 185, 6947–6959. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Chen, H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Shin, J.H.; Shin, D.W.; Noh, M. Interleukin-17A inhibits adipocyte differentiation in human mesenchymal stem cells and regulates pro-inflammatory responses in adipocytes. Biochem. Pharmacol. 2009, 77, 1835–1844. [Google Scholar] [CrossRef]
- He, B.; Wu, L.; Xie, W.; Shao, Y.; Jiang, J.; Zhao, Z.; Cui, D. The imbalance of Th17/Treg cells is involved in the progression of nonalcoholic fatty liver disease in mice. BMC Immunol. 2017, 18, 33. [Google Scholar] [CrossRef] [Green Version]
- Bain, C.C.; Montgomery, J.; Scott, C.L.; Kel, J.M.; Girard-Madoux, M.J.H.; Martens, L. TGFbetaR signalling controls CD103(+)CD11b(+) dendritic cell development in the intestine. Nat. Commun. 2017, 8, 620. [Google Scholar] [CrossRef] [Green Version]
- Festa, A.; D’Agostino, R., Jr.; Williams, K.; Karter, A.J.; Mayer-Davis, E.J.; Tracy, R.P.; Haffner, S.M. The relation of body fat mass and distribution to markers of chronic inflammation. Int. J. Obes. 2001, 25, 1407–1415. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; He, C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018, 44, 38–50. [Google Scholar] [CrossRef]
- Borst, S.E. The Role of TNF-α in Insulin Resistance. Endocrine 2004, 23, 177–182. [Google Scholar] [CrossRef]
- Fjeldborg, K.; Pedersen, S.B.; Møller, H.J.; Christiansen, T.; Bennetzen, M.; Richelsen, B. Human adipose tissue macrophages are enhanced but changed to an anti-inflammatory profile in obesity. J. Immunol. Res. 2014, 2014, 309548. [Google Scholar] [CrossRef]
- Moraes-Vieira, P.; Yore, M.M.; Dwyer, P.M.; Syed, I.; Aryal, P.; Kahn, B.B. RBP4 Activates antigen-presenting cells, leading to adipose tissue inflammation and systemic insulin resistance. Cell Metab. 2014, 19, 512–526. [Google Scholar] [CrossRef] [Green Version]
- Noël, W.; Raes, G.; Hassanzadeh Ghassabeh, G.; De Baetselier, P.; Beschin, A. Alternatively activated macrophages during parasite infections. Trends Parasitol. 2004, 20, 126–133. [Google Scholar] [CrossRef]
- Lee, Y.; Song, Y.-S.; Fang, C.-H.; So, B.-I.; Park, J.-Y.; Joo, H.-W.; Park, I.-H.; Shen, G.-Y.; Shin, J.-H.; Kim, H.; et al. Anti-obesity effects of granulocyte-colony stimulating factor in otsuka-long-evans-tokushima fatty rats. PLoS ONE 2014, 9, e105603. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional Profiling of the Human Monocyte-to-Macrophage Differentiation and Polarization: New Molecules and Patterns of Gene Expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef] [Green Version]
- Durant, L.; Watford, W.T.; Ramos, H.L.; Laurence, A.; Vahedi, G.; Wei, L.; Takahashi, H.; Sun, H.-W.; Kanno, Y.; Powrie, F.; et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 2010, 32, 605–615. [Google Scholar] [CrossRef] [Green Version]
- Harris, T.J.; Grosso, J.F.; Yen, H.-R.; Xin, H.; Kortylewski, M.; Albesiano, E.; Hipkiss, E.L.; Getnet, D.; Goldberg, M.V.; Maris, C.H.; et al. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J. Immunol. 2007, 179, 4313–4317. [Google Scholar] [CrossRef] [Green Version]
- Chawla, A.; Barak, Y.; Nagy, L.; Liao, D.; Tontonoz, P.; Evans, R.M. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat. Med. 2001, 7, 48–52. [Google Scholar] [CrossRef]
- Liao, X.; Sharma, N.; Kapadia, F.; Zhou, G.; Lu, Y.; Hong, H.; Paruchuri, K.; Mahabeleshwar, G.H.; Dalmas, E.; Venteclef, N.; et al. Krüppel-like factor 4 regulates macrophage polarization. J. Clin. Investig. 2011, 121, 2736–2749. [Google Scholar] [CrossRef] [Green Version]
- Tang, P.; Virtue, S.; Goie, J.Y.G.; Png, C.W.; Guo, J.; Li, Y.; Jiao, H.; Chua, Y.L.; Campbell, M.; Moreno-Navarrete, J.M.; et al. Regulation of adipogenic differentiation and adipose tissue inflammation by interferon regulatory factor 3. Cell Death Differ. 2021, 28, 3022–3035. [Google Scholar] [CrossRef]
- Mao, Y.; Luo, W.; Zhang, L.; Wu, W.; Yuan, L.; Xu, H.; Song, J.; Fujiwara, K.; Abe, J.-I.; Lemaire, S.A.; et al. STING–IRF3 Triggers Endothelial Inflammation in Response to Free Fatty Acid-Induced Mitochondrial Damage in Diet-Induced Obesity. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 920–929. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiran, S.; Rakib, A.; Kodidela, S.; Kumar, S.; Singh, U.P. High-Fat Diet-Induced Dysregulation of Immune Cells Correlates with Macrophage Phenotypes and Chronic Inflammation in Adipose Tissue. Cells 2022, 11, 1327. https://doi.org/10.3390/cells11081327
Kiran S, Rakib A, Kodidela S, Kumar S, Singh UP. High-Fat Diet-Induced Dysregulation of Immune Cells Correlates with Macrophage Phenotypes and Chronic Inflammation in Adipose Tissue. Cells. 2022; 11(8):1327. https://doi.org/10.3390/cells11081327
Chicago/Turabian StyleKiran, Sonia, Ahmed Rakib, Sunitha Kodidela, Santosh Kumar, and Udai P. Singh. 2022. "High-Fat Diet-Induced Dysregulation of Immune Cells Correlates with Macrophage Phenotypes and Chronic Inflammation in Adipose Tissue" Cells 11, no. 8: 1327. https://doi.org/10.3390/cells11081327