
Store-Operated Calcium Entry and Its Implications in Cancer Stem Cells

 ,
,  , and
, and 
Abstract
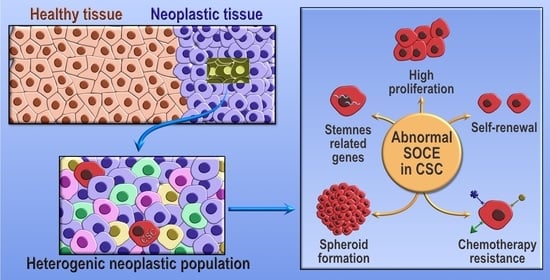
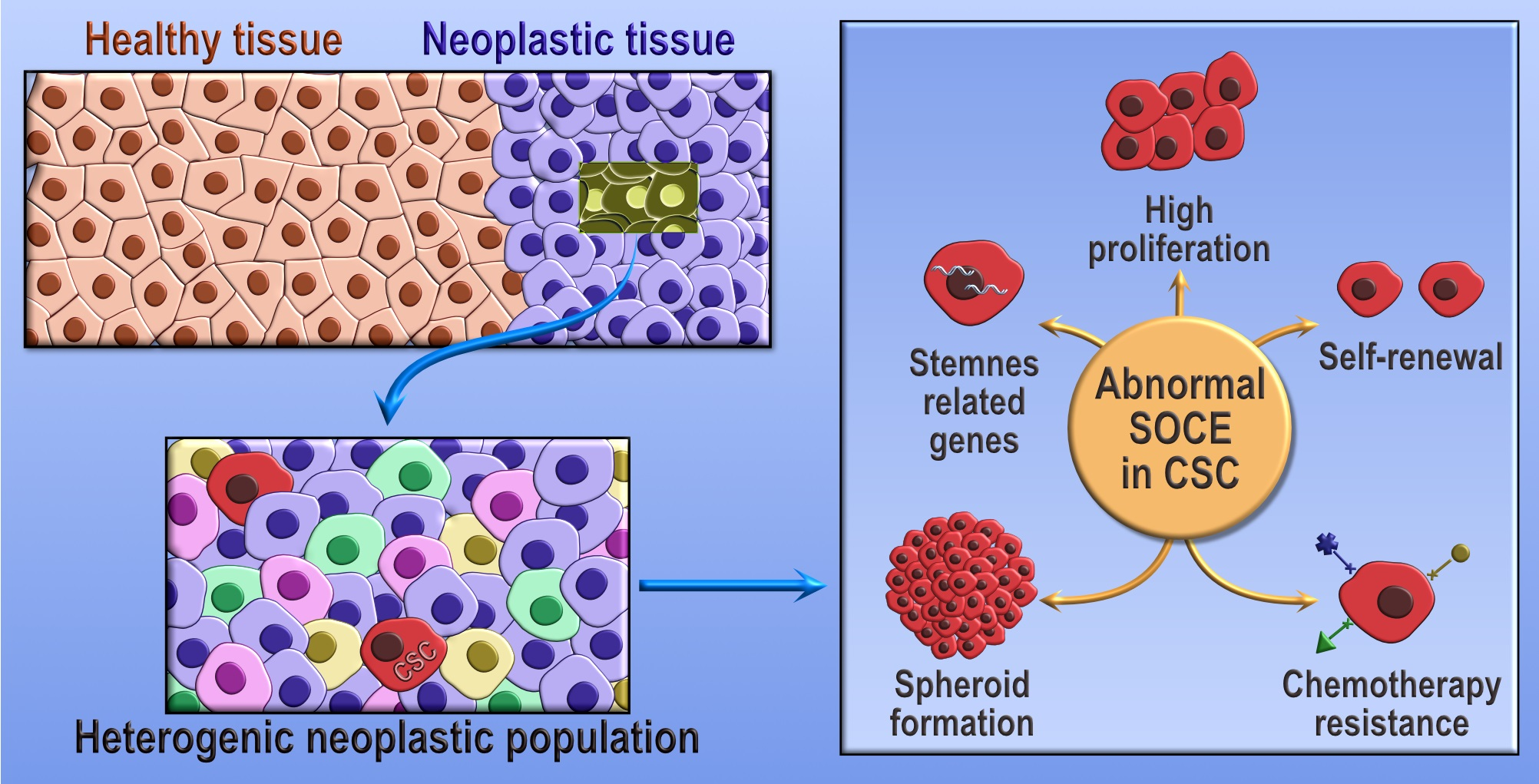
1. Introduction
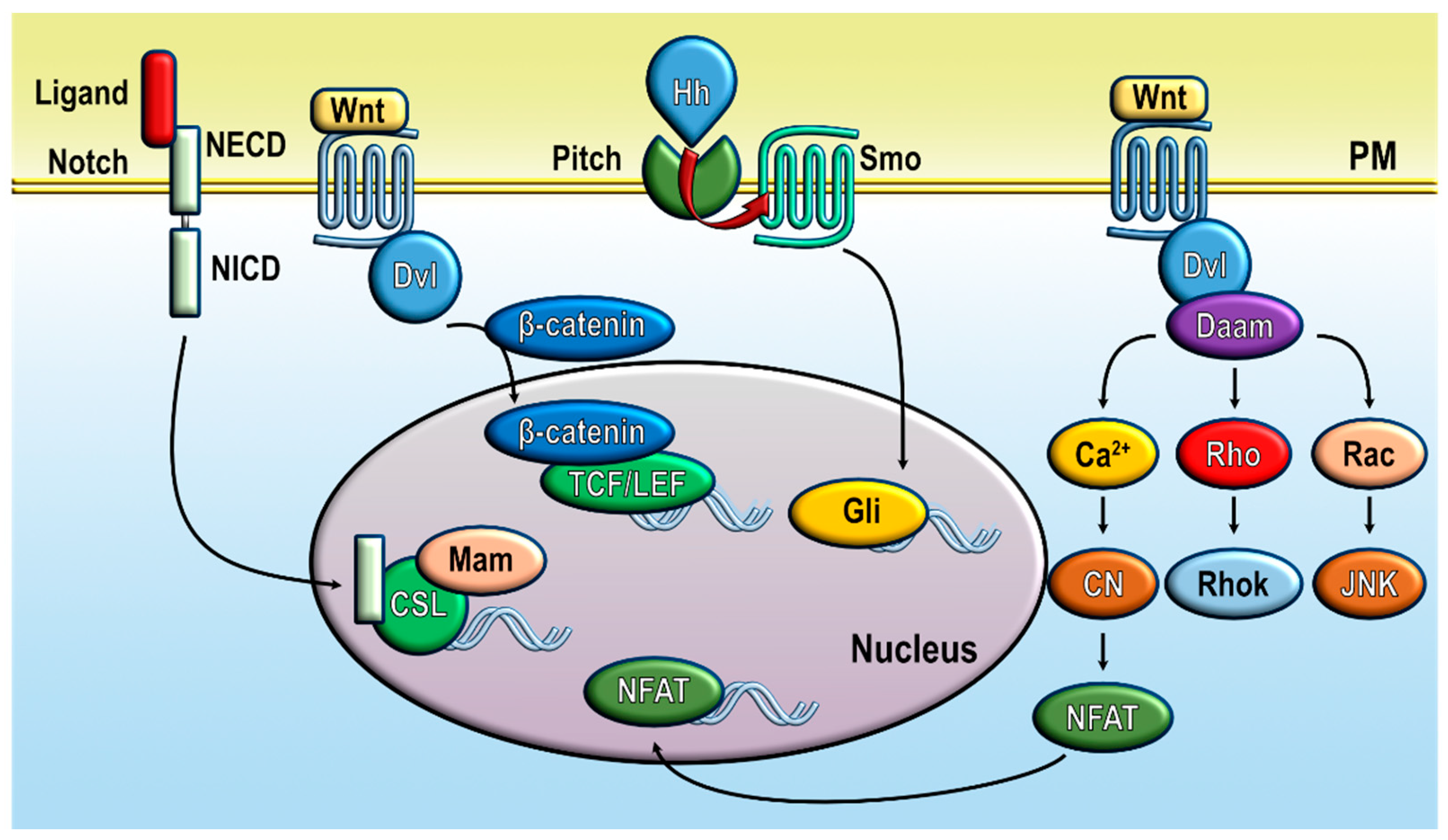
2. Calcium Signaling in Cancer Stem Cells and Cancer Hallmarks
3. Store-Operated Calcium Entry in Cancer Stem Cells and Cancer Hallmarks
4. Functional Role of Orai in Cancer Stem Cells and Cancer Hallmarks
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramalho-Santos, M.; Willenbring, H. On the origin of the term “stem cell”. Cell Stem Cell 2007, 1, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Jaenisch, R. Stem Cells, Genome Editing, and the Path to Translational Medicine. Cell 2018, 175, 615–632. [Google Scholar] [CrossRef] [PubMed]
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef] [PubMed]
- Neuzil, J.; Stantic, M.; Zobalova, R.; Chladova, J.; Wang, X.; Prochazka, L.; Dong, L.; Andera, L.; Ralph, S.J. Tumour-initiating cells vs. ‘cancer stem’ cells and CD133: What’s in the name? Biochem. Biophys. Res. Commun. 2007, 355, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Kabakov, A.; Yakimova, A.; Matchuk, O. Molecular Chaperones in Cancer Stem Cells: Determinants of Stemness and Potential Targets for Antitumor Therapy. Cells 2020, 9, 892. [Google Scholar] [CrossRef]
- Li, L.; Neaves, W.B. Normal stem cells and cancer stem cells: The niche matters. Cancer Res. 2006, 66, 4553–4557. [Google Scholar] [CrossRef]
- Rahman, M.; Deleyrolle, L.; Vedam-Mai, V.; Azari, H.; Abd-El-Barr, M.; Reynolds, B.A. The cancer stem cell hypothesis: Failures and pitfalls. Neurosurgery 2011, 68, 531–545. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Hollande, E.; de St-Front, V.T.; Louet-Hermitte, P.; Bara, J.; Pequignot, J.; Estival, A.; Clemente, F. Differentiation features of human pancreatic tumor cells maintained in nude mice and in culture: Immunocytochemical and ultrastructural studies. Int. J. Cancer 1984, 34, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Miyauchi, M.; Takekoshi, T.; Zhao, M.; Kudo, Y.; Ogawa, I.; Kitagawa, S.; Fujita, M.; Takata, T. Reduced expression of CD44 variant 9 is related to lymph node metastasis and poor survival in squamous cell carcinoma of tongue. Oral Oncol. 2000, 36, 545–549. [Google Scholar] [CrossRef]
- Grotenhuis, B.A.; Dinjens, W.N.; Wijnhoven, B.P.; Sonneveld, P.; Sacchetti, A.; Franken, P.F.; van Dekken, H.; Tilanus, H.W.; van Lanschot, J.J.; Fodde, R. Barrett’s oesophageal adenocarcinoma encompasses tumour-initiating cells that do not express common cancer stem cell markers. J. Pathol. 2010, 221, 379–389. [Google Scholar] [CrossRef]
- Yang, Z.F.; Ngai, P.; Ho, D.W.; Yu, W.C.; Ng, M.N.; Lau, C.K.; Li, M.L.; Tam, K.H.; Lam, C.T.; Poon, R.T.; et al. Identification of local and circulating cancer stem cells in human liver cancer. Hepatology 2008, 47, 919–928. [Google Scholar] [CrossRef]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea—A paradigm shift. Cancer Res. 2006, 66, 1883–1890. [Google Scholar] [CrossRef]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauss, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Cho, Y.; Lee, H.W.; Kang, H.G.; Kim, H.Y.; Kim, S.J.; Chun, K.H. Cleaved CD44 intracellular domain supports activation of stemness factors and promotes tumorigenesis of breast cancer. Oncotarget 2015, 6, 8709–8721. [Google Scholar] [CrossRef] [PubMed]
- Screaton, G.R.; Bell, M.V.; Jackson, D.G.; Cornelis, F.B.; Gerth, U.; Bell, J.I. Genomic structure of DNA encoding the lymphocyte homing receptor CD44 reveals at least 12 alternatively spliced exons. Proc. Natl. Acad. Sci. USA 1992, 89, 12160–12164. [Google Scholar] [CrossRef] [PubMed]
- Gunthert, U.; Hofmann, M.; Rudy, W.; Reber, S.; Zoller, M.; Haussmann, I.; Matzku, S.; Wenzel, A.; Ponta, H.; Herrlich, P. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell 1991, 65, 13–24. [Google Scholar] [CrossRef]
- Pierres, M.; Naquet, P.; Barbet, J.; Marchetto, S.; Marics, I.; Devaux, C.; Barad, M.; Hyman, R.; Rougon, G. Evidence that murine hematopoietic cell subset marker J11d is attached to a glycosyl-phosphatidylinositol membrane anchor. Eur. J. Immunol. 1987, 17, 1781–1785. [Google Scholar] [CrossRef] [PubMed]
- Ricardo, S.; Vieira, A.F.; Gerhard, R.; Leitao, D.; Pinto, R.; Cameselle-Teijeiro, J.F.; Milanezi, F.; Schmitt, F.; Paredes, J. Breast cancer stem cell markers CD44, CD24 and ALDH1: Expression distribution within intrinsic molecular subtype. J. Clin. Pathol. 2011, 64, 937–946. [Google Scholar] [CrossRef]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2017, 7, 13856. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Marcato, P.; Dean, C.A.; Pan, D.; Araslanova, R.; Gillis, M.; Joshi, M.; Helyer, L.; Pan, L.; Leidal, A.; Gujar, S.; et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells 2011, 29, 32–45. [Google Scholar] [CrossRef]
- Ohi, Y.; Umekita, Y.; Yoshioka, T.; Souda, M.; Rai, Y.; Sagara, Y.; Tanimoto, A. Aldehyde dehydrogenase 1 expression predicts poor prognosis in triple-negative breast cancer. Histopathology 2011, 59, 776–780. [Google Scholar] [CrossRef]
- Zhou, L.; Li, K.; Luo, Y.; Tian, L.; Wang, M.; Li, C.; Huang, Q. Novel prognostic markers for patients with triple-negative breast cancer. Hum. Pathol. 2013, 44, 2180–2187. [Google Scholar] [CrossRef]
- Wilson, J.J.; Kovall, R.A. Crystal structure of the CSL-Notch-Mastermind ternary complex bound to DNA. Cell 2006, 124, 985–996. [Google Scholar] [CrossRef]
- Wang, R.; Sun, Q.; Wang, P.; Liu, M.; Xiong, S.; Luo, J.; Huang, H.; Du, Q.; Geller, D.A.; Cheng, B. Notch and Wnt/beta-catenin signaling pathway play important roles in activating liver cancer stem cells. Oncotarget 2016, 7, 5754–5768. [Google Scholar] [CrossRef]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, Y.; Zhang, Y.; Li, X.; Liu, C.; Huang, S.; Xu, D.; Wu, Y. T-cell factor (TCF/LEF1) binding elements (TBEs) of FasL (Fas ligand or CD95 ligand) bind and cluster Fas (CD95) and form complexes with the TCF-4 and b-catenin transcription factors in vitro and in vivo which result in triggering cell death and/or cell activation. Cell. Mol. Neurobiol. 2016, 36, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Hooper, R.; Zaidi, M.R.; Soboloff, J. The heterogeneity of store-operated calcium entry in melanoma. Sci. China Life Sci. 2016, 59, 764–769. [Google Scholar] [CrossRef] [PubMed][Green Version]
- van Amerongen, R. Alternative Wnt pathways and receptors. Cold Spring Harb. Perspect. Biol. 2012, 4, a007914. [Google Scholar] [CrossRef]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/beta-Catenin and NF-kappaB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef]
- Hodeify, R.; Yu, F.; Courjaret, R.; Nader, N.; Dib, M.; Sun, L.; Adap, E.; Hubrack, S.; Machaca, K. Regulation and Role of Store-Operated Ca2+ Entry in Cellular Proliferation. Calcium Entry Channels Non-Excit. Cells 2018, 2017, 215–240. [Google Scholar] [CrossRef]
- Qin, J.J.; Nag, S.; Wang, W.; Zhou, J.; Zhang, W.D.; Wang, H.; Zhang, R. NFAT as cancer target: Mission possible? Biochim. Biophys. Acta 2014, 1846, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 2012, 16, S17–S27. [Google Scholar] [CrossRef]
- Lin, R.J.; Kuo, M.W.; Yang, B.C.; Tsai, H.H.; Chen, K.; Huang, J.R.; Lee, Y.S.; Yu, A.L.; Yu, J. B3GALT5 knockout alters gycosphingolipid profile and facilitates transition to human naive pluripotency. Proc. Natl. Acad. Sci. USA 2020, 117, 27435–27444. [Google Scholar] [CrossRef]
- Zubeldia, I.G.; Bleau, A.M.; Redrado, M.; Serrano, D.; Agliano, A.; Gil-Puig, C.; Vidal-Vanaclocha, F.; Lecanda, J.; Calvo, A. Epithelial to mesenchymal transition and cancer stem cell phenotypes leading to liver metastasis are abrogated by the novel TGFbeta1-targeting peptides P17 and P144. Exp. Cell Res. 2013, 319, 12–22. [Google Scholar] [CrossRef]
- Cocola, C.; Molgora, S.; Piscitelli, E.; Veronesi, M.C.; Greco, M.; Bragato, C.; Moro, M.; Crosti, M.; Gray, B.; Milanesi, L.; et al. FGF2 and EGF Are Required for Self-Renewal and Organoid Formation of Canine Normal and Tumor Breast Stem Cells. J. Cell Biochem. 2017, 118, 570–584. [Google Scholar] [CrossRef]
- Dong, J.; Aulestia, F.J.; Assad Kahn, S.; Zeniou, M.; Dubois, L.G.; El-Habr, E.A.; Daubeuf, F.; Tounsi, N.; Cheshier, S.H.; Frossard, N.; et al. Bisacodyl and its cytotoxic activity on human glioblastoma stem-like cells. Implication of inositol 1,4,5-triphosphate receptor dependent calcium signaling. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1018–1027. [Google Scholar] [CrossRef]
- Marciel, M.P.; Khadka, V.S.; Deng, Y.; Kilicaslan, P.; Pham, A.; Bertino, P.; Lee, K.; Chen, S.; Glibetic, N.; Hoffmann, F.W.; et al. Selenoprotein K deficiency inhibits melanoma by reducing calcium flux required for tumor growth and metastasis. Oncotarget 2018, 9, 13407–13422. [Google Scholar] [CrossRef]
- Lu, H.; Chen, I.; Shimoda, L.A.; Park, Y.; Zhang, C.; Tran, L.; Zhang, H.; Semenza, G.L. Chemotherapy-Induced Ca2+ Release Stimulates Breast Cancer Stem Cell Enrichment. Cell Rep. 2017, 18, 1946–1957. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cruickshanks, N.; Yuan, F.; Wang, B.; Pahuski, M.; Wulfkuhle, J.; Gallagher, I.; Koeppel, A.F.; Hatef, S.; Papanicolas, C.; et al. Targetable T-type Calcium Channels Drive Glioblastoma. Cancer Res. 2017, 77, 3479–3490. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, J.W.; Kim, D.K.; Choi, D.K.; Lee, S.; Yu, J.H.; Kwon, O.B.; Lee, J.; Lee, D.S.; Kim, J.H.; et al. Calcium Channels as Novel Therapeutic Targets for Ovarian Cancer Stem Cells. Int. J. Mol. Sci. 2020, 21, 2327. [Google Scholar] [CrossRef] [PubMed]
- Niklasson, M.; Maddalo, G.; Sramkova, Z.; Mutlu, E.; Wee, S.; Sekyrova, P.; Schmidt, L.; Fritz, N.; Dehnisch, I.; Kyriatzis, G.; et al. Membrane-Depolarizing Channel Blockers Induce Selective Glioma Cell Death by Impairing Nutrient Transport and Unfolded Protein/Amino Acid Responses. Cancer Res. 2017, 77, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, S.; Zhao, W.; Duan, J.; Wang, Z.; Chen, H.; Tian, Y.; Wang, D.; Zhao, J.; An, T.; et al. Mechanistic Exploration of Cancer Stem Cell Marker Voltage-Dependent Calcium Channel alpha2delta1 Subunit-mediated Chemotherapy Resistance in Small-Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 2148–2158. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, X.; Zhao, W.; Yang, Y.; Zhang, Z. Calcium channel alpha2delta1 subunit is a functional marker and therapeutic target for tumor-initiating cells in non-small cell lung cancer. Cell Death Dis. 2021, 12, 257. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, W.; Yang, X.; An, G.; Zhao, W. The alpha2delta1 subunit of the voltage-gated calcium channel acts as a potential candidate for breast cancer tumor initial cells biomarker. Cancer Biomark. 2021, 31, 295–305. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, L.; Han, H.; Jin, K.; Lin, N.; Guo, T.; Chen, Y.; Cheng, H.; Lu, F.; Fang, W.; et al. 1B50-1, a mAb raised against recurrent tumor cells, targets liver tumor-initiating cells by binding to the calcium channel alpha2delta1 subunit. Cancer Cell 2013, 23, 541–556. [Google Scholar] [CrossRef]
- Huang, C.; Li, Y.; Zhao, W.; Zhang, A.; Lu, C.; Wang, Z.; Liu, L. alpha2delta1 may be a potential marker for cancer stem cell in laryngeal squamous cell carcinoma. Cancer Biomark. 2019, 24, 97–107. [Google Scholar] [CrossRef]
- Hirata, N.; Yamada, S.; Yanagida, S.; Ono, A.; Yasuhiko, Y.; Nishida, M.; Kanda, Y. Lysophosphatidic Acid Promotes the Expansion of Cancer Stem Cells via TRPC3 Channels in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 1967. [Google Scholar] [CrossRef]
- Liu, K.; Xu, S.H.; Chen, Z.; Zeng, Q.X.; Li, Z.J.; Chen, Z.M. TRPM7 overexpression enhances the cancer stem cell-like and metastatic phenotypes of lung cancer through modulation of the Hsp90alpha/uPA/MMP2 signaling pathway. BMC Cancer 2018, 18, 1167. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Inoue, K.; Leng, T.; Guo, S.; Xiong, Z.G. TRPM7 channels regulate glioma stem cell through STAT3 and Notch signaling pathways. Cell. Signal. 2014, 26, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Middelbeek, J.; Visser, D.; Henneman, L.; Kamermans, A.; Kuipers, A.J.; Hoogerbrugge, P.M.; Jalink, K.; van Leeuwen, F.N. TRPM7 maintains progenitor-like features of neuroblastoma cells: Implications for metastasis formation. Oncotarget 2015, 6, 8760–8776. [Google Scholar] [CrossRef] [PubMed]
- Santoni, G.; Amantini, C. The Transient Receptor Potential Vanilloid Type-2(TRPV2) Ion Channels in Neurogenesis andGliomagenesis: Cross-Talk between TranscriptionFactors and Signaling Molecules. Cancers 2019, 11, 322. [Google Scholar] [CrossRef] [PubMed]
- Shiozaki, A.; Kudou, M.; Ichikawa, D.; Fujiwara, H.; Shimizu, H.; Ishimoto, T.; Arita, T.; Kosuga, T.; Konishi, H.; Komatsu, S.; et al. Esophageal cancer stem cells are suppressed by tranilast, a TRPV2 channel inhibitor. J. Gastroenterol. 2018, 53, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.B.; Nabissi, M.; Amantini, C.; Farfariello, V.; Ricci-Vitiani, L.; di Martino, S.; Pallini, R.; Larocca, L.M.; Caprodossi, S.; Santoni, M.; et al. The transient receptor potential vanilloid-2 cation channel impairs glioblastoma stem-like cell proliferation and promotes differentiation. Int. J. Cancer 2012, 131, E1067–E1077. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Liberati, S.; Santoni, M.; Ricci-Vitiani, L.; Pallini, R.; Santoni, G. Cannabidiol stimulates Aml-1a-dependent glial differentiation and inhibits glioma stem-like cells proliferation by inducing autophagy in a TRPV2-dependent manner. Int. J. Cancer 2015, 137, 1855–1869. [Google Scholar] [CrossRef]
- Hu, Z.; Cao, X.; Fang, Y.; Liu, G.; Xie, C.; Qian, K.; Lei, X.; Cao, Z.; Du, H.; Cheng, X.; et al. Transient receptor potential vanilloid-type 2 targeting on stemness in liver cancer. Biomed. Pharmacother. 2018, 105, 697–706. [Google Scholar] [CrossRef]
- Santoni, G.; Nabissi, M.; Amantini, C.; Santoni, M.; Ricci-Vitiani, L.; Pallini, R.; Maggi, F.; Morelli, M.B. ERK Phosphorylation Regulates the Aml1/Runx1 Splice Variants and the TRP Channels Expression during the Differentiation of Glioma Stem Cell Lines. Cells 2021, 10, 2052. [Google Scholar] [CrossRef]
- Redondo, P.C.; Rosado, J.A.; Pariente, J.A.; Salido, G.M. Collaborative effect of SERCA and PMCA in cytosolic calcium homeostasis in human platelets. J. Physiol. Biochem. 2005, 61, 507–516. [Google Scholar] [CrossRef]
- Elaib, Z.; Saller, F.; Bobe, R. The Calcium Entry-Calcium Refilling Coupling. Adv. Exp. Med. Biol. 2016, 898, 333–352. [Google Scholar] [CrossRef] [PubMed]
- Lariccia, V.; Piccirillo, S.; Preziuso, A.; Amoroso, S.; Magi, S. Cracking the code of sodium/calcium exchanger (NCX) gating: Old and new complexities surfacing from the deep web of secondary regulations. Cell Calcium 2020, 87, 102169. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Kim, S.W.; Jeon, J.Y.; Jo, A.R.; Choi, H.J.; Kim, J.; Lee, H.G.; Kim, Y.; Mills, G.B.; Noh, S.H.; et al. Survival of Cancer Stem-Like Cells Under Metabolic Stress via CaMK2alpha-mediated Upregulation of Sarco/Endoplasmic Reticulum Calcium ATPase Expression. Clin. Cancer Res. 2018, 24, 1677–1690. [Google Scholar] [CrossRef] [PubMed]
- Robil, N.; Petel, F.; Kilhoffer, M.C.; Haiech, J. Glioblastoma and calcium signaling—Analysis of calcium toolbox expression. Int. J. Dev. Biol. 2015, 59, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W.; Bird, G.S. Cytoplasmic calcium oscillations and store-operated calcium influx. J. Physiol. 2008, 586, 3055–3059. [Google Scholar] [CrossRef]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Jardin, I.; Rosado, J.A. STIM and calcium channel complexes in cancer. Biochim. Biophys. Acta 2016, 1863, 1418–1426. [Google Scholar] [CrossRef]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef]
- Berna-Erro, A.; Jardin, I.; Salido, G.M.; Rosado, J.A. Role of STIM2 in cell function and physiopathology. J. Physiol. 2017, 595, 3111–3128. [Google Scholar] [CrossRef]
- Grabmayr, H.; Romanin, C.; Fahrner, M. STIM Proteins: An Ever-Expanding Family. Int. J. Mol. Sci. 2020, 22, 378. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, M.; Romanin, C. The many states of STIM1. Elife 2021, 10, e75174. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Pan, L.J.; Zhang, Z.M. Functional interactions among STIM1, Orai1 and TRPC1 on the activation of SOCs in HL-7702 cells. Amino Acids 2009, 39, 195–204. [Google Scholar] [CrossRef]
- Desai, P.N.; Zhang, X.; Wu, S.; Janoshazi, A.; Bolimuntha, S.; Putney, J.W.; Trebak, M. Multiple types of calcium channels arising from alternative translation initiation of the Orai1 message. Sci. Signal. 2015, 8, ra74. [Google Scholar] [CrossRef]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Zhou, Y.; Ramachandran, S.; Oh-Hora, M.; Rao, A.; Hogan, P.G. Pore architecture of the ORAI1 store-operated calcium channel. Proc. Natl. Acad. Sci. USA 2010, 107, 4896–4901. [Google Scholar] [CrossRef]
- Mignen, O.; Thompson, J.L.; Shuttleworth, T.J. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J. Physiol. 2008, 586, 419–425. [Google Scholar] [CrossRef]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef]
- Butorac, C.; Krizova, A.; Derler, I. Review: Structure and Activation Mechanisms of CRAC Channels. Adv. Exp. Med. Biol. 2020, 1131, 547–604. [Google Scholar] [CrossRef]
- Qiu, R.; Lewis, R.S. Structural features of STIM and Orai underlying store-operated calcium entry. Curr. Opin. Cell Biol. 2019, 57, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Shuttleworth, T.J. Arachidonic acid, ARC channels, and Orai proteins. Cell Calcium 2009, 45, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Cantonero, C.; Sanchez-Collado, J.; Gonzalez-Nunez, M.A.; Salido, G.M.; Lopez, J.J.; Jardin, I.; Rosado, J.A. Store-independent Orai1-mediated Ca2+ entry and cancer. Cell Calcium 2019, 80, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Chen, L. Recent progress in structural studies on canonical TRP ion channels. Cell Calcium 2019, 83, 102075. [Google Scholar] [CrossRef]
- Lopez, J.J.; Jardin, I.; Albarran, L.; Sanchez-Collado, J.; Cantonero, C.; Salido, G.M.; Smani, T.; Rosado, J.A. Molecular Basis and Regulation of Store-Operated Calcium Entry. Adv. Exp. Med. Biol. 2020, 1131, 445–469. [Google Scholar] [CrossRef]
- Huang, G.N.; Zeng, W.; Kim, J.Y.; Yuan, J.P.; Han, L.; Muallem, S.; Worley, P.F. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat. Cell Biol. 2006, 8, 1003–1010. [Google Scholar] [CrossRef]
- Yuan, J.P.; Zeng, W.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 2007, 9, 636–645. [Google Scholar] [CrossRef]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Ambudkar, I.S. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J. Biol. Chem. 2008, 283, 12935–12940. [Google Scholar] [CrossRef]
- Lee, K.P.; Choi, S.; Hong, J.H.; Ahuja, M.; Graham, S.; Ma, R.; So, I.; Shin, D.M.; Muallem, S.; Yuan, J.P. Molecular determinants mediating gating of Transient Receptor Potential Canonical (TRPC) channels by stromal interaction molecule 1 (STIM1). J. Biol. Chem. 2014, 289, 6372–6382. [Google Scholar] [CrossRef]
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Orai1 mediates the interaction between STIM1 and hTRPC1 and regulates the mode of activation of hTRPC1-forming Ca2+ channels. J. Biol. Chem. 2008, 283, 25296–25304. [Google Scholar] [CrossRef] [PubMed]
- Zitt, C.; Zobel, A.; Obukhov, A.G.; Harteneck, C.; Kalkbrenner, F.; Luckhoff, A.; Schultz, G. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron 1996, 16, 1189–1196. [Google Scholar] [CrossRef]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Swaim, W.; Ambudkar, I.S. Local Ca2+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLoS Biol. 2011, 9, e1001025. [Google Scholar] [CrossRef] [PubMed]
- Stathopulos, P.B.; Zheng, L.; Li, G.Y.; Plevin, M.J.; Ikura, M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 2008, 135, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Muik, M.; Frischauf, I.; Derler, I.; Fahrner, M.; Bergsmann, J.; Eder, P.; Schindl, R.; Hesch, C.; Polzinger, B.; Fritsch, R.; et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J. Biol. Chem. 2008, 283, 8014–8022. [Google Scholar] [CrossRef]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef]
- Derler, I.; Fahrner, M.; Muik, M.; Lackner, B.; Schindl, R.; Groschner, K.; Romanin, C. A CRAC modulatory domain (CMD) within STIM1 mediates fast Ca2+-dependent inactivation of ORAI1 channels. J. Biol. Chem. 2009, 284, 24933–24938. [Google Scholar] [CrossRef]
- Lee, K.P.; Yuan, J.P.; Zeng, W.; So, I.; Worley, P.F.; Muallem, S. Molecular determinants of fast Ca2+-dependent inactivation and gating of the Orai channels. Proc. Natl. Acad. Sci. USA 2009, 106, 14687–14692. [Google Scholar] [CrossRef]
- Albarran, L.; Lopez, J.J.; Jardin, I.; Sanchez-Collado, J.; Berna-Erro, A.; Smani, T.; Camello, P.J.; Salido, G.M.; Rosado, J.A. EFHB is a Novel Cytosolic Ca2+ Sensor That Modulates STIM1-SARAF Interaction. Cell Physiol. Biochem. 2018, 51, 1164–1178. [Google Scholar] [CrossRef]
- Jha, A.; Ahuja, M.; Maleth, J.; Moreno, C.M.; Yuan, J.P.; Kim, M.S.; Muallem, S. The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function. J. Cell Biol. 2013, 202, 71–79. [Google Scholar] [CrossRef]
- Palty, R.; Raveh, A.; Kaminsky, I.; Meller, R.; Reuveny, E. SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell 2012, 149, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Ueyama, T.; Lange, I.; Feske, S.; Saito, N. Protein kinase C-induced phosphorylation of Orai1 regulates the intracellular Ca2+ level via the store-operated Ca2+ channel. J. Biol. Chem. 2010, 285, 25720–25730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Pathak, T.; Yoast, R.; Emrich, S.; Xin, P.; Nwokonko, R.M.; Johnson, M.; Wu, S.; Delierneux, C.; Gueguinou, M.; et al. A calcium/cAMP signaling loop at the ORAI1 mouth drives channel inactivation to shape NFAT induction. Nat. Commun. 2019, 10, 1971. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Collado, J.; Lopez, J.J.; Jardin, I.; Berna-Erro, A.; Camello, P.J.; Cantonero, C.; Smani, T.; Salido, G.M.; Rosado, J.A. Orai1alpha, but not Orai1beta, co-localizes with TRPC1 and is required for its plasma membrane location and activation in HeLa cells. Cell Mol. Life Sci. 2022, 79, 33. [Google Scholar] [CrossRef]
- Li, Y.; Guo, B.; Xie, Q.; Ye, D.; Zhang, D.; Zhu, Y.; Chen, H.; Zhu, B. STIM1 Mediates Hypoxia-Driven Hepatocarcinogenesis via Interaction with HIF-1. Cell Rep. 2015, 12, 388–395. [Google Scholar] [CrossRef]
- Aulestia, F.J.; Neant, I.; Dong, J.; Haiech, J.; Kilhoffer, M.C.; Moreau, M.; Leclerc, C. Quiescence status of glioblastoma stem-like cells involves remodelling of Ca2+ signalling and mitochondrial shape. Sci. Rep. 2018, 8, 9731. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, H.; Zheng, L.; Zhou, Y.; Wu, L.; Xu, Y.; Zhang, X.; Yan, G.; Sheng, H.; Xin, R.; et al. FGF19/SOCE/NFATc2 signaling circuit facilitates the self-renewal of liver cancer stem cells. Theranostics 2021, 11, 5045–5060. [Google Scholar] [CrossRef]
- Yoast, R.E.; Emrich, S.M.; Zhang, X.; Xin, P.; Johnson, M.T.; Fike, A.J.; Walter, V.; Hempel, N.; Yule, D.I.; Sneyd, J.; et al. The native ORAI channel trio underlies the diversity of Ca2+ signaling events. Nat. Commun. 2020, 11, 2444. [Google Scholar] [CrossRef]
- Lee, S.H.; Rigas, N.K.; Lee, C.R.; Bang, A.; Srikanth, S.; Gwack, Y.; Kang, M.K.; Kim, R.H.; Park, N.H.; Shin, K.H. Orai1 promotes tumor progression by enhancing cancer stemness via NFAT signaling in oral/oropharyngeal squamous cell carcinoma. Oncotarget 2016, 7, 43239–43255. [Google Scholar] [CrossRef]
- Singh, A.K.; Roy, N.K.; Bordoloi, D.; Padmavathi, G.; Banik, K.; Khwairakpam, A.D.; Kunnumakkara, A.B.; Sukumar, P. Orai-1 and Orai-2 regulate oral cancer cell migration and colonisation by suppressing Akt/mTOR/NF-kappaB signalling. Life Sci. 2020, 261, 118372. [Google Scholar] [CrossRef]
- Tang, B.D.; Xia, X.; Lv, X.F.; Yu, B.X.; Yuan, J.N.; Mai, X.Y.; Shang, J.Y.; Zhou, J.G.; Liang, S.J.; Pang, R.P. Inhibition of Orai1-mediated Ca2+ entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J. Cell. Mol. Med. 2017, 21, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC6 Channels Are Required for Proliferation, Migration and Invasion of Breast Cancer Cell Lines by Modulation of Orai1 and Orai3 Surface Exposure. Cancers 2018, 10, 331. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Chen, M.; Huang, J.; Zhang, F.; Lv, Z.; Jia, Y.; Cui, Y.Z.; Sun, L.Z.; Wang, Y.; Tang, Y.; et al. ORAI2 Promotes Gastric Cancer Tumorigenicity and Metastasis through PI3K/Akt Signaling and MAPK-Dependent Focal Adhesion Disassembly. Cancer Res. 2021, 81, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Collado, J.; Lopez, J.J.; Cantonero, C.; Jardin, I.; Regodon, S.; Redondo, P.C.; Gordillo, J.; Smani, T.; Salido, G.M.; Rosado, J.A. Orai2 Modulates Store-Operated Ca(2+) Entry and Cell Cycle Progression in Breast Cancer Cells. Cancers 2021, 14, 114. [Google Scholar] [CrossRef] [PubMed]
- Diez-Bello, R.; Jardin, I.; Salido, G.M.; Rosado, J.A. Orai1 and Orai2 mediate store-operated calcium entry that regulates HL60 cell migration and FAK phosphorylation. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1064–1070. [Google Scholar] [CrossRef]
- Hasna, J.; Hague, F.; Rodat-Despoix, L.; Geerts, D.; Leroy, C.; Tulasne, D.; Ouadid-Ahidouch, H.; Kischel, P. Orai3 calcium channel and resistance to chemotherapy in breast cancer cells: The p53 connection. Cell Death Differ. 2018, 25, 693–707. [Google Scholar] [CrossRef]
- Benzerdjeb, N.; Sevestre, H.; Ahidouch, A.; Ouadid-Ahidouch, H. Orai3 is a predictive marker of metastasis and survival in resectable lung adenocarcinoma. Oncotarget 2016, 7, 81588–81597. [Google Scholar] [CrossRef]
- Arora, S.; Tanwar, J.; Sharma, N.; Saurav, S.; Motiani, R.K. Orai3 Regulates Pancreatic Cancer Metastasis by Encoding a Functional Store Operated Calcium Entry Channel. Cancers 2021, 13, 5937. [Google Scholar] [CrossRef]
- Dubois, C.; Vanden Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef]
- Tanwar, J.; Arora, S.; Motiani, R.K. Orai3: Oncochannel with therapeutic potential. Cell Calcium 2020, 90, 102247. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, S.B.; Monteith, G.R. ORAI channels and cancer. Cell Calcium 2018, 74, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Store-Operated Ca2+ Entry in Breast Cancer Cells: Remodeling and Functional Role. Int. J. Mol. Sci. 2018, 19, 4053. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Collado, J.; Jardin, I.; Lopez, J.J.; Ronco, V.; Salido, G.M.; Dubois, C.; Prevarskaya, N.; Rosado, J.A. Role of Orai3 in the Pathophysiology of Cancer. Int. J. Mol. Sci. 2021, 22, 11426. [Google Scholar] [CrossRef]
- Vashisht, A.; Trebak, M.; Motiani, R.K. STIM and Orai proteins as novel targets for cancer therapy. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C457–C469. [Google Scholar] [CrossRef]
- Goswamee, P.; Pounardjian, T.; Giovannucci, D.R. Arachidonic acid-induced Ca2+ entry and migration in a neuroendocrine cancer cell line. Cancer Cell Int. 2018, 18, 30. [Google Scholar] [CrossRef]
- Fiorio Pla, A.; Grange, C.; Antoniotti, S.; Tomatis, C.; Merlino, A.; Bussolati, B.; Munaron, L. Arachidonic acid-induced Ca2+ entry is involved in early steps of tumor angiogenesis. Mol. Cancer Res. 2008, 6, 535–545. [Google Scholar] [CrossRef]
- Baron, S.; Vangheluwe, P.; Sepulveda, M.R.; Wuytack, F.; Raeymaekers, L.; Vanoevelen, J. The secretory pathway Ca2+-ATPase 1 is associated with cholesterol-rich microdomains of human colon adenocarcinoma cells. Biochim. Biophys. Acta 2010, 1798, 1512–1521. [Google Scholar] [CrossRef]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef]
- Feng, M.Y.; Rao, R. New insights into store-independent Ca2+ entry: Secretory pathway calcium ATPase 2 in normal physiology and cancer. Int. J. Oral. Sci. 2013, 5, 71–74. [Google Scholar] [CrossRef]
- Chantome, A.; Potier-Cartereau, M.; Clarysse, L.; Fromont, G.; Marionneau-Lambot, S.; Gueguinou, M.; Pages, J.C.; Collin, C.; Oullier, T.; Girault, A.; et al. Pivotal role of the lipid Raft SK3-Orai1 complex in human cancer cell migration and bone metastases. Cancer Res. 2013, 73, 4852–4861. [Google Scholar] [CrossRef] [PubMed]
- Gueguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantome, A.; Haelters, J.P.; Jaffres, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: A novel opportunity to modulate anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget 2016, 7, 36168–36184. [Google Scholar] [CrossRef] [PubMed]
- Badaoui, M.; Mimsy-Julienne, C.; Saby, C.; Van Gulick, L.; Peretti, M.; Jeannesson, P.; Morjani, H.; Ouadid-Ahidouch, H. Collagen type 1 promotes survival of human breast cancer cells by overexpressing Kv10.1 potassium and Orai1 calcium channels through DDR1-dependent pathway. Oncotarget 2018, 9, 24653–24671. [Google Scholar] [CrossRef] [PubMed]
- Hammadi, M.; Chopin, V.; Matifat, F.; Dhennin-Duthille, I.; Chasseraud, M.; Sevestre, H.; Ouadid-Ahidouch, H. Human ether a-gogo K(+) channel 1 (hEag1) regulates MDA-MB-231 breast cancer cell migration through Orai1-dependent calcium entry. J. Cell Physiol. 2012, 227, 3837–3846. [Google Scholar] [CrossRef]
- Tiffner, A.; Hopl, V.; Schober, R.; Sallinger, M.; Grabmayr, H.; Hoglinger, C.; Fahrner, M.; Lunz, V.; Maltan, L.; Frischauf, I.; et al. Orai1 Boosts SK3 Channel Activation. Cancers 2021, 13, 6357. [Google Scholar] [CrossRef]
- Peretti, M.; Badaoui, M.; Girault, A.; Van Gulick, L.; Mabille, M.P.; Tebbakha, R.; Sevestre, H.; Morjani, H.; Ouadid-Ahidouch, H. Original association of ion transporters mediates the ECM-induced breast cancer cell survival: Kv10.1-Orai1-SPCA2 partnership. Sci. Rep. 2019, 9, 1175. [Google Scholar] [CrossRef]
- Daya, H.A.; Kouba, S.; Ouled-Haddou, H.; Benzerdjeb, N.; Telliez, M.S.; Dayen, C.; Sevestre, H.; Garcon, L.; Hague, F.; Ouadid-Ahidouch, H. Orai3-Mediates Cisplatin-Resistance in Non-Small Cell Lung Cancer Cells by Enriching Cancer Stem Cell Population through PI3K/AKT Pathway. Cancers 2021, 13, 2314. [Google Scholar] [CrossRef]
- Terrie, E.; Deliot, N.; Benzidane, Y.; Harnois, T.; Cousin, L.; Bois, P.; Oliver, L.; Arnault, P.; Vallette, F.; Constantin, B.; et al. Store-Operated Calcium Channels Control Proliferation and Self-Renewal of Cancer Stem Cells from Glioblastoma. Cancers 2021, 13, 3428. [Google Scholar] [CrossRef]
- Schmidt, S.; Liu, G.; Liu, G.; Yang, W.; Honisch, S.; Pantelakos, S.; Stournaras, C.; Honig, A.; Lang, F. Enhanced Orai1 and STIM1 expression as well as store operated Ca2+ entry in therapy resistant ovary carcinoma cells. Oncotarget 2014, 5, 4799–4810. [Google Scholar] [CrossRef]
- Flourakis, M.; Lehen’kyi, V.; Beck, B.; Raphael, M.; Vandenberghe, M.; Abeele, F.V.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010, 1, e75. [Google Scholar] [CrossRef]
- Faouzi, M.; Kischel, P.; Hague, F.; Ahidouch, A.; Benzerdjeb, N.; Sevestre, H.; Penner, R.; Ouadid-Ahidouch, H. ORAI3 silencing alters cell proliferation and cell cycle progression via c-myc pathway in breast cancer cells. Biochim. Biophys. Acta 2013, 1833, 752–760. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Ca2+ Pumps and Exchangers | |||||
|---|---|---|---|---|---|
| Protein | Expression/Functional Change in CSC | CSC Type | Role in CSC | Signaling Pathway Activated | Ref. |
| SERCA | Overexpression | Breast cancer stem cells | Cell survival in glucose-deprived conditions | Decrease [Ca2+]c and avoid Ca2+-dependent apoptosis during glucose deprivation | [73] |
| ER Ca2+ channels | |||||
| Protein | Expression/Functional Change in CSC | CSC Type | Role in CSC | Signaling Pathway Activated | Ref. |
| RyR1 | HIF-depended activation | Breast cancer stem cells | Chemoresistance | PYK2/SRC/STAT3 signaling pathway | [50] |
| IP3R | Channel activation | Melanoma stem cells | Stemness maintenance | ND | [49] |
| Channel activation | Glioblastoma stem cell | Cell self-renewal Chemoresistance | ND | [48] | |
| Non-SOCE channels | |||||
| Protein | Expression/Functional Change in CSC | CSC Type | Role in CSC | Signaling Pathway Activated | Ref. |
| VGCC | |||||
| L- and T-type | Overexpression | Ovarian cancer stem cells | Tumor spheres formation Apoptosis resistance Stemness maintenance | Increase the transcription of Oct, Nanog, and Sox2 via ERK1/2 and AKT signaling pathways | [53] |
| T-type calcium channel | Overexpression (Cav3.2) | Glioblastoma stem cells | Apoptosis resistance Chemoresistance Stemness maintenance | Increase cell survival via AKT/mTOR pathways | [52] |
| Overexpression | Glioblastoma stem cells | Apoptosis resistance | Stimulate Na+-dependent nutrient transport | [54] | |
| α2δ1 subunit | Overexpression | Small cell lung cancer stem cells | Chemoresistance | MEK/ERK signal pathway?? | [55] |
| α2δ1 subunit | Overexpression | Non-small cell lung cancer stem cells | Chemoresistance Cell Survival Stemness maintenance | Notch3 activation via Ca2+-Calcineurin/NFATc2 signaling pathway | [56] |
| Overexpression | Breast cancer stem cells | Stemness maintenance Cell self-renewal | ND | [57] | |
| Overexpression | Hepatocellular cancer stem cells | Cell self-renewal Cell survival Stemness maintenance | ERK1/2 MAPK signaling pathway | [58] | |
| Overexpression | Laryngeal squamous cancer stem cells | Tumor spheres formation Chemoresistance Tumorigenesis Stemness maintenance | ND | [59] | |
| Protein | Expression/Functional Change in CSC | CSC Type | Role in CSC | Signaling Pathway Activated | Ref. |
| TRP Channels | |||||
| TRPC3 | Overexpression | Breast cancer stem cells | Cell self-renewal | Increase IL-8 secretion via LPA/LPAR3/TRPC3 pathway | [60] |
| TRPM7 | Overexpression | Lung cancer stem cells | Tumor spheres formation Stemness maintenance | Hsp90α/uPA/MMP2 signaling pathway | [61] |
| Channel activation | Glioblastoma stem cells | Stemness maintenance Cell proliferation, migration and invasion | STAT3 and Notch signaling pathways | [62] | |
| Overexpression | Neuroblastoma stem cells | Stemness maintenance | ND | [63] | |
| TRPA1 | Channel activation Overexpression | Glioma stem cells | Cell differentiation Apoptotic cell death Stemness loss | ND | [69] |
| TRPV1 | Channel activation Overexpression | Glioma stem cells | Cell differentiation Apoptotic cell death Stemness loss | ND | [69] |
| TRPV2 | Overexpression | Esophageal squamous cancer stem cells | Stemness maintenance Cell proliferation | ND | [65] |
| Channel activation Overexpression | Glioblastoma stem cells | Stem cell differentiation Reduce self-renewal capacity Apoptotic cell death | AKT-PI3K/RPS6KBI/PTEN signaling pathway | [64,66,67] | |
| Channel activation Overexpression | Liver cancer stem cells | Impair tumor spheres formation and self-renewal capacity Stemness loss | ND | [68] | |
| SOCE channels | |||||
| Protein | Expression/Functional Change in CSC | CSC Type | Role in CSC | Signaling Pathway Activated | Ref. |
| Orai1 | Overexpression | oral/oropharyngeal squamous cancer stem cells | Tumor spheres formation Cell self-renewal Stemness maintenance | NFAT signaling pathway | [119] |
| Overexpression | Glioblastoma stem cell | Tumor spheres formation Cell self-renewal Stemness maintenance | ND | [148] | |
| Orai3 | Overexpression | Non-small cell lung cancer stem cells | Chemoresistance Stemness maintenance | PI3K/AKT signaling pathway | [147] |
| SOC Channels | Channel activation | Glioblastoma stem cell | Cell proliferation | Up-regulation of CDKN1A and G0S2 and the down-regulation of CCNB1 genes | [116] |
| Channel activation | Liver cancer stem cells | Stemness maintenance Tumor spheres formation Cell self-renewal | FGF19/SOCE/NFATc2 signaling pathway | [117] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jardin, I.; Lopez, J.J.; Sanchez-Collado, J.; Gomez, L.J.; Salido, G.M.; Rosado, J.A. Store-Operated Calcium Entry and Its Implications in Cancer Stem Cells. Cells 2022, 11, 1332. https://doi.org/10.3390/cells11081332
Jardin I, Lopez JJ, Sanchez-Collado J, Gomez LJ, Salido GM, Rosado JA. Store-Operated Calcium Entry and Its Implications in Cancer Stem Cells. Cells. 2022; 11(8):1332. https://doi.org/10.3390/cells11081332
Chicago/Turabian StyleJardin, Isaac, Jose J. Lopez, Jose Sanchez-Collado, Luis J. Gomez, Gines M. Salido, and Juan A. Rosado. 2022. "Store-Operated Calcium Entry and Its Implications in Cancer Stem Cells" Cells 11, no. 8: 1332. https://doi.org/10.3390/cells11081332
APA StyleJardin, I., Lopez, J. J., Sanchez-Collado, J., Gomez, L. J., Salido, G. M., & Rosado, J. A. (2022). Store-Operated Calcium Entry and Its Implications in Cancer Stem Cells. Cells, 11(8), 1332. https://doi.org/10.3390/cells11081332