
A Novel Attention-Mechanism Based Cox Survival Model by Exploiting Pan-Cancer Empirical Genomic Information

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset Preparation
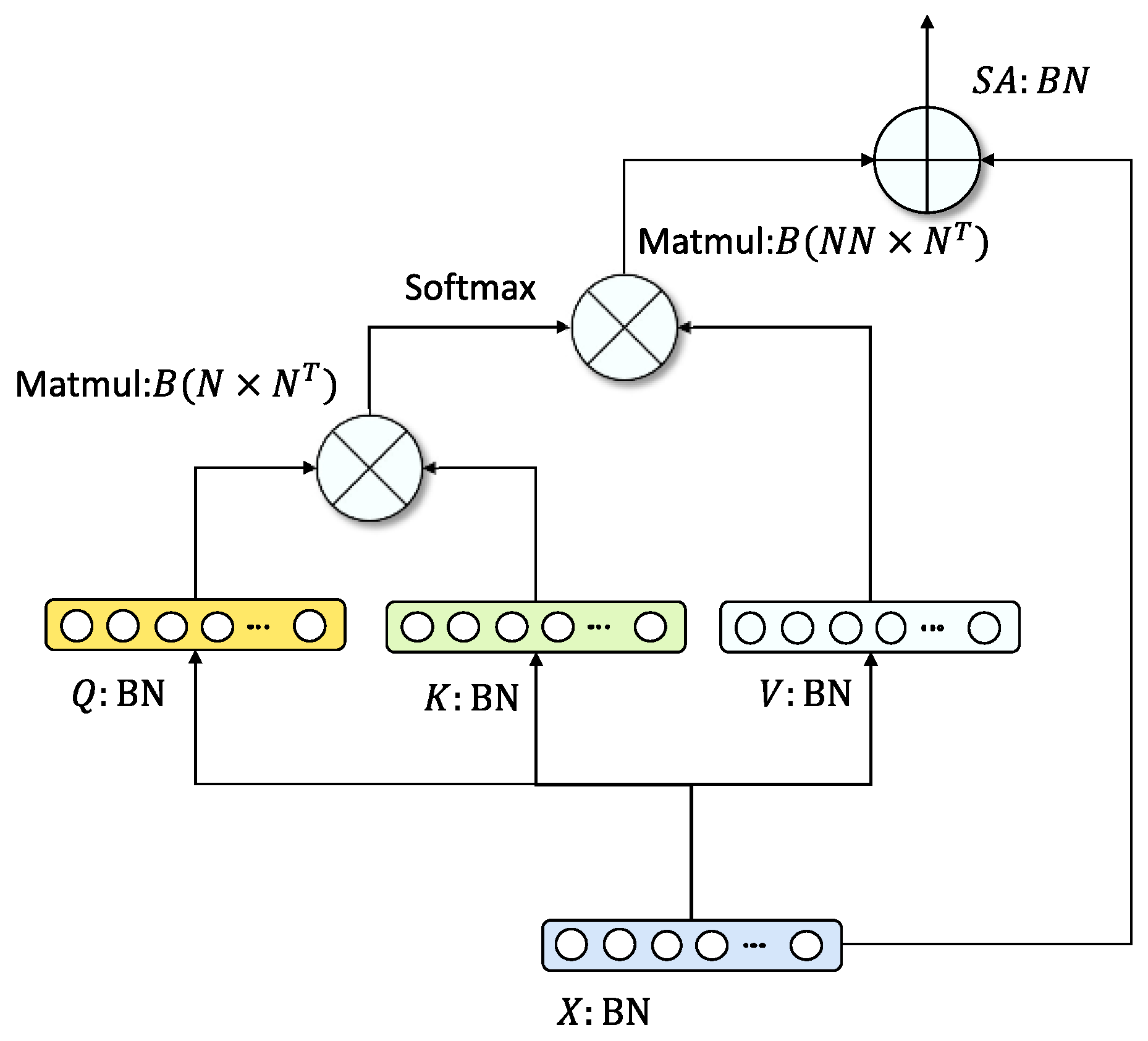
2.2. Dimensionality Reduction Pretraining Using GAN
2.3. Survival Analysis Based on Transfer Learning
2.4. Experiment Settings
2.5. Evaluation Metric
3. Results
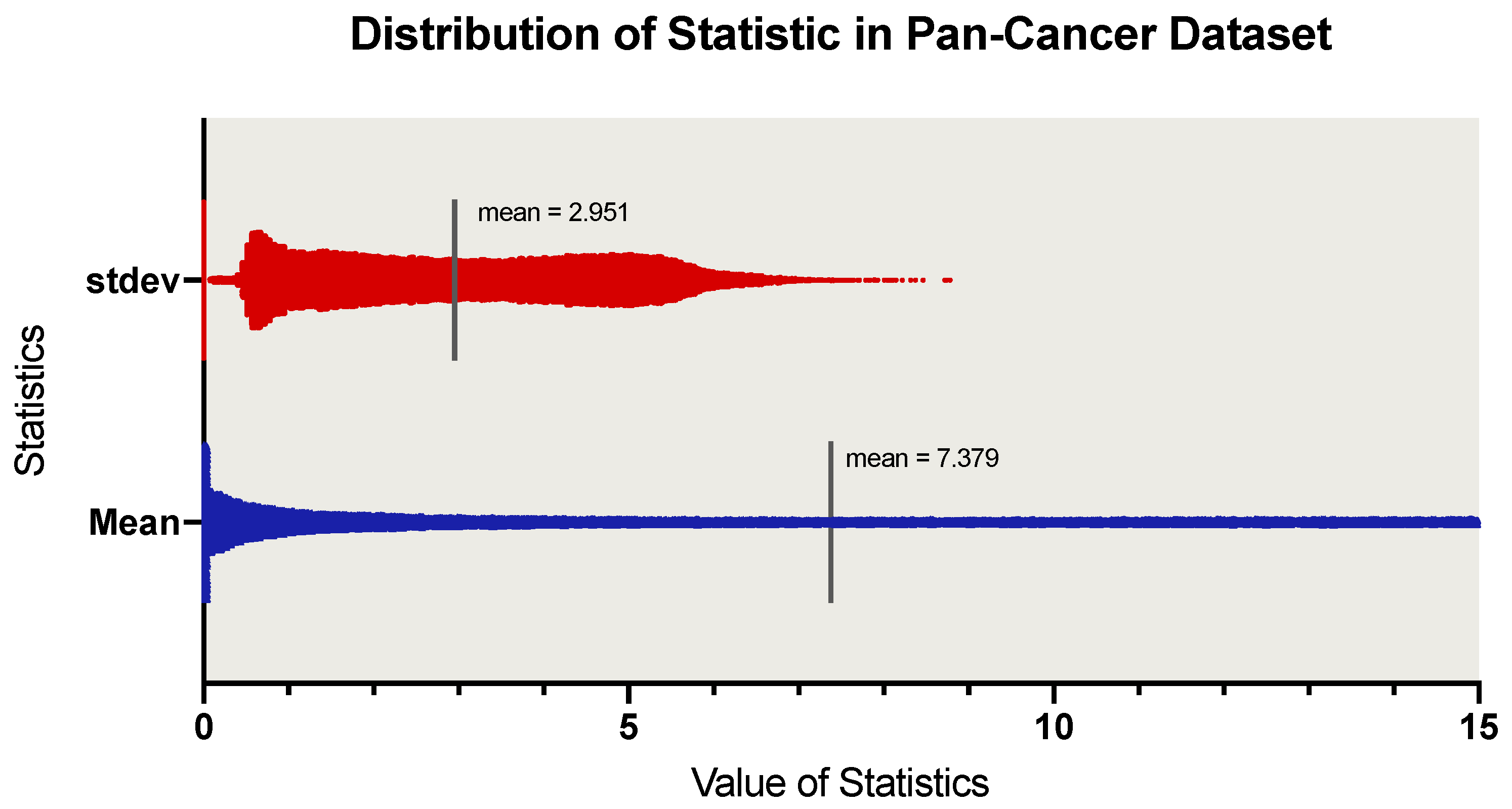
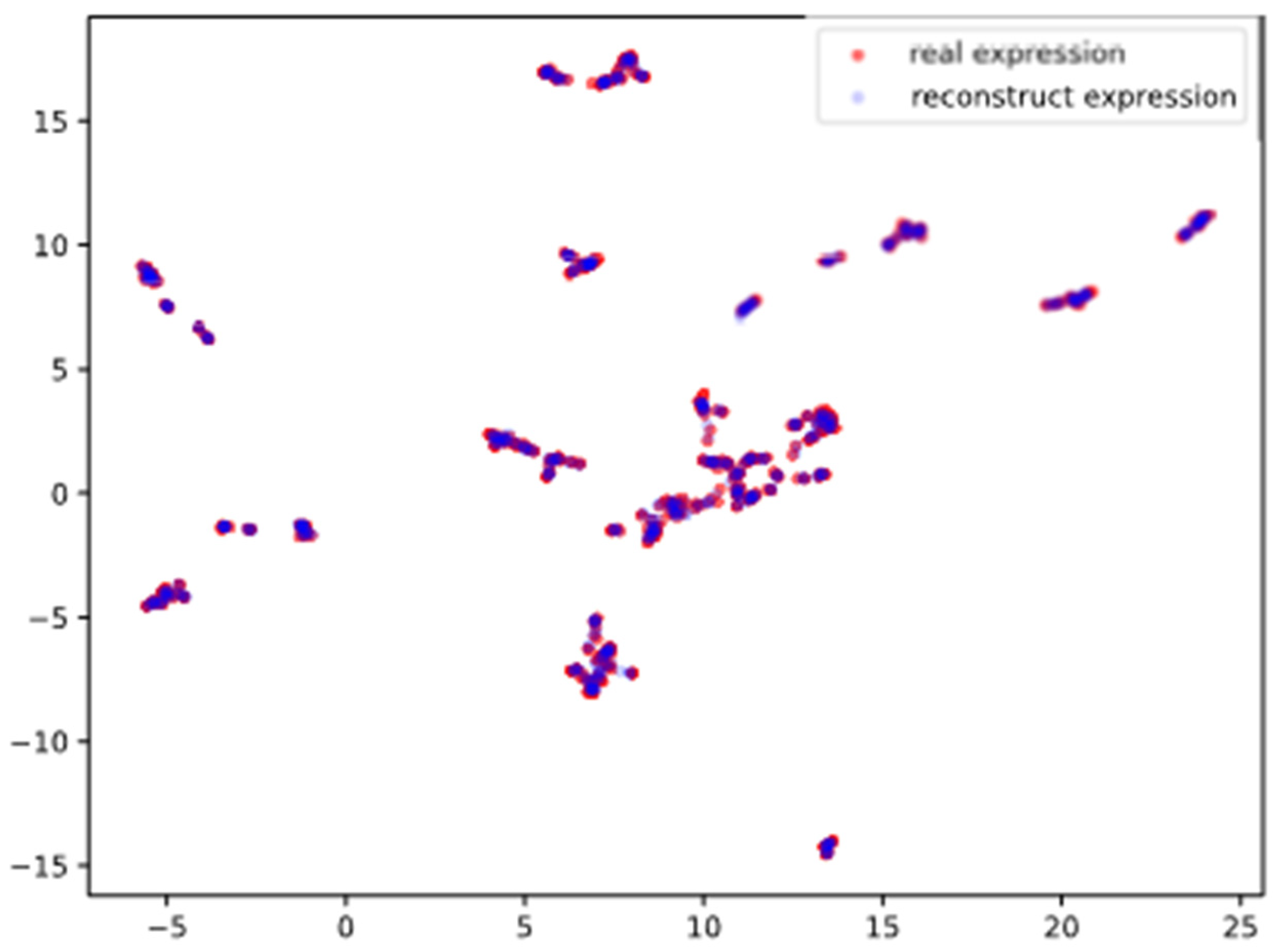
3.1. Performance of Dimensionality Reduction
- Generator can reconstruct that are consistent with ;
- The reconstruction of the generator is based on the latent encoding , indicating that the encoder of the generator can effectively generate , which retains rich features that can represent .
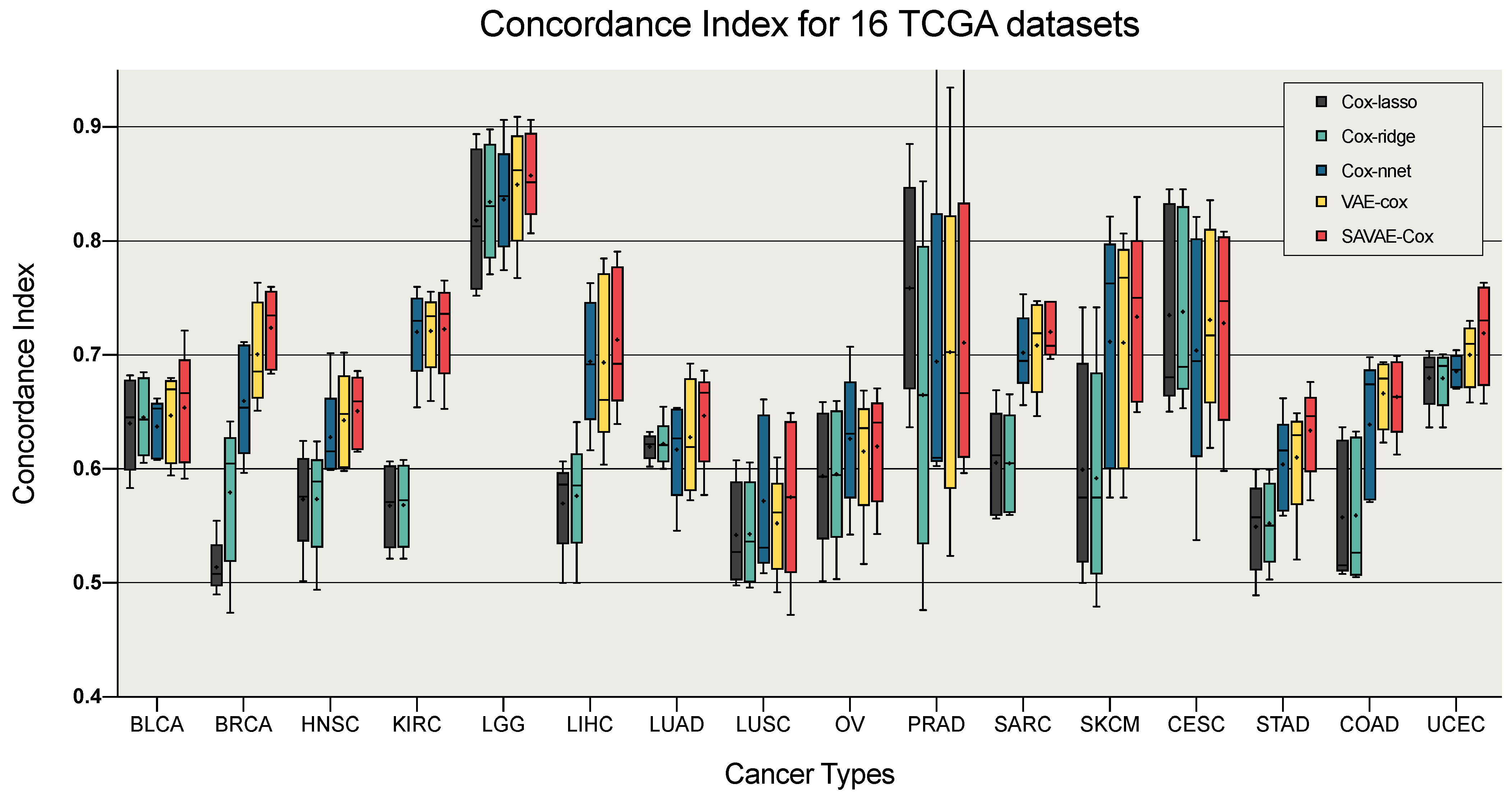
3.2. Performance of Survival Analysis
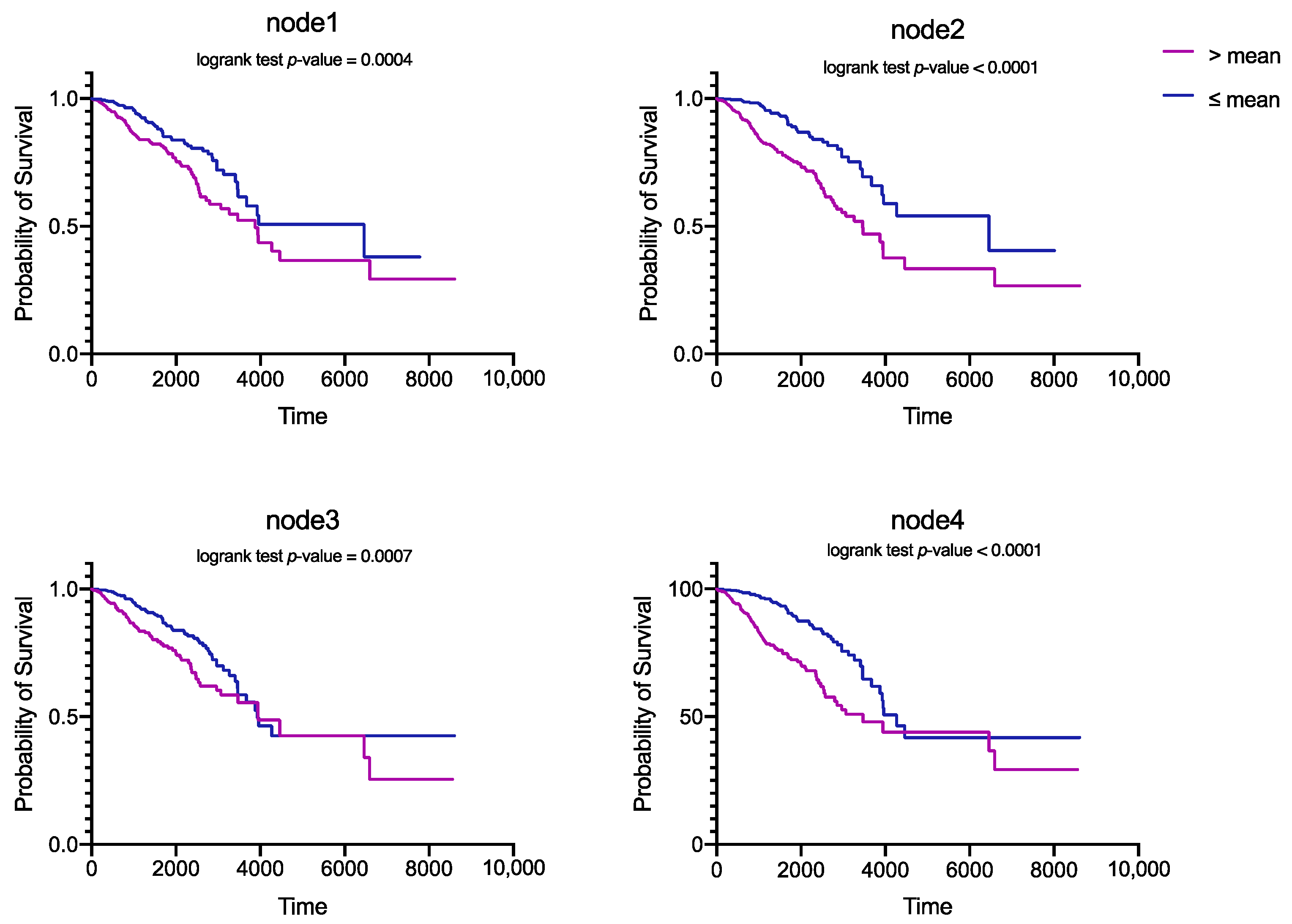
3.3. Feature Analysis of SAVAE-Cox
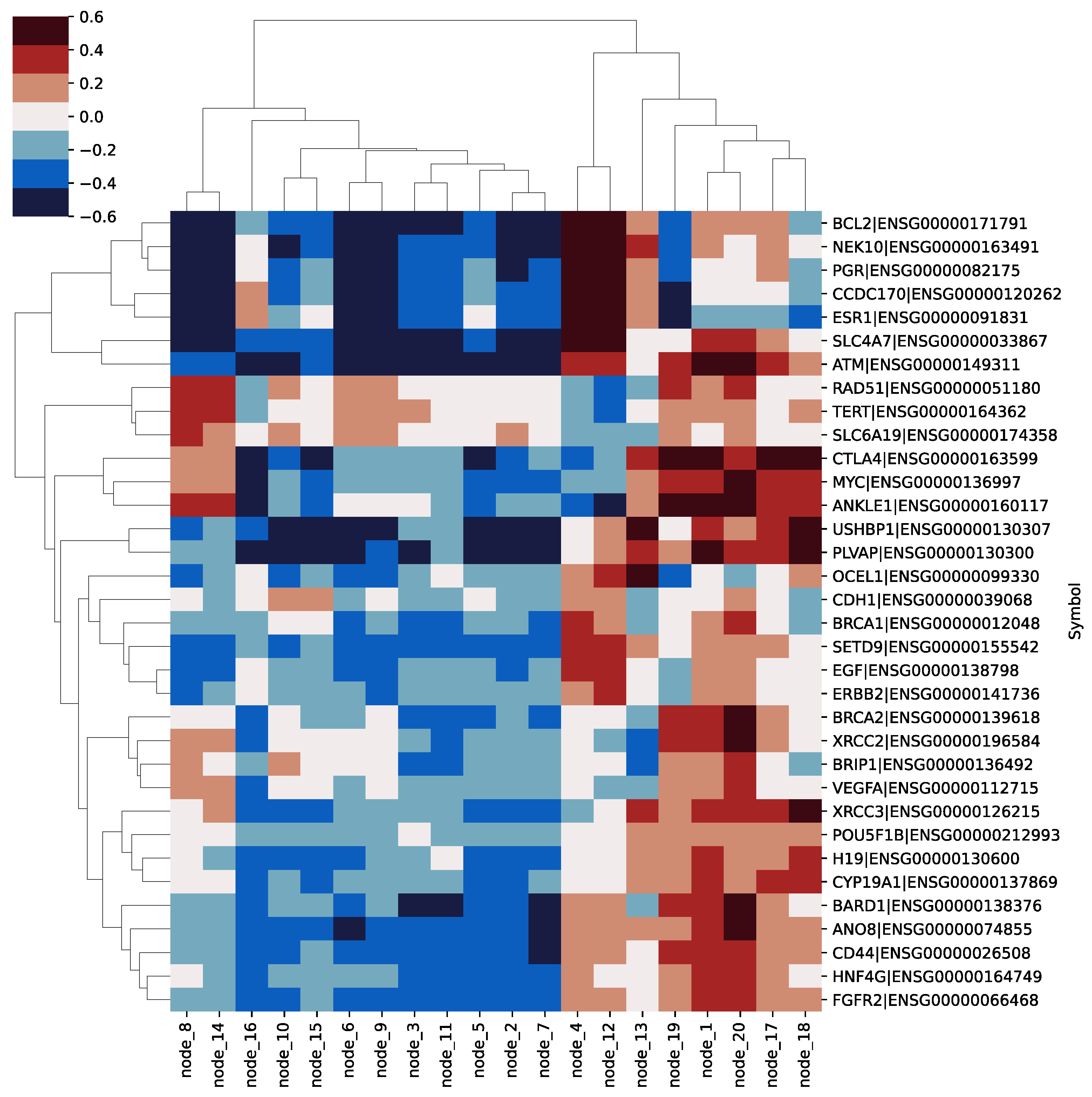
3.4. Biological Function Analysis of Hidden Nodes
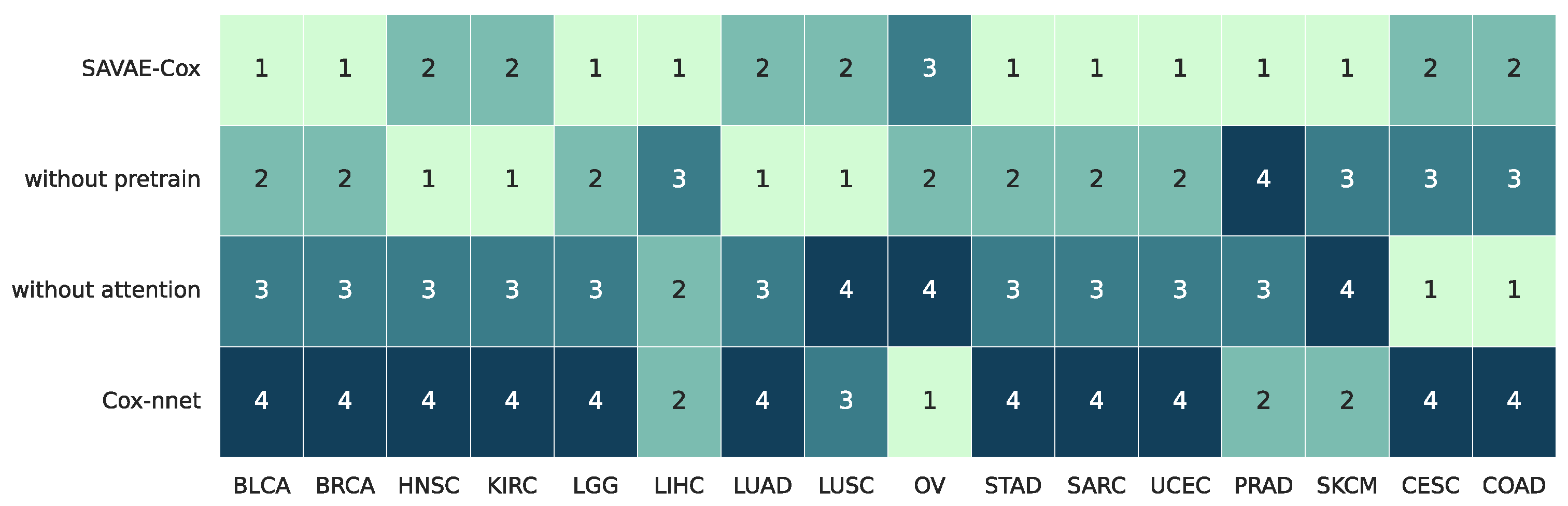
3.5. Ablation Study for SAVAE-Cox
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Descriptions and Download Details of the Datasets
Appendix A.2. Hyperparameter Selection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Survival Analysis Models | Learning Rate | Epoch | Batch Size | |
|---|---|---|---|---|
| dimensionality reduction | Cox-Chi2 | 0.0005 | 15 | 1024 |
| Cox-Pearson | 0.001 | 15 | 1024 | |
| Cox-MIC | 0.0005 | 15 | 1024 | |
| Cox-PCA | 0.0005 | 15 | 1024 | |
| Cox-AE | 0.001 | 15 | 1024 | |
| Cox-dnoiseAE | 0.0005 | 15 | 1024 | |
| Comparative Experiment | Cox-lasso | 0.0005 | 15 | 1024 |
| Cox-ridge | 0.001 | 15 | 1024 | |
| Cox-nnet | 0.001 | 15 | 1024 | |
| VAECox | 0.001 | 15 | 1024 | |
| Ablation Study | Without pretrain | 0.001 | 20 | 512 |
| Without attention | 0.001 | 20 | 512 | |
| Ours | SAVAE-Cox | 0.001 | 20 | 512 |
References
- Nicholson, R.I.; Gee, J.M.W.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, 9–15. [Google Scholar]
- Cox, D.R. Regression models and life-tables. J. R. Stat. Soc. Ser. B 1972, 34, 187–202. [Google Scholar]
- Broder, S.; Subramanian, G.; Venter, J.C. The human genome. Pharm. Search Individ. Ther. 2002, 9–34. [Google Scholar]
- Lussier, Y.A.; Li, H. Breakthroughs in genomics data integration for predicting clinical outcome. J. Biomed. Inform. 2012, 45, 1199. [Google Scholar]
- Valdes-Mora, F.; Handler, K.; Law, A.M.; Salomon, R.; Oakes, S.R.; Ormandy, C.J.; Gallego-Ortega, D. Single-cell transcriptomics in cancer immunobiology: The future of precision oncology. Front. Immunol. 2018, 9, 2582. [Google Scholar]
- Nagy, Á.; Munkácsy, G.; Győrffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 2021, 11, 6047. [Google Scholar]
- Ding, Z. The application of support vector machine in survival analysis. In Proceedings of the 2011 2nd International Conference on Artificial Intelligence, Management Science and Electronic Commerce (AIMSEC), Zhengzhou, China, 8–10 August 2011; pp. 6816–6819. [Google Scholar]
- Evers, L.; Messow, C.-M. Sparse kernel methods for high-dimensional survival data. Bioinformatics 2008, 24, 1632–1638. [Google Scholar]
- Bin, R.D. Boosting in Cox regression: A comparison between the likelihood-based and the model-based approaches with focus on the R-packages CoxBoost and mboost. Comput. Stat. 2016, 31, 513–531. [Google Scholar] [CrossRef]
- Ishwaran, H.; Kogalur, U.B.; Blackstone, E.H.; Lauer, M.S. Random survival forests. Ann. Appl. Stat. 2008, 2, 841–860. [Google Scholar]
- Meng, X.; Zhang, X.; Wang, G.; Zhang, Y.; Shi, X.; Dai, H.; Wang, Z.; Wang, X. Exploiting full Resolution Feature Context for Liver Tumor and Vessel Segmentation via Fusion Encoder: Application to Liver Tumor and Vessel 3D reconstruction. arXiv 2021, arXiv:2111.13299. [Google Scholar]
- Song, T.; Zhang, X.; Ding, M.; Rodriguez-Paton, A.; Wang, S.; Wang, G. DeepFusion: A deep learning based multi-scale feature fusion method for predicting drug-target interactions. Methods 2022, in press. [Google Scholar] [CrossRef]
- Faraggi, D.; Simon, R. A neural network model for survival data. Stat. Med. 1995, 14, 73–82. [Google Scholar]
- Ching, T.; Zhu, X.; Garmire, L.X. Cox-nnet: An artificial neural network method for prognosis prediction of high-throughput omics data. PLoS Comput. Biol. 2018, 14, e1006076. [Google Scholar]
- Katzman, J.L.; Shaham, U.; Cloninger, A.; Bates, J.; Jiang, T.; Kluger, Y. DeepSurv: Personalized treatment recommender system using a Cox proportional hazards deep neural network. BMC Med. Res. Methodol. 2018, 18, 24. [Google Scholar]
- Huang, Z.; Zhan, X.; Xiang, S.; Johnson, T.S.; Helm, B.; Yu, C.Y.; Zhang, J.; Salama, P.; Rizkalla, M.; Han, Z. SALMON: Survival analysis learning with multi-omics neural networks on breast cancer. Front. Genet. 2019, 10, 166. [Google Scholar]
- Kim, S.; Kim, K.; Choe, J.; Lee, I.; Kang, J. Improved survival analysis by learning shared genomic information from pan-cancer data. Bioinformatics 2020, 36, i389–i398. [Google Scholar]
- Ramirez, R.; Chiu, Y.-C.; Zhang, S.; Ramirez, J.; Chen, Y.; Huang, Y.; Jin, Y.-F. Prediction and interpretation of cancer survival using graph convolution neural networks. Methods 2021, 192, 120–130. [Google Scholar]
- Huang, Z.; Johnson, T.S.; Han, Z.; Helm, B.; Cao, S.; Zhang, C.; Salama, P.; Rizkalla, M.; Yu, C.Y.; Cheng, J. Deep learning-based cancer survival prognosis from RNA-seq data: Approaches and evaluations. BMC Med. Genom. 2020, 13, 41. [Google Scholar]
- Rehman, M.U.; Tayara, H.; Chong, K.T. DCNN-4mC: Densely connected neural network based N4-methylcytosine site prediction in multiple species. Comput. Struct. Biotechnol. J. 2021, 19, 6009–6019. [Google Scholar]
- Chen, J.; Wang, W.H.; Shi, X. Differential privacy protection against membership inference attack on machine learning for genomic data. In Proceedings of the BIOCOMPUTING 2021: Proceedings of the Pacific Symposium, Kohala Coast, HI, USA, 3–7 January 2021; pp. 26–37. [Google Scholar]
- Torada, L.; Lorenzon, L.; Beddis, A.; Isildak, U.; Pattini, L.; Mathieson, S.; Fumagalli, M. ImaGene: A convolutional neural network to quantify natural selection from genomic data. BMC Bioinform. 2019, 20, 337. [Google Scholar]
- Hao, J.; Kosaraju, S.C.; Tsaku, N.Z.; Song, D.H.; Kang, M. PAGE-Net: Interpretable and integrative deep learning for survival analysis using histopathological images and genomic data. In Proceedings of the Pacific Symposium on Biocomputing, Kohala Coast, HI, USA, 3–7 January 2020; pp. 355–366. [Google Scholar]
- Jeong, S.; Kim, J.-Y.; Kim, N. GMStool: GWAS-based marker selection tool for genomic prediction from genomic data. Sci. Rep. 2020, 10, 19653. [Google Scholar] [PubMed]
- Rehman, M.U.; Hong, K.J.; Tayara, H.; Chong, K. m6A-NeuralTool: Convolution neural tool for RNA N6-Methyladenosine site identification in different species. IEEE Access 2021, 9, 17779–17786. [Google Scholar]
- Ramirez, R.; Chiu, Y.-C.; Hererra, A.; Mostavi, M.; Ramirez, J.; Chen, Y.; Huang, Y.; Jin, Y.-F. Classification of cancer types using graph convolutional neural networks. Front. Phys. 2020, 8, 203. [Google Scholar] [PubMed]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial nets. Adv. Neural Inf. Processing Syst. 2014, 27. Available online: https://proceedings.neurips.cc/paper/2014/file/5ca3e9b122f61f8f06494c97b1afccf3-Paper.pdf (accessed on 15 March 2022).
- Repecka, D.; Jauniskis, V.; Karpus, L.; Rembeza, E.; Rokaitis, I.; Zrimec, J.; Poviloniene, S.; Laurynenas, A.; Viknander, S.; Abuajwa, W. Expanding functional protein sequence spaces using generative adversarial networks. Nat. Mach. Intell. 2021, 3, 324–333. [Google Scholar]
- Lin, E.; Mukherjee, S.; Kannan, S. A deep adversarial variational autoencoder model for dimensionality reduction in single-cell RNA sequencing analysis. BMC Bioinform. 2020, 21, 64. [Google Scholar]
- Jiang, X.; Zhao, J.; Qian, W.; Song, W.; Lin, G.N. A generative adversarial network model for disease gene prediction with RNA-seq data. IEEE Access 2020, 8, 37352–37360. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. Adv. Neural Inf. Processing Syst. 2017, 30, 5998–6008. [Google Scholar]
- Kingma, D.P.; Welling, M. Auto-encoding variational bayes. arXiv 2013, arXiv:1312.6114, preprint. [Google Scholar]
- Gulrajani, I.; Ahmed, F.; Arjovsky, M.; Dumoulin, V.; Courville, A.C. Improved training of wasserstein gans. Adv. Neural Inf. Processing Syst. 2017, 30. Available online: https://www.semanticscholar.org/paper/Improved-Training-of-Wasserstein-GANs-Gulrajani-Ahmed/edf73ab12595c6709f646f542a0d2b33eb20a3f4 (accessed on 15 March 2022).
- Raykar, V.C.; Steck, H.; Krishnapuram, B.; Dehing-Oberije, C.; Lambin, P. On ranking in survival analysis: Bounds on the concordance index. In Proceedings of the Proceedings of the 20th International Conference on Neural Information Processing Systems, Vancouver, BC, Canada, 3–6 December 2007; pp. 1209–1216. [Google Scholar]
- Callagy, G.M.; Webber, M.J.; Pharoah, P.D.; Caldas, C. Meta-analysis confirms BCL2 is an independent prognostic marker in breast cancer. BMC Cancer 2008, 8, 153. [Google Scholar]
- Bryan, M.S.; Argos, M.; Andrulis, I.L.; Hopper, J.L.; Chang-Claude, J.; Malone, K.E.; John, E.M.; Gammon, M.D.; Daly, M.B.; Terry, M.B. Germline variation and breast cancer incidence: A gene-based association study and whole-genome prediction of early-onset breast cancer. Cancer Epidemiol. Prev. Biomark. 2018, 27, 1057–1064. [Google Scholar]
- Kunc, M.; Biernat, W.; Senkus-Konefka, E. Estrogen receptor-negative progesterone receptor-positive breast cancer–“Nobody’s land “or just an artifact? Cancer Treat. Rev. 2018, 67, 78–87. [Google Scholar]
- Jiang, P.; Li, Y.; Poleshko, A.; Medvedeva, V.; Baulina, N.; Zhang, Y.; Zhou, Y.; Slater, C.M.; Pellegrin, T.; Wasserman, J. The protein encoded by the CCDC170 breast cancer gene functions to organize the golgi-microtubule network. EBioMedicine 2017, 22, 28–43. [Google Scholar]
- Holst, F.; Stahl, P.R.; Ruiz, C.; Hellwinkel, O.; Jehan, Z.; Wendland, M.; Lebeau, A.; Terracciano, L.; Al-Kuraya, K.; Jänicke, F. Estrogen receptor alpha (ESR1) gene amplification is frequent in breast cancer. Nat. Genet. 2007, 39, 655–660. [Google Scholar]
- Chen, W.; Zhong, R.; Ming, J.; Zou, L.; Zhu, B.; Lu, X.; Ke, J.; Zhang, Y.; Liu, L.; Miao, X. The SLC4A7 variant rs4973768 is associated with breast cancer risk: Evidence from a case–control study and a meta-analysis. Breast Cancer Res. Treat. 2012, 136, 847–857. [Google Scholar]
- Ahmed, M.; Rahman, N. ATM and breast cancer susceptibility. Oncogene 2006, 25, 5906–5911. [Google Scholar]
- Wiegmans, A.P.; Al-Ejeh, F.; Chee, N.; Yap, P.-Y.; Gorski, J.J.; Da Silva, L.; Bolderson, E.; Chenevix-Trench, G.; Anderson, R.; Simpson, P.T. Rad51 supports triple negative breast cancer metastasis. Oncotarget 2014, 5, 3261. [Google Scholar]
- Chen, X.; Shao, Q.; Hao, S.; Zhao, Z.; Wang, Y.; Guo, X.; He, Y.; Gao, W.; Mao, H. CTLA-4 positive breast cancer cells suppress dendritic cells maturation and function. Oncotarget 2017, 8, 13703. [Google Scholar]
- Xu, J.; Chen, Y.; Olopade, O.I. MYC and breast cancer. Genes Cancer 2010, 1, 629–640. [Google Scholar]
- Corso, G.; Intra, M.; Trentin, C.; Veronesi, P.; Galimberti, V. CDH1 germline mutations and hereditary lobular breast cancer. Fam. Cancer 2016, 15, 215–219. [Google Scholar]
- Rosen, E.M.; Fan, S.; Pestell, R.G.; Goldberg, I.D. BRCA1 gene in breast cancer. J. Cell. Physiol. 2003, 196, 19–41. [Google Scholar]
- Chrysogelos, S.A.; Dickson, R.B. EGF receptor expression, regulation, and function in breast cancer. Breast Cancer Res. Treat. 1994, 29, 29–40. [Google Scholar]
- Revillion, F.; Bonneterre, J.; Peyrat, J. ERBB2 oncogene in human breast cancer and its clinical significance. Eur. J. Cancer 1998, 34, 791–808. [Google Scholar]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar]
- Park, D.; Lesueur, F.; Nguyen-Dumont, T.; Pertesi, M.; Odefrey, F.; Hammet, F.; Neuhausen, S.L.; John, E.M.; Andrulis, I.L.; Terry, M.B. Rare mutations in XRCC2 increase the risk of breast cancer. Am. J. Hum. Genet. 2012, 90, 734–739. [Google Scholar]
- Smith, T.R.; Miller, M.S.; Lohman, K.; Lange, E.M.; Case, L.D.; Mohrenweiser, H.W.; Hu, J.J. Polymorphisms of XRCC1 and XRCC3 genes and susceptibility to breast cancer. Cancer Lett. 2003, 190, 183–190. [Google Scholar]
- Lottin, S.; Adriaenssens, E.; Dupressoir, T.; Berteaux, N.; Montpellier, C.; Coll, J.; Dugimont, T.; Curgy, J.J. Overexpression of an ectopic H19 gene enhances the tumorigenic properties of breast cancer cells. Carcinogenesis 2002, 23, 1885–1895. [Google Scholar]
- Long, J.-R.; Kataoka, N.; Shu, X.-O.; Wen, W.; Gao, Y.-T.; Cai, Q.; Zheng, W. Genetic polymorphisms of the CYP19A1 gene and breast cancer survival. Cancer Epidemiol. Prev. Biomark. 2006, 15, 2115–2122. [Google Scholar]
- Ratajska, M.; Antoszewska, E.; Piskorz, A.; Brozek, I.; Borg, Å.; Kusmierek, H.; Biernat, W.; Limon, J. Cancer predisposing BARD1 mutations in breast–ovarian cancer families. Breast Cancer Res. Treat. 2012, 131, 89–97. [Google Scholar]
- Fletcher, M.N.; Castro, M.A.; Wang, X.; De Santiago, I.; O’Reilly, M.; Chin, S.-F.; Rueda, O.M.; Caldas, C.; Ponder, B.A.; Markowetz, F. Master regulators of FGFR2 signalling and breast cancer risk. Nat. Commun. 2013, 4, 2464. [Google Scholar] [PubMed]



| Cancer Type | Data Attribute | ||
|---|---|---|---|
| Total Samples | Censored Samples | Time Range | |
| PANCAN | 9895 | # | # |
| BLCA | 397 | 227 | 13–5050 |
| BRCA | 1031 | 896 | 1–8605 |
| HNSC | 489 | 302 | 1–6417 |
| KIRC | 504 | 347 | 2–4537 |
| LGG | 491 | 302 | 1–6423 |
| LIHC | 359 | 183 | 1–3765 |
| LUAD | 491 | 290 | 4–7248 |
| LUSC | 463 | 327 | 1–5287 |
| OV | 351 | 95 | 8–5481 |
| STAD | 345 | 227 | 1–3720 |
| CESC | 283 | 215 | 2–6408 |
| COAD | 415 | 239 | 1–4270 |
| SARC | 253 | 116 | 15–5723 |
| UCEC | 524 | 404 | 1–6859 |
| PRAD | 477 | 289 | 23–5024 |
| SKCM | 312 | 239 | 14–1785 |
| Cancer Type | Dimensionality Reduction Method | ||||||
|---|---|---|---|---|---|---|---|
| AE | Denoise-AE | Chi2 | Pearson | MIC | PCA | SAVAE | |
| BLCA | 0.642 | 0.643 | 0.582 | 0.552 | 0.624 | 0.545 | 0.654 |
| BRCA | 0.704 | 0.709 | 0.651 | 0.500 | 0.492 | 0.488 | 0.724 |
| HNSC | 0.649 | 0.642 | 0.522 | 0.590 | 0.531 | 0.489 | 0.651 |
| KIRC | 0.725 | 0.731 | 0.620 | 0.698 | 0.673 | 0.547 | 0.723 |
| LGG | 0.844 | 0.843 | 0.712 | 0.820 | 0.786 | 0.673 | 0.857 |
| LIHC | 0.704 | 0.696 | 0.467 | 0.627 | 0.401 | 0.423 | 0.713 |
| LUAD | 0.617 | 0.635 | 0.627 | 0.595 | 0.571 | 0.570 | 0.647 |
| LUSC | 0.552 | 0.559 | 0.605 | 0.534 | 0.529 | 0.496 | 0.575 |
| OV | 0.608 | 0.621 | 0.550 | 0.512 | 0.517 | 0.471 | 0.620 |
| STAD | 0.602 | 0.616 | 0.556 | 0.572 | 0.531 | 0.476 | 0.610 |
| CESC | 0.690 | 0.722 | 0.724 | 0.565 | 0.598 | 0.398 | 0.663 |
| COAD | 0.631 | 0.638 | 0.489 | 0.533 | 0.521 | 0.496 | 0.728 |
| SARC | 0.698 | 0.700 | 0.558 | 0.647 | 0.637 | 0.511 | 0.720 |
| UCEC | 0.677 | 0.701 | 0.640 | 0.591 | 0.611 | 0.472 | 0.698 |
| PRAD | 0.724 | 0.649 | 0.687 | 0.751 | 0.586 | 0.606 | 0.774 |
| SKCM | 0.684 | 0.655 | 0.863 | 0.652 | 0.531 | 0.512 | 0.734 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, X.; Wang, X.; Zhang, X.; Zhang, C.; Zhang, Z.; Zhang, K.; Wang, S. A Novel Attention-Mechanism Based Cox Survival Model by Exploiting Pan-Cancer Empirical Genomic Information. Cells 2022, 11, 1421. https://doi.org/10.3390/cells11091421
Meng X, Wang X, Zhang X, Zhang C, Zhang Z, Zhang K, Wang S. A Novel Attention-Mechanism Based Cox Survival Model by Exploiting Pan-Cancer Empirical Genomic Information. Cells. 2022; 11(9):1421. https://doi.org/10.3390/cells11091421
Chicago/Turabian StyleMeng, Xiangyu, Xun Wang, Xudong Zhang, Chaogang Zhang, Zhiyuan Zhang, Kuijie Zhang, and Shudong Wang. 2022. "A Novel Attention-Mechanism Based Cox Survival Model by Exploiting Pan-Cancer Empirical Genomic Information" Cells 11, no. 9: 1421. https://doi.org/10.3390/cells11091421
APA StyleMeng, X., Wang, X., Zhang, X., Zhang, C., Zhang, Z., Zhang, K., & Wang, S. (2022). A Novel Attention-Mechanism Based Cox Survival Model by Exploiting Pan-Cancer Empirical Genomic Information. Cells, 11(9), 1421. https://doi.org/10.3390/cells11091421