
Proteostasis Perturbations and Their Roles in Causing Sterile Inflammation and Autoinflammatory Diseases


Abstract
1. Introduction
2. Proteostasis Sensors and Their Roles in Triggering Sterile Inflammation
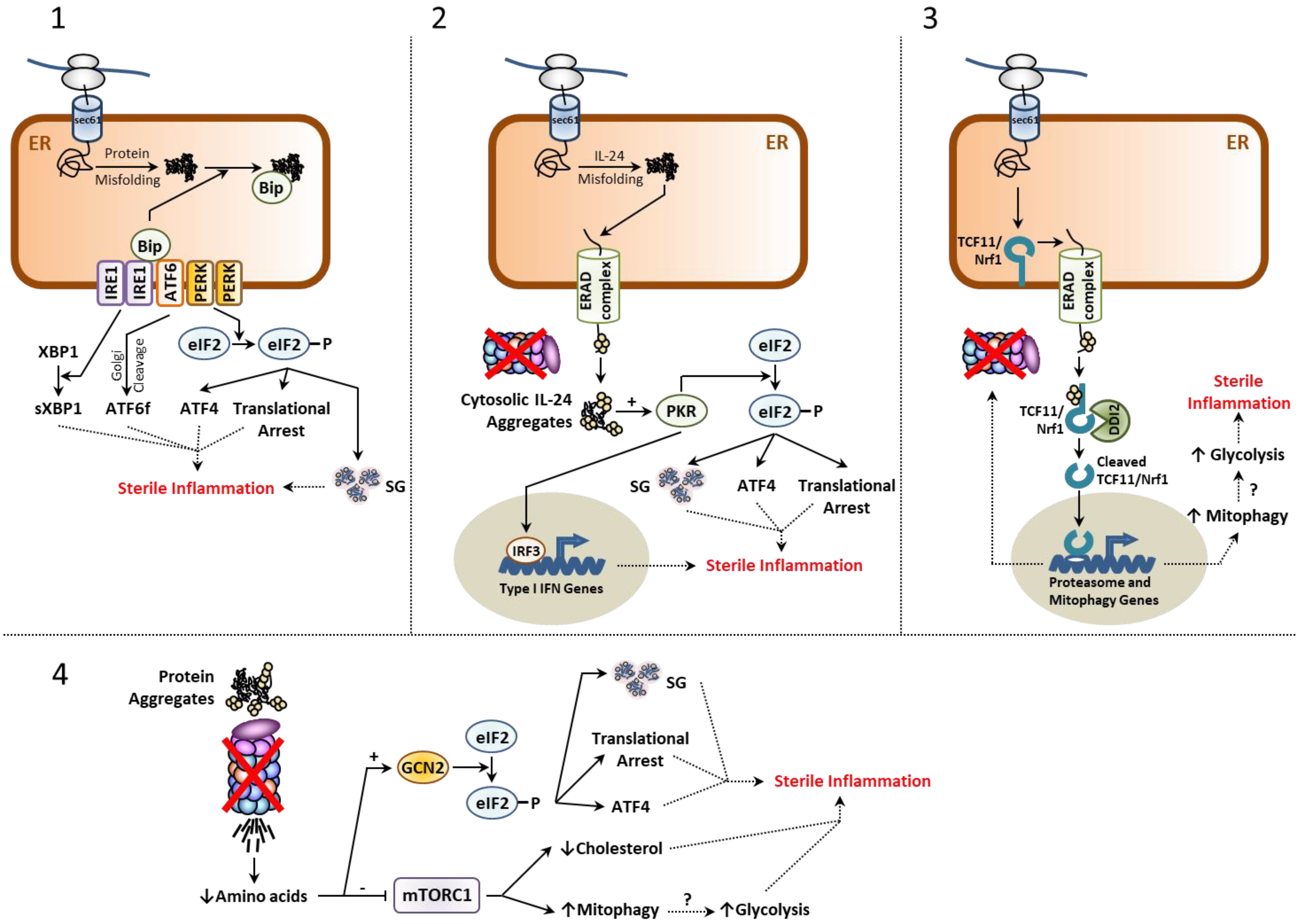
2.1. The Unfolded Protein Response (UPR)
- Arresting global protein biosynthesis;
2.2. The Integrated Stress Response (ISR)
2.3. The mTORC1 Signaling Complex
2.4. Stress-Induced Granulation
2.5. The TCF11/Nrf1-NGLY1-DDI2 Axis
2.6. Pathogen Recognition Receptors (PRR)
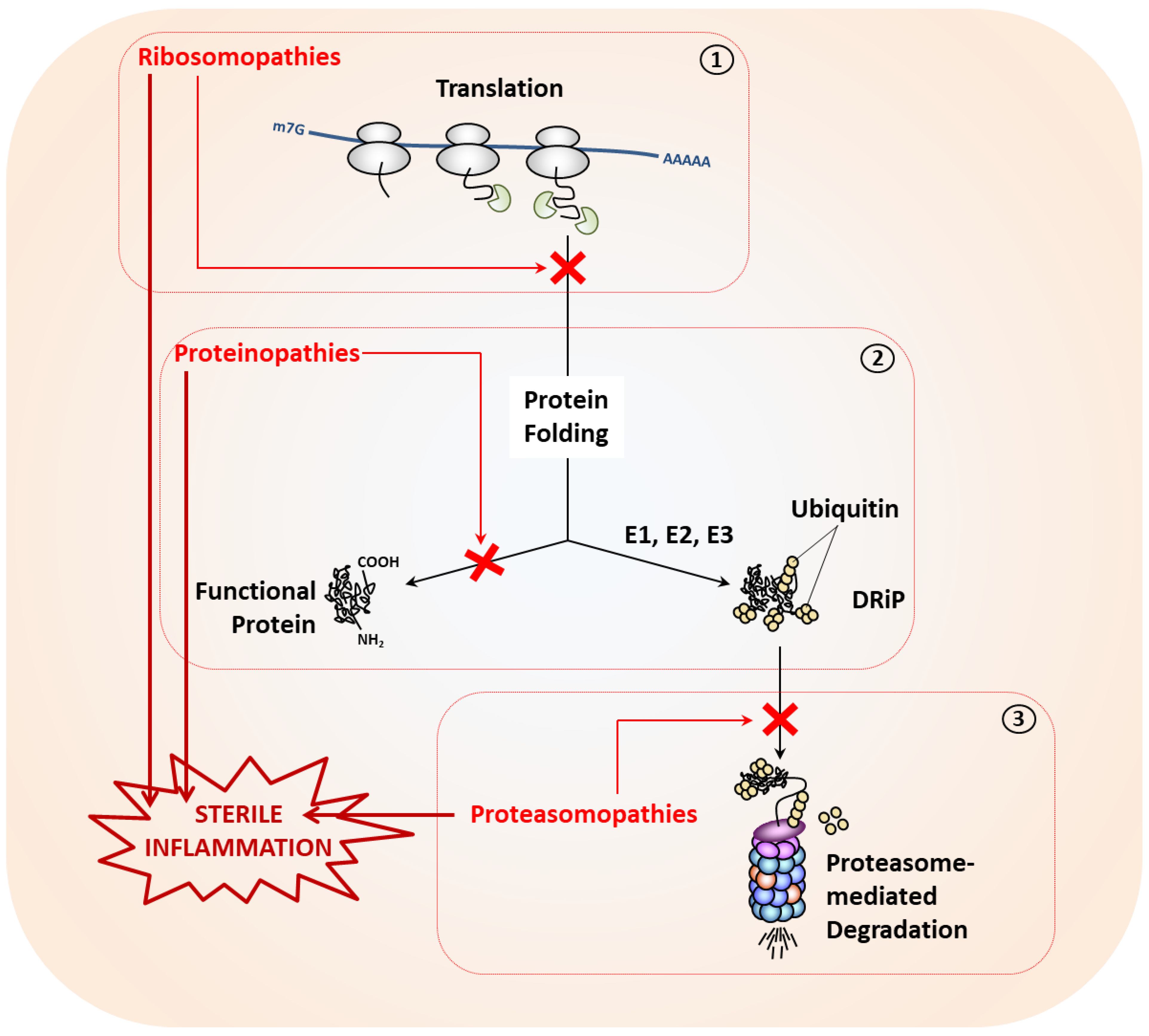
3. Causes of Proteostasis Perturbations and Associated Autoinflammatory Syndromes
- Protein aggregation or;
- Protein depletion.
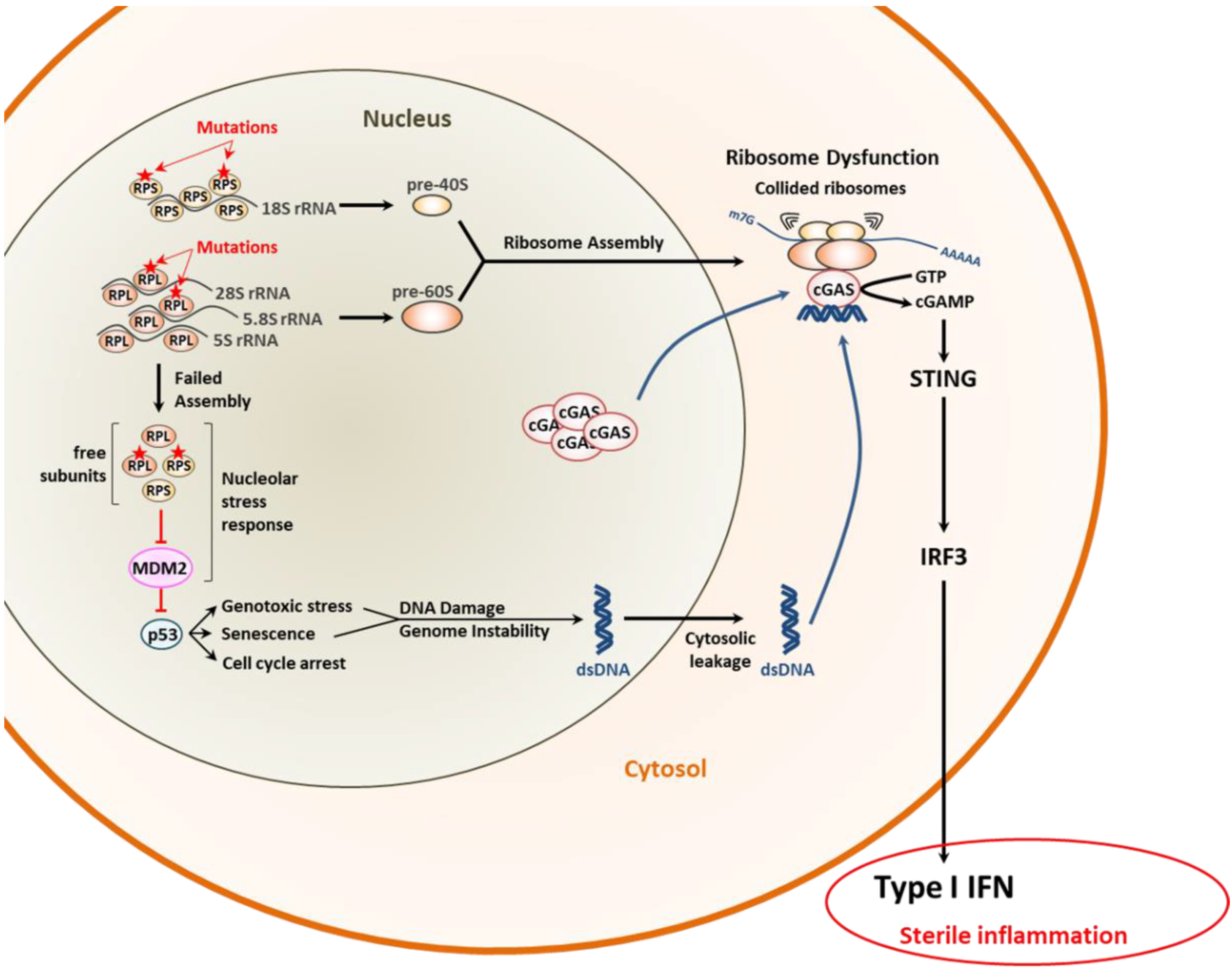
3.1. Proteostasis Perturbations Caused by Translation Deficiency
3.2. Proteostasis Perturbations Caused by Protein Misfolding
- (i)
- UPS components or;
- (ii)
- Highly translated host (or viral) proteins.
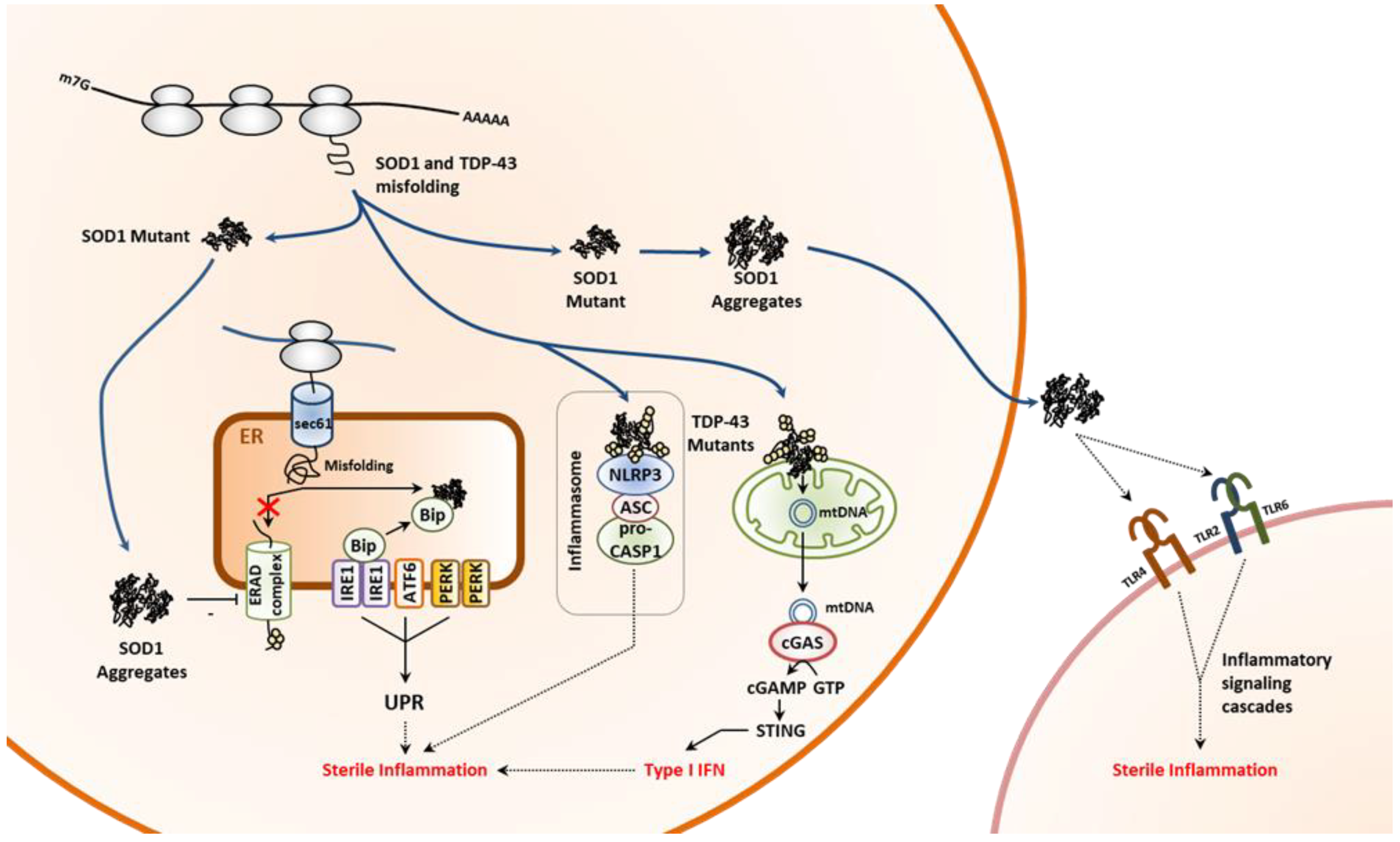
3.2.1. Non-Neurodegenerative Proteinopathies
3.2.2. Neurodegenerative Proteinopathies
3.3. Proteostasis Perturbations Caused by Impaired Protein Degradation
- The ubiquitin–proteasome system (UPS) and;
- The autophagy–lysosomal system [179].
3.3.1. The UPS and its Dysfunctions
3.3.2. Proteasome-Associated Autoinflammatory Syndromes (PRAAS)
3.3.3. Neurodevelopmental Disorders (NDD) Caused by Proteasome Variants
3.3.4. Disorders due to Deficient DUB and/or E3 Ubiquitin Ligases
3.3.5. The Autophagy-Lysosomal System and Its Defects
4. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- A Series of Essays on Inflammation and Its Varieties. Essay, I. The Natural History of the Disease. Med. Chir. Rev. 1846, 4, 251–253.
- Plytycz, B.; Seljelid, R. From inflammation to sickness: Historical perspective. Arch. Immunol. Exp. (Warsz) 2003, 51, 105–109. [Google Scholar] [PubMed]
- Cavaillon, J.M. Once upon a time, inflammation. J. Venom. Anim. Toxins Incl. Trop. Dis. 2021, 27, e20200147. [Google Scholar] [CrossRef] [PubMed]
- Diamond, G.; Legarda, D.; Ryan, L.K. The innate immune response of the respiratory epithelium. Immunol. Rev. 2000, 173, 27–38. [Google Scholar] [CrossRef]
- Shishido, S.N.; Varahan, S.; Yuan, K.; Li, X.; Fleming, S.D. Humoral innate immune response and disease. Clin. Immunol. 2012, 144, 142–158. [Google Scholar] [CrossRef]
- Koenderman, L.; Buurman, W.; Daha, M.R. The innate immune response. Immunol. Lett. 2014, 162, 95–102. [Google Scholar] [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Skoberne, M.; Beignon, A.S.; Bhardwaj, N. Danger signals: A time and space continuum. Trends Mol. Med. 2004, 10, 251–257. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef]
- Sok, S.P.M.; Ori, D.; Nagoor, N.H.; Kawai, T. Sensing Self and Non-Self DNA by Innate Immune Receptors and Their Signaling Pathways. Crit. Rev. Immunol. 2018, 38, 279–301. [Google Scholar] [CrossRef]
- Thaiss, C.A.; Levy, M.; Itav, S.; Elinav, E. Integration of Innate Immune Signaling. Trends Immunol. 2016, 37, 84–101. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [PubMed]
- Capobianchi, M.R.; Uleri, E.; Caglioti, C.; Dolei, A. Type I IFN family members: Similarity, differences and interaction. Cytokine Growth Factor Rev. 2015, 26, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Nace, G.; Evankovich, J.; Eid, R.; Tsung, A. Dendritic cells and damage-associated molecular patterns: Endogenous danger signals linking innate and adaptive immunity. J. Innate Immun. 2012, 4, 6–15. [Google Scholar] [CrossRef]
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef]
- Jounai, N.; Kobiyama, K.; Takeshita, F.; Ishii, K.J. Recognition of damage-associated molecular patterns related to nucleic acids during inflammation and vaccination. Front. Cell Infect. Microbiol. 2012, 2, 168. [Google Scholar] [CrossRef]
- Denning, N.L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Allam, R.; Darisipudi, M.N.; Tschopp, J.; Anders, H.J. Histones trigger sterile inflammation by activating the NLRP3 inflammasome. Eur. J. Immunol. 2013, 43, 3336–3342. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res. Rev. 2015, 24, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Franklin, E.C.; Holman, H.R.; Muller-Eberhard, H.J.; Kunkel, H.G. An unusual protein component of high molecular weight in the serum of certain patients with rheumatoid arthritis. J. Exp. Med. 1957, 105, 425–438. [Google Scholar] [CrossRef]
- Mellors, R.C.; Heimer, R.; Corcos, J.; Korngold, L. Cellular Origin of Rheumatoid Factor. J. Exp. Med. 1959, 110, 875–886. [Google Scholar] [CrossRef]
- Blom, A.B.; Radstake, T.R.; Holthuysen, A.E.; Sloetjes, A.W.; Pesman, G.J.; Sweep, F.G.; van de Loo, F.A.; Joosten, L.A.; Barrera, P.; van Lent, P.L.; et al. Increased expression of Fcgamma receptors II and III on macrophages of rheumatoid arthritis patients results in higher production of tumor necrosis factor alpha and matrix metalloproteinase. Arthritis Rheum 2003, 48, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Radstake, T.R.; Blom, A.B.; Sloetjes, A.W.; van Gorselen, E.O.; Pesman, G.J.; Engelen, L.; Torensma, R.; van den Berg, W.B.; Figdor, C.G.; van Lent, P.L.; et al. Increased FcgammaRII expression and aberrant tumour necrosis factor alpha production by mature dendritic cells from patients with active rheumatoid arthritis. Ann. Rheum. Dis. 2004, 63, 1556–1563. [Google Scholar] [CrossRef]
- Brehm, A.; Kruger, E. Dysfunction in protein clearance by the proteasome: Impact on autoinflammatory diseases. Semin. Immunopathol. 2015, 37, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Anton, L.C.; Yewdell, J.W. Translating DRiPs: MHC class I immunosurveillance of pathogens and tumors. J. Leukoc. Biol. 2014, 95, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Mancini, R.; Fagioli, C.; Fra, A.M.; Maggioni, C.; Sitia, R. Degradation of unassembled soluble Ig subunits by cytosolic proteasomes: Evidence that retrotranslocation and degradation are coupled events. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2000, 14, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Chillaron, J.; Haas, I.G. Dissociation from BiP and retrotranslocation of unassembled immunoglobulin light chains are tightly coupled to proteasome activity. Mol. Biol. Cell 2000, 11, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Mayer, T.U.; Braun, T.; Jentsch, S. Role of the proteasome in membrane extraction of a short-lived ER-transmembrane protein. EMBO J. 1998, 17, 3251–3257. [Google Scholar] [CrossRef] [PubMed]
- Ebstein, F.; Poli Harlowe, M.C.; Studencka-Turski, M.; Kruger, E. Contribution of the Unfolded Protein Response (UPR) to the Pathogenesis of Proteasome-Associated Autoinflammatory Syndromes (PRAAS). Front. Immunol. 2019, 10, 2756. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Dekker, J.; Strous, G.J. Covalent oligomerization of rat gastric mucin occurs in the rough endoplasmic reticulum, is N-glycosylation-dependent, and precedes initial O-glycosylation. J. Biol. Chem. 1990, 265, 18116–18122. [Google Scholar] [CrossRef]
- Pahl, H.L.; Baeuerle, P.A. A novel signal transduction pathway from the endoplasmic reticulum to the nucleus is mediated by transcription factor NF-kappa B. EMBO J. 1995, 14, 2580–2588. [Google Scholar] [CrossRef]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef]
- Jaud, M.; Philippe, C.; Di Bella, D.; Tang, W.; Pyronnet, S.; Laurell, H.; Mazzolini, L.; Rouault-Pierre, K.; Touriol, C. Translational Regulations in Response to Endoplasmic Reticulum Stress in Cancers. Cells 2020, 9, 540. [Google Scholar] [CrossRef]
- Deng, J.; Lu, P.D.; Zhang, Y.; Scheuner, D.; Kaufman, R.J.; Sonenberg, N.; Harding, H.P.; Ron, D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 2004, 24, 10161–10168. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Niinuma, Y.; Nomura, Y. Activation signal of nuclear factor-kappa B in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2. Biol. Pharm. Bull. 2003, 26, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; Munch, C.; Hanssum, A.; Bertolotti, A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol. Cell 2012, 48, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Qiu, H.; Garcia-Barrio, M.; Anderson, J.; Hinnebusch, A.G. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell 2000, 6, 269–279. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Dar, A.C.; Dever, T.E.; Sicheri, F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell 2005, 122, 887–900. [Google Scholar] [CrossRef]
- Clemens, M.J.; Hershey, J.W.; Hovanessian, A.C.; Jacobs, B.C.; Katze, M.G.; Kaufman, R.J.; Lengyel, P.; Samuel, C.E.; Sen, G.C.; Williams, B.R. PKR: Proposed nomenclature for the RNA-dependent protein kinase induced by interferon. J. Interferon Res. 1993, 13, 241. [Google Scholar] [CrossRef]
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, N.S.; Kerr, I.M.; Williams, B.R.; Hovanessian, A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef]
- Gilfoy, F.D.; Mason, P.W. West Nile virus-induced interferon production is mediated by the double-stranded RNA-dependent protein kinase PKR. J. Virol. 2007, 81, 11148–11158. [Google Scholar] [CrossRef]
- Barry, G.; Breakwell, L.; Fragkoudis, R.; Attarzadeh-Yazdi, G.; Rodriguez-Andres, J.; Kohl, A.; Fazakerley, J.K. PKR acts early in infection to suppress Semliki Forest virus production and strongly enhances the type I interferon response. J. Gen. Virol. 2009, 90, 1382–1391. [Google Scholar] [CrossRef]
- McAllister, C.S.; Toth, A.M.; Zhang, P.; Devaux, P.; Cattaneo, R.; Samuel, C.E. Mechanisms of protein kinase PKR-mediated amplification of beta interferon induction by C protein-deficient measles virus. J. Virol. 2010, 84, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Schulz, O.; Pichlmair, A.; Rehwinkel, J.; Rogers, N.C.; Scheuner, D.; Kato, H.; Takeuchi, O.; Akira, S.; Kaufman, R.J.; Reis e Sousa, C. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe 2010, 7, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Fowlkes, V.; Handy, I.; Patel, C.V.; Patel, R.C. Essential role of PACT-mediated PKR activation in tunicamycin-induced apoptosis. J. Mol. Biol. 2009, 385, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Pyo, C.W.; Lee, S.H.; Choi, S.Y. Oxidative stress induces PKR-dependent apoptosis via IFN-gamma activation signaling in Jurkat T cells. Biochem. Biophys. Res. Commun. 2008, 377, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Murtha-Riel, P.; Davies, M.V.; Scherer, B.J.; Choi, S.Y.; Hershey, J.W.; Kaufman, R.J. Expression of a phosphorylation-resistant eukaryotic initiation factor 2 alpha-subunit mitigates heat shock inhibition of protein synthesis. J. Biol. Chem. 1993, 268, 12946–12951. [Google Scholar] [CrossRef]
- Ito, T.; Yang, M.; May, W.S. RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J. Biol. Chem. 1999, 274, 15427–15432. [Google Scholar] [CrossRef]
- Patel, C.V.; Handy, I.; Goldsmith, T.; Patel, R.C. PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J. Biol. Chem. 2000, 275, 37993–37998. [Google Scholar] [CrossRef]
- Ito, T.; Warnken, S.P.; May, W.S. Protein synthesis inhibition by flavonoids: Roles of eukaryotic initiation factor 2alpha kinases. Biochem. Biophys. Res. Commun. 1999, 265, 589–594. [Google Scholar] [CrossRef]
- Davidson, S.; Yu, C.H.; Steiner, A.; Ebstein, F.; Baker, P.J.; Jarur-Chamy, V.; Hrovat Schaale, K.; Laohamonthonkul, P.; Kong, K.; Calleja, D.J.; et al. Protein kinase R is an innate immune sensor of proteotoxic stress via accumulation of cytoplasmic IL-24. Sci. Immunol. 2022, 7, eabi6763. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener 2019, 14, 32. [Google Scholar] [CrossRef]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2018, 11, 480. [Google Scholar] [CrossRef] [PubMed]
- Marchal, J.A.; Lopez, G.J.; Peran, M.; Comino, A.; Delgado, J.R.; Garcia-Garcia, J.A.; Conde, V.; Aranda, F.M.; Rivas, C.; Esteban, M.; et al. The impact of PKR activation: From neurodegeneration to cancer. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Chukwurah, E.; Farabaugh, K.T.; Guan, B.J.; Ramakrishnan, P.; Hatzoglou, M. A tale of two proteins: PACT and PKR and their roles in inflammation. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ingrand, S.; Barrier, L.; Lafay-Chebassier, C.; Fauconneau, B.; Page, G.; Hugon, J. The oxindole/imidazole derivative C16 reduces in vivo brain PKR activation. FEBS Lett. 2007, 581, 4473–4478. [Google Scholar] [CrossRef] [PubMed]
- Goberdhan, D.C.; Wilson, C.; Harris, A.L. Amino Acid Sensing by mTORC1: Intracellular Transporters Mark the Spot. Cell Metab. 2016, 23, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Broer, S.; Broer, A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem. J. 2017, 474, 1935–1963. [Google Scholar] [CrossRef] [PubMed]
- Condon, K.J.; Sabatini, D.M. Nutrient regulation of mTORC1 at a glance. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Isidor, B.; Ebstein, F.; Hurst, A.; Vincent, M.; Bader, I.; Rudy, N.L.; Cogne, B.; Mayr, J.; Brehm, A.; Bupp, C.; et al. Stankiewicz-Isidor syndrome: Expanding the clinical and molecular phenotype. Genet. Med. 2021. [Google Scholar] [CrossRef]
- York, A.G.; Williams, K.J.; Argus, J.P.; Zhou, Q.D.; Brar, G.; Vergnes, L.; Gray, E.E.; Zhen, A.; Wu, N.C.; Yamada, D.H.; et al. Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I IFN Signaling. Cell 2015, 163, 1716–1729. [Google Scholar] [CrossRef]
- Soto-Heredero, G.; Gomez de Las Heras, M.M.; Gabande-Rodriguez, E.; Oller, J.; Mittelbrunn, M. Glycolysis—A key player in the inflammatory response. FEBS J. 2020, 287, 3350–3369. [Google Scholar] [CrossRef]
- Allaman, I.; Gavillet, M.; Belanger, M.; Laroche, T.; Viertl, D.; Lashuel, H.A.; Magistretti, P.J. Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: Impact on neuronal viability. J. Neurosci. 2010, 30, 3326–3338. [Google Scholar] [CrossRef] [PubMed]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, M.; Taylor, J.P.; Parker, R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 2013, 154, 727–736. [Google Scholar] [CrossRef]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef]
- Ash, P.E.; Vanderweyde, T.E.; Youmans, K.L.; Apicco, D.J.; Wolozin, B. Pathological stress granules in Alzheimer’s disease. Brain Res. 2014, 1584, 52–58. [Google Scholar] [CrossRef]
- Vanderweyde, T.; Apicco, D.J.; Youmans-Kidder, K.; Ash, P.E.A.; Cook, C.; Lummertz da Rocha, E.; Jansen-West, K.; Frame, A.A.; Citro, A.; Leszyk, J.D.; et al. Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Rep. 2016, 15, 1455–1466. [Google Scholar] [CrossRef]
- McAleese, K.E.; Walker, L.; Erskine, D.; Thomas, A.J.; McKeith, I.G.; Attems, J. TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain Pathol. 2017, 27, 472–479. [Google Scholar] [CrossRef]
- St-Amour, I.; Turgeon, A.; Goupil, C.; Planel, E.; Hebert, S.S. Co-occurrence of mixed proteinopathies in late-stage Huntington’s disease. Acta Neuropathol. 2018, 135, 249–265. [Google Scholar] [CrossRef]
- Coudert, L.; Nonaka, T.; Bernard, E.; Hasegawa, M.; Schaeffer, L.; Leblanc, P. Phosphorylated and aggregated TDP-43 with seeding properties are induced upon mutant Huntingtin (mHtt) polyglutamine expression in human cellular models. Cell Mol. Life Sci 2019, 76, 2615–2632. [Google Scholar] [CrossRef] [PubMed]
- Ryan, V.H.; Fawzi, N.L. Physiological, Pathological, and Targetable Membraneless Organelles in Neurons. Trends Neurosci. 2019, 42, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Dudman, J.; Qi, X. Stress Granule Dysregulation in Amyotrophic Lateral Sclerosis. Front. Cell Neurosci. 2020, 14, 598517. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Kedersha, N.; Anderson, P.; Ivanov, P. Molecular mechanisms of stress granule assembly and disassembly. Biochim Biophys Acta Mol. Cell Res. 2021, 1868, 118876. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Mathieu, C.; Kolaitis, R.M.; Zhang, P.; Messing, J.; Yurtsever, U.; Yang, Z.; Wu, J.; Li, Y.; Pan, Q.; et al. G3BP1 Is a Tunable Switch that Triggers Phase Separation to Assemble Stress Granules. Cell 2020, 181, 325–345.e28. [Google Scholar] [CrossRef]
- Sanders, D.W.; Kedersha, N.; Lee, D.S.W.; Strom, A.R.; Drake, V.; Riback, J.A.; Bracha, D.; Eeftens, J.M.; Iwanicki, A.; Wang, A.; et al. Competing Protein-RNA Interaction Networks Control Multiphase Intracellular Organization. Cell 2020, 181, 306–324.e28. [Google Scholar] [CrossRef]
- Guillen-Boixet, J.; Kopach, A.; Holehouse, A.S.; Wittmann, S.; Jahnel, M.; Schlussler, R.; Kim, K.; Trussina, I.; Wang, J.; Mateju, D.; et al. RNA-Induced Conformational Switching and Clustering of G3BP Drive Stress Granule Assembly by Condensation. Cell 2020, 181, 346–361.e17. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Mazroui, R.; Di Marco, S.; Kaufman, R.J.; Gallouzi, I.E. Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell 2007, 18, 2603–2618. [Google Scholar] [CrossRef]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; Boczek, E.E.; Maharana, S.; Lee, H.O.; Carra, S.; Hyman, A.A.; Alberti, S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; Xu, Y.; Reineke, L.C.; Bhattacharya, A.; Tyryshkin, A.; Shin, J.N.; Eissa, N.T. Post-Transcriptional Regulation of Alpha One Antitrypsin by a Proteasome Inhibitor. Int. J. Mol. Sci. 2020, 21, 4318. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.W.; Onomoto, K.; Wakimoto, M.; Onoguchi, K.; Ishidate, F.; Fujiwara, T.; Yoneyama, M.; Kato, H.; Fujita, T. Leader-Containing Uncapped Viral Transcript Activates RIG-I in Antiviral Stress Granules. PLoS Pathog. 2016, 12, e1005444. [Google Scholar] [CrossRef] [PubMed]
- Onomoto, K.; Jogi, M.; Yoo, J.S.; Narita, R.; Morimoto, S.; Takemura, A.; Sambhara, S.; Kawaguchi, A.; Osari, S.; Nagata, K.; et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS ONE 2012, 7, e43031. [Google Scholar] [CrossRef]
- Yoneyama, M.; Jogi, M.; Onomoto, K. Regulation of antiviral innate immune signaling by stress-induced RNA granules. J. Biochem. 2016, 159, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Sun, H.; Yin, L.; Li, J.; Mei, S.; Xu, F.; Wu, C.; Liu, X.; Zhao, F.; Zhang, D.; et al. PKR-dependent cytosolic cGAS foci are necessary for intracellular DNA sensing. Sci Signal. 2019, 12. [Google Scholar] [CrossRef]
- Liu, Z.S.; Cai, H.; Xue, W.; Wang, M.; Xia, T.; Li, W.J.; Xing, J.Q.; Zhao, M.; Huang, Y.J.; Chen, S.; et al. G3BP1 promotes DNA binding and activation of cGAS. Nat. Immunol. 2019, 20, 18–28. [Google Scholar] [CrossRef]
- Kim, S.S.; Sze, L.; Liu, C.; Lam, K.P. The stress granule protein G3BP1 binds viral dsRNA and RIG-I to enhance interferon-beta response. J. Biol. Chem. 2019, 294, 6430–6438. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef]
- Steffen, J.; Seeger, M.; Koch, A.; Kruger, E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Tomlin, F.M.; Gerling-Driessen, U.I.M.; Liu, Y.C.; Flynn, R.A.; Vangala, J.R.; Lentz, C.S.; Clauder-Muenster, S.; Jakob, P.; Mueller, W.F.; Ordonez-Rueda, D.; et al. Inhibition of NGLY1 Inactivates the Transcription Factor Nrf1 and Potentiates Proteasome Inhibitor Cytotoxicity. ACS Cent. Sci. 2017, 3, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Irie, T.; Hirayama, S.; Sakurai, Y.; Yashiroda, H.; Naguro, I.; Ichijo, H.; Hamazaki, J.; Murata, S. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Huang, R.; Fujihira, H.; Suzuki, T.; Yan, N. N-glycanase NGLY1 regulates mitochondrial homeostasis and inflammation through NRF1. J. Exp. Med. 2018, 215, 2600–2616. [Google Scholar] [CrossRef] [PubMed]
- Widenmaier, S.B.; Snyder, N.A.; Nguyen, T.B.; Arduini, A.; Lee, G.Y.; Arruda, A.P.; Saksi, J.; Bartelt, A.; Hotamisligil, G.S. NRF1 Is an ER Membrane Sensor that Is Central to Cholesterol Homeostasis. Cell 2017, 171, 1094–1109.e15. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous alpha-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef]
- Stefanova, N.; Fellner, L.; Reindl, M.; Masliah, E.; Poewe, W.; Wenning, G.K. Toll-like receptor 4 promotes alpha-synuclein clearance and survival of nigral dopaminergic neurons. Am. J. Pathol. 2011, 179, 954–963. [Google Scholar] [CrossRef]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef]
- Dzamko, N.; Gysbers, A.; Perera, G.; Bahar, A.; Shankar, A.; Gao, J.; Fu, Y.; Halliday, G.M. Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol. 2017, 133, 303–319. [Google Scholar] [CrossRef]
- Kim, C.; Rockenstein, E.; Spencer, B.; Kim, H.K.; Adame, A.; Trejo, M.; Stafa, K.; Lee, H.J.; Lee, S.J.; Masliah, E. Antagonizing Neuronal Toll-like Receptor 2 Prevents Synucleinopathy by Activating Autophagy. Cell Rep. 2015, 13, 771–782. [Google Scholar] [CrossRef]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Lehnardt, S.; Massillon, L.; Follett, P.; Jensen, F.E.; Ratan, R.; Rosenberg, P.A.; Volpe, J.J.; Vartanian, T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 8514–8519. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rube, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T.; et al. TLR2 is a primary receptor for Alzheimer’s amyloid beta peptide to trigger neuroinflammatory activation. J. Immunol. 2012, 188, 1098–1107. [Google Scholar] [CrossRef]
- Scholtzova, H.; Chianchiano, P.; Pan, J.; Sun, Y.; Goni, F.; Mehta, P.D.; Wisniewski, T. Amyloid beta and Tau Alzheimer’s disease related pathology is reduced by Toll-like receptor 9 stimulation. Acta Neuropathol. Commun. 2014, 2, 101. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 activates microglia through NF-kappaB and NLRP3 inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef]
- Kadhim, H.; Deltenre, P.; Martin, J.J.; Sebire, G. In-situ expression of Interleukin-18 and associated mediators in the human brain of sALS patients: Hypothesis for a role for immune-inflammatory mechanisms. Med. Hypotheses 2016, 86, 14–17. [Google Scholar] [CrossRef]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef]
- Kampen, K.R.; Sulima, S.O.; Vereecke, S.; De Keersmaecker, K. Hallmarks of ribosomopathies. Nucleic Acids Res. 2020, 48, 1013–1028. [Google Scholar] [CrossRef]
- Berman, I.R.; Iliescu, H.; Stachura, I. Pulmonary effects of blood container materials. Surg. Forum 1977, 28, 182–184. [Google Scholar]
- Da Costa, L.; Leblanc, T.; Mohandas, N. Diamond-Blackfan anemia. Blood 2020, 136, 1262–1273. [Google Scholar] [CrossRef] [PubMed]
- Ellis, S.R. Nucleolar stress in Diamond Blackfan anemia pathophysiology. Biochim. Biophys. Acta 2014, 1842, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Uechi, T.; Kenmochi, N. Guarding the ‘translation apparatus’: Defective ribosome biogenesis and the p53 signaling pathway. Wiley Interdiscip. Rev. RNA 2011, 2, 507–522. [Google Scholar] [CrossRef]
- Kang, J.; Brajanovski, N.; Chan, K.T.; Xuan, J.; Pearson, R.B.; Sanij, E. Ribosomal proteins and human diseases: Molecular mechanisms and targeted therapy. Signal. Transduct. Target. 2021, 6, 323. [Google Scholar] [CrossRef] [PubMed]
- Kapralova, K.; Jahoda, O.; Koralkova, P.; Gursky, J.; Lanikova, L.; Pospisilova, D.; Divoky, V.; Horvathova, M. Oxidative DNA Damage, Inflammatory Signature, and Altered Erythrocytes Properties in Diamond-Blackfan Anemia. Int. J. Mol. Sci. 2020, 21, 9652. [Google Scholar] [CrossRef] [PubMed]
- Pesciotta, E.N.; Lam, H.S.; Kossenkov, A.; Ge, J.; Showe, L.C.; Mason, P.J.; Bessler, M.; Speicher, D.W. In-Depth, Label-Free Analysis of the Erythrocyte Cytoplasmic Proteome in Diamond Blackfan Anemia Identifies a Unique Inflammatory Signature. PLoS ONE 2015, 10, e0140036. [Google Scholar] [CrossRef] [PubMed]
- Gazda, H.T.; Kho, A.T.; Sanoudou, D.; Zaucha, J.M.; Kohane, I.S.; Sieff, C.A.; Beggs, A.H. Defective ribosomal protein gene expression alters transcription, translation, apoptosis, and oncogenic pathways in Diamond-Blackfan anemia. Stem Cells 2006, 24, 2034–2044. [Google Scholar] [CrossRef]
- Danilova, N.; Wilkes, M.; Bibikova, E.; Youn, M.Y.; Sakamoto, K.M.; Lin, S. Innate immune system activation in zebrafish and cellular models of Diamond Blackfan Anemia. Sci. Rep. 2018, 8, 5165. [Google Scholar] [CrossRef]
- Girardi, T.; Vereecke, S.; Sulima, S.O.; Khan, Y.; Fancello, L.; Briggs, J.W.; Schwab, C.; de Beeck, J.O.; Verbeeck, J.; Royaert, J.; et al. The T-cell leukemia-associated ribosomal RPL10 R98S mutation enhances JAK-STAT signaling. Leukemia 2018, 32, 809–819. [Google Scholar] [CrossRef]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef]
- Frisch, S.M.; MacFawn, I.P. Type I interferons and related pathways in cell senescence. Aging Cell 2020, 19, e13234. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Juszkiewicz, S.; Blears, D.; Bajpe, P.K.; Han, Z.; Faull, P.; Mitter, R.; Stewart, A.; Snijders, A.P.; Hegde, R.S.; et al. Translation stress and collided ribosomes are co-activators of cGAS. Mol. Cell 2021, 81, 2808–2822.e10. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W.; Nicchitta, C.V. The DRiP hypothesis decennial: Support, controversy, refinement and extension. Trends Immunol. 2006, 27, 368–373. [Google Scholar] [CrossRef]
- Schubert, U.; Anton, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Wegele, H.; Muller, L.; Buchner, J. Hsp70 and Hsp90—A relay team for protein folding. Rev. Physiol. Biochem. Pharm. 2004, 151, 1–44. [Google Scholar] [CrossRef]
- Mishra, A.; Godavarthi, S.K.; Maheshwari, M.; Goswami, A.; Jana, N.R. The ubiquitin ligase E6-AP is induced and recruited to aggresomes in response to proteasome inhibition and may be involved in the ubiquitination of Hsp70-bound misfolded proteins. J. Biol. Chem. 2009, 284, 10537–10545. [Google Scholar] [CrossRef]
- Mishra, R.; Amanullah, A.; Upadhyay, A.; Dhiman, R.; Dubey, A.R.; Singh, S.; Prasad, A.; Mishra, A. Ubiquitin ligase LRSAM1 suppresses neurodegenerative diseases linked aberrant proteins induced cell death. Int. J. Biochem. Cell Biol. 2020, 120, 105697. [Google Scholar] [CrossRef]
- Inda, C.; Bolaender, A.; Wang, T.; Gandu, S.R.; Koren, J., 3rd. Stressing Out Hsp90 in Neurotoxic Proteinopathies. Curr. Top. Med. Chem. 2016, 16, 2829–2838. [Google Scholar] [CrossRef]
- Olzscha, H. Posttranslational modifications and proteinopathies: How guardians of the proteome are defeated. Biol. Chem. 2019, 400, 895–915. [Google Scholar] [CrossRef]
- Mear, J.P.; Schreiber, K.L.; Munz, C.; Zhu, X.; Stevanovic, S.; Rammensee, H.G.; Rowland-Jones, S.L.; Colbert, R.A. Misfolding of HLA-B27 as a result of its B pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. J. Immunol. 1999, 163, 6665–6670. [Google Scholar]
- Dangoria, N.S.; DeLay, M.L.; Kingsbury, D.J.; Mear, J.P.; Uchanska-Ziegler, B.; Ziegler, A.; Colbert, R.A. HLA-B27 misfolding is associated with aberrant intermolecular disulfide bond formation (dimerization) in the endoplasmic reticulum. J. Biol. Chem. 2002, 277, 23459–23468. [Google Scholar] [CrossRef] [PubMed]
- Colbert, R.A. HLA-B27 misfolding: A solution to the spondyloarthropathy conundrum? Mol. Med. Today 2000, 6, 224–230. [Google Scholar] [CrossRef]
- Colbert, R.A.; Tran, T.M.; Layh-Schmitt, G. HLA-B27 misfolding and ankylosing spondylitis. Mol. Immunol 2014, 57, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Kerbiriou, M.; Le Drevo, M.A.; Ferec, C.; Trouve, P. Coupling cystic fibrosis to endoplasmic reticulum stress: Differential role of Grp78 and ATF6. Biochim. Biophys. Acta 2007, 1772, 1236–1249. [Google Scholar] [CrossRef]
- Trouve, P.; Ferec, C.; Genin, E. The Interplay between the Unfolded Protein Response, Inflammation and Infection in Cystic Fibrosis. Cells 2021, 10, 2980. [Google Scholar] [CrossRef]
- Segeritz, C.P.; Rashid, S.T.; de Brito, M.C.; Serra, M.P.; Ordonez, A.; Morell, C.M.; Kaserman, J.E.; Madrigal, P.; Hannan, N.R.F.; Gatto, L.; et al. hiPSC hepatocyte model demonstrates the role of unfolded protein response and inflammatory networks in alpha1-antitrypsin deficiency. J. Hepatol. 2018, 69, 851–860. [Google Scholar] [CrossRef]
- Lawless, M.W.; Greene, C.M.; Mulgrew, A.; Taggart, C.C.; O’Neill, S.J.; McElvaney, N.G. Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z alpha 1-antitrypsin deficiency. J. Immunol. 2004, 172, 5722–5726. [Google Scholar] [CrossRef]
- Carroll, T.P.; Greene, C.M.; O’Connor, C.A.; Nolan, A.M.; O’Neill, S.J.; McElvaney, N.G. Evidence for unfolded protein response activation in monocytes from individuals with alpha-1 antitrypsin deficiency. J. Immunol. 2010, 184, 4538–4546. [Google Scholar] [CrossRef]
- Saraiva, M.J.; Magalhaes, J.; Ferreira, N.; Almeida, M.R. Transthyretin deposition in familial amyloidotic polyneuropathy. Curr. Med. Chem. 2012, 19, 2304–2311. [Google Scholar] [CrossRef]
- Quarta, C.C.; Buxbaum, J.N.; Shah, A.M.; Falk, R.H.; Claggett, B.; Kitzman, D.W.; Mosley, T.H.; Butler, K.R.; Boerwinkle, E.; Solomon, S.D. The amyloidogenic V122I transthyretin variant in elderly black Americans. N. Engl. J. Med. 2015, 372, 21–29. [Google Scholar] [CrossRef]
- Kanda, Y.; Goodman, D.S.; Canfield, R.E.; Morgan, F.J. The amino acid sequence of human plasma prealbumin. J. Biol. Chem. 1974, 249, 6796–6805. [Google Scholar] [CrossRef]
- Sousa, M.M.; Saraiva, M.J. Neurodegeneration in familial amyloid polyneuropathy: From pathology to molecular signaling. Prog. Neurobiol. 2003, 71, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, E.P.; Guimaraes-Costa, A.B.; Bandeira-Melo, C.; Chimelli, L.; Waddington-Cruz, M.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. Inflammatory profiling of patients with familial amyloid polyneuropathy. BMC Neurol. 2019, 19, 146. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.M.; Du Yan, S.; Fernandes, R.; Guimaraes, A.; Stern, D.; Saraiva, M.J. Familial amyloid polyneuropathy: Receptor for advanced glycation end products-dependent triggering of neuronal inflammatory and apoptotic pathways. J. Neurosci. 2001, 21, 7576–7586. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, E.P.; Ledo, J.H.; Barbosa, G.; Sobrinho, M.; Diniz, L.; Fonseca, A.C.; Gomes, F.; Romao, L.; Lima, F.R.; Palhano, F.L.; et al. Activated microglia mediate synapse loss and short-term memory deficits in a mouse model of transthyretin-related oculoleptomeningeal amyloidosis. Cell Death Dis. 2013, 4, e789. [Google Scholar] [CrossRef][Green Version]
- Azevedo, E.P.; Guimaraes-Costa, A.B.; Torezani, G.S.; Braga, C.A.; Palhano, F.L.; Kelly, J.W.; Saraiva, E.M.; Foguel, D. Amyloid fibrils trigger the release of neutrophil extracellular traps (NETs), causing fibril fragmentation by NET-associated elastase. J. Biol. Chem. 2012, 287, 37206–37218. [Google Scholar] [CrossRef]
- Williams, D.B.; Windebank, A.J. Motor neuron disease (amyotrophic lateral sclerosis). Mayo Clin. Proc. 1991, 66, 54–82. [Google Scholar] [CrossRef]
- Rowland, L.P. Amyotrophic lateral sclerosis. Curr. Opin. Neurol. 1994, 7, 310–315. [Google Scholar] [CrossRef]
- Chia, R.; Chio, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Maurel, C.; Dangoumau, A.; Marouillat, S.; Brulard, C.; Chami, A.; Hergesheimer, R.; Corcia, P.; Blasco, H.; Andres, C.R.; Vourc’h, P. Causative Genes in Amyotrophic Lateral Sclerosis and Protein Degradation Pathways: A Link to Neurodegeneration. Mol. Neurobiol. 2018, 55, 6480–6499. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front. Biosci. 2008, 13, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. TDP-43: Gumming up neurons through protein-protein and protein-RNA interactions. Trends Biochem. Sci. 2012, 37, 237–247. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Cook, C.N.; Wu, Y.; Odeh, H.M.; Gendron, T.F.; Jansen-West, K.; Del Rosso, G.; Yue, M.; Jiang, P.; Gomes, E.; Tong, J.; et al. C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci Transl Med. 2020, 12. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Muller, K.; Marroquin, N.; Nordin, F.; Hubers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.S.; Beers, D.R.; Zhao, W.; Appel, S.H. Microglia in ALS: The good, the bad, and the resting. J. Neuroimmune Pharm. 2009, 4, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Shahheydari, H.; Ragagnin, A.; Walker, A.K.; Toth, R.P.; Vidal, M.; Jagaraj, C.J.; Perri, E.R.; Konopka, A.; Sultana, J.M.; Atkin, J.D. Protein Quality Control and the Amyotrophic Lateral Sclerosis/Frontotemporal Dementia Continuum. Front. Mol. Neurosci. 2017, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008, 22, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Henkel, J.S.; Zhang, W.; Urushitani, M.; Julien, J.P.; Appel, S.H. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 2010, 58, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Magrane, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef]
- Varshavsky, A. The Ubiquitin System, Autophagy, and Regulated Protein Degradation. Annu Rev. Biochem. 2017, 86, 123–128. [Google Scholar] [CrossRef]
- Cetin, G.; Klafack, S.; Studencka-Turski, M.; Kruger, E.; Ebstein, F. The Ubiquitin-Proteasome System in Immune Cells. Biomolecules 2021, 11, 60. [Google Scholar] [CrossRef]
- Pickart, C.M.; Fushman, D. Polyubiquitin chains: Polymeric protein signals. Curr. Opin. Chem. Biol. 2004, 8, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Mizushima, T.; Saeki, Y. The proteasome: Molecular machinery and pathophysiological roles. Biol. Chem. 2012, 393, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef] [PubMed]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, B. Proteasomes. Essays Biochem. 2005, 41, 31–48. [Google Scholar] [CrossRef]
- Greene, E.R.; Dong, K.C.; Martin, A. Understanding the 26S proteasome molecular machine from a structural and conformational dynamics perspective. Curr. Opin. Struct. Biol. 2020, 61, 33–41. [Google Scholar] [CrossRef]
- Ebstein, F.; Kloetzel, P.M.; Kruger, E.; Seifert, U. Emerging roles of immunoproteasomes beyond MHC class I antigen processing. Cell Mol. Life Sci. 2012, 69, 2543–2558. [Google Scholar] [CrossRef]
- Tubio-Santamaria, N.; Ebstein, F.; Heidel, F.H.; Kruger, E. Immunoproteasome Function in Normal and Malignant Hematopoiesis. Cells 2021, 10, 1577. [Google Scholar] [CrossRef]
- Seifert, U.; Bialy, L.P.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schroter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Ebstein, F.; Voigt, A.; Lange, N.; Warnatsch, A.; Schroter, F.; Prozorovski, T.; Kuckelkorn, U.; Aktas, O.; Seifert, U.; Kloetzel, P.M.; et al. Immunoproteasomes are important for proteostasis in immune responses. Cell 2013, 152, 935–937. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.S.; Kim, K.H.; Tschida, B.; Sachs, Z.; Noble-Orcutt, K.E.; Moriarity, B.S.; Ai, T.; Ding, R.; Williams, J.; Chen, L.; et al. mTORC1 Coordinates Protein Synthesis and Immunoproteasome Formation via PRAS40 to Prevent Accumulation of Protein Stress. Mol. Cell 2016, 61, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Niewerth, D.; Kaspers, G.J.; Assaraf, Y.G.; van Meerloo, J.; Kirk, C.J.; Anderl, J.; Blank, J.L.; van de Ven, P.M.; Zweegman, S.; Jansen, G.; et al. Interferon-gamma-induced upregulation of immunoproteasome subunit assembly overcomes bortezomib resistance in human hematological cell lines. J. Hematol. Oncol. 2014, 7, 7. [Google Scholar] [CrossRef]
- Opitz, E.; Koch, A.; Klingel, K.; Schmidt, F.; Prokop, S.; Rahnefeld, A.; Sauter, M.; Heppner, F.L.; Volker, U.; Kandolf, R.; et al. Impairment of immunoproteasome function by beta5i/LMP7 subunit deficiency results in severe enterovirus myocarditis. PLoS Pathog. 2011, 7, e1002233. [Google Scholar] [CrossRef] [PubMed]
- Kruger, E.; Kloetzel, P.M. Immunoproteasomes at the interface of innate and adaptive immune responses: Two faces of one enzyme. Curr. Opin. Immunol. 2012, 24, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Goetzke, C.C.; Ebstein, F.; Kallinich, T. Role of Proteasomes in Inflammation. J. Clin. Med. 2021, 10, 1783. [Google Scholar] [CrossRef] [PubMed]
- Chondrogianni, N.; Gonos, E.S. Proteasome function determines cellular homeostasis and the rate of aging. Adv. Exp. Med. Biol. 2010, 694, 38–46. [Google Scholar] [CrossRef]
- Keller, J.N.; Huang, F.F.; Markesbery, W.R. Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience 2000, 98, 149–156. [Google Scholar] [CrossRef]
- Tydlacka, S.; Wang, C.E.; Wang, X.; Li, S.; Li, X.J. Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J. Neurosci. 2008, 28, 13285–13295. [Google Scholar] [CrossRef]
- Giannini, C.; Kloss, A.; Gohlke, S.; Mishto, M.; Nicholson, T.P.; Sheppard, P.W.; Kloetzel, P.M.; Dahlmann, B. Poly-Ub-substrate-degradative activity of 26S proteasome is not impaired in the aging rat brain. PLoS ONE 2013, 8, e64042. [Google Scholar] [CrossRef]
- Keller, J.N.; Hanni, K.B.; Markesbery, W.R. Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 2000, 75, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Lopez Salon, M.; Morelli, L.; Castano, E.M.; Soto, E.F.; Pasquini, J.M. Defective ubiquitination of cerebral proteins in Alzheimer’s disease. J. Neurosci. Res. 2000, 62, 302–310. [Google Scholar] [CrossRef]
- Jenner, P.; Olanow, C.W. Understanding cell death in Parkinson’s disease. Ann. Neurol. 1998, 44, 72–84. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- McNaught, K.S.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 2003, 179, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Ansar, M.; Ebstein, F.; Ozkoc, H.; Paracha, S.A.; Iwaszkiewicz, J.; Gesemann, M.; Zoete, V.; Ranza, E.; Santoni, F.A.; Sarwar, M.T.; et al. Biallelic variants in PSMB1 encoding the proteasome subunit beta6 cause impairment of proteasome function, microcephaly, intellectual disability, developmental delay and short stature. Hum. Mol. Genet. 2020, 29, 1132–1143. [Google Scholar] [CrossRef]
- Brehm, A.; Liu, Y.; Sheikh, A.; Marrero, B.; Omoyinmi, E.; Zhou, Q.; Montealegre, G.; Biancotto, A.; Reinhardt, A.; Almeida de Jesus, A.; et al. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J. Clin. Investig. 2015, 125, 4196–4211. [Google Scholar] [CrossRef]
- Verhoeven, D.; Schonenberg-Meinema, D.; Ebstein, F.; Papendorf, J.J.; Baars, P.A.; van Leeuwen, E.M.M.; Jansen, M.H.; Lankester, A.C.; van der Burg, M.; Florquin, S.; et al. Hematopoietic stem cell transplantation in a patient with proteasome-associated auto-inflammatory syndrome (PRAAS). J. Allergy Clin. Immunol. 2021. [Google Scholar] [CrossRef]
- Kitamura, A.; Maekawa, Y.; Uehara, H.; Izumi, K.; Kawachi, I.; Nishizawa, M.; Toyoshima, Y.; Takahashi, H.; Standley, D.M.; Tanaka, K.; et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J. Clin. Investig. 2011, 121, 4150–4160. [Google Scholar] [CrossRef]
- Arima, K.; Kinoshita, A.; Mishima, H.; Kanazawa, N.; Kaneko, T.; Mizushima, T.; Ichinose, K.; Nakamura, H.; Tsujino, A.; Kawakami, A.; et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc. Natl Acad Sci USA 2011, 108, 14914–14919. [Google Scholar] [CrossRef]
- Liu, Y.; Ramot, Y.; Torrelo, A.; Paller, A.S.; Si, N.; Babay, S.; Kim, P.W.; Sheikh, A.; Lee, C.C.; Chen, Y.; et al. Mutations in proteasome subunit beta type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2012, 64, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Xing, C.; DeMartino, G.N.; Mizrachi, D.; Hernandez, M.D.; Sousa, A.B.; Martinez de Villarreal, L.; dos Santos, H.G.; Garg, A. PSMB8 encoding the beta5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am. J. Hum. Genet. 2010, 87, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Zheng, Y.; Feng, C.; Yang, T.; Geng, S. A Chinese case of Nakajo-Nishimura syndrome with novel compound heterozygous mutations of the PSMB8 gene. BMC Med. Genet. 2020, 21, 126. [Google Scholar] [CrossRef]
- Patel, P.N.; Hunt, R.; Pettigrew, Z.J.; Shirley, J.B.; Vogel, T.P.; de Guzman, M.M. Successful treatment of chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome with tofacitinib. Pediatr. Derm. 2021, 38, 528–529. [Google Scholar] [CrossRef] [PubMed]
- Boyadzhiev, M.; Marinov, L.; Boyadzhiev, V.; Iotova, V.; Aksentijevich, I.; Hambleton, S. Disease course and treatment effects of a JAK inhibitor in a patient with CANDLE syndrome. Pediatr. Rheumatol. Online J. 2019, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki-Nakashimada, M.A.; Santos-Chavez, E.E.; de Jesus, A.A.; Rivas-Larrauri, F.; Guzman-Martinez, M.N.; Goldbach-Mansky, R.; Espinosa-Padilla, S.; Saez-de-Ocariz, M.D.; Orozco-Covarrubias, L.; Blancas-Galicia, L. Systemic Autoimmunity in a Patient With CANDLE Syndrome. J. Investig. Allergol. Clin. Immunol. 2019, 29, 75–76. [Google Scholar] [CrossRef]
- Cardis, M.A.; Montealegre Sanchez, G.A.; Goldbach-Mansky, R.; Richard Lee, C.C.; Cowen, E.W. Recurrent fevers, progressive lipodystrophy, and annular plaques in a child. J. Am. Acad. Dermatol. 2019, 80, 291–295. [Google Scholar] [CrossRef]
- Kataoka, S.; Kawashima, N.; Okuno, Y.; Muramatsu, H.; Miwata, S.; Narita, K.; Hamada, M.; Murakami, N.; Taniguchi, R.; Ichikawa, D.; et al. Successful treatment of a novel type I interferonopathy due to a de novo PSMB9 gene mutation with a Janus kinase inhibitor. J. Allergy Clin. Immunol. 2021. [Google Scholar] [CrossRef]
- Kanazawa, N.; Hemmi, H.; Kinjo, N.; Ohnishi, H.; Hamazaki, J.; Mishima, H.; Kinoshita, A.; Mizushima, T.; Hamada, S.; Hamada, K.; et al. Heterozygous missense variant of the proteasome subunit beta-type 9 causes neonatal-onset autoinflammation and immunodeficiency. Nat. Commun. 2021, 12, 6819. [Google Scholar] [CrossRef]
- Sarrabay, G.; Mechin, D.; Salhi, A.; Boursier, G.; Rittore, C.; Crow, Y.; Rice, G.; Tran, T.A.; Cezar, R.; Duffy, D.; et al. PSMB10, the last immunoproteasome gene missing for PRAAS. J. Allergy Clin. Immunol. 2019. [Google Scholar] [CrossRef]
- Poli, M.C.; Ebstein, F.; Nicholas, S.K.; de Guzman, M.M.; Forbes, L.R.; Chinn, I.K.; Mace, E.M.; Vogel, T.P.; Carisey, A.F.; Benavides, F.; et al. Heterozygous Truncating Variants in POMP Escape Nonsense-Mediated Decay and Cause a Unique Immune Dysregulatory Syndrome. Am. J. Hum. Genet. 2018, 102, 1126–1142. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, A.; Ramos, P.C.; Dohmen, R.J.; Lucas, N.; Lee-Kirsch, M.A.; Becker, B.; de Laffolie, J.; Cunha, T.; Niehues, T.; Salzer, U.; et al. Curative Treatment of POMP-Related Autoinflammation and Immune Dysregulation (PRAID) by Hematopoietic Stem Cell Transplantation. J. Clin. Immunol. 2021, 41, 1664–1667. [Google Scholar] [CrossRef] [PubMed]
- de Jesus, A.A.; Brehm, A.; VanTries, R.; Pillet, P.; Parentelli, A.S.; Montealegre Sanchez, G.A.; Deng, Z.; Paut, I.K.; Goldbach-Mansky, R.; Kruger, E. Novel proteasome assembly chaperone mutations in PSMG2/PAC2 cause the autoinflammatory interferonopathy CANDLE/PRAAS4. J. Allergy Clin. Immunol. 2019, 143, 1939–1943.e8. [Google Scholar] [CrossRef] [PubMed]
- Kury, S.; Besnard, T.; Ebstein, F.; Khan, T.N.; Gambin, T.; Douglas, J.; Bacino, C.A.; Craigen, W.J.; Sanders, S.J.; Lehmann, A.; et al. De Novo Disruption of the Proteasome Regulatory Subunit PSMD12 Causes a Syndromic Neurodevelopmental Disorder. Am. J. Hum. Genet. 2017, 100, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Khalil, R.; Kenny, C.; Hill, R.S.; Mochida, G.H.; Nasir, R.; Partlow, J.N.; Barry, B.J.; Al-Saffar, M.; Egan, C.; Stevens, C.R.; et al. PSMD12 haploinsufficiency in a neurodevelopmental disorder with autistic features. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 736–745. [Google Scholar] [CrossRef]
- Yan, K.; Zhang, J.; Lee, P.Y.; Tao, P.; Wang, J.; Wang, S.; Zhou, Q.; Dong, M. Haploinsufficiency of PSMD12 causes proteasome dysfunction and subclinical autoinflammation. Arthritis Rheumatol. 2022. [Google Scholar] [CrossRef]
- Kroll-Hermi, A.; Ebstein, F.; Stoetzel, C.; Geoffroy, V.; Schaefer, E.; Scheidecker, S.; Bar, S.; Takamiya, M.; Kawakami, K.; Zieba, B.A.; et al. Proteasome subunit PSMC3 variants cause neurosensory syndrome combining deafness and cataract due to proteotoxic stress. EMBO Mol. Med. 2020, 12, e11861. [Google Scholar] [CrossRef]
- Kitano, Y.; Matsunaga, E.; Morimoto, T.; Okada, N.; Sano, S. A syndrome with nodular erythema, elongated and thickened fingers, and emaciation. Arch. Derm. 1985, 121, 1053–1056. [Google Scholar] [CrossRef]
- Tanaka, M.; Miyatani, N.; Yamada, S.; Miyashita, K.; Toyoshima, I.; Sakuma, K.; Tanaka, K.; Yuasa, T.; Miyatake, T.; Tsubaki, T. Hereditary lipo-muscular atrophy with joint contracture, skin eruptions and hyper-gamma-globulinemia: A new syndrome. Intern. Med. 1993, 32, 42–45. [Google Scholar] [CrossRef][Green Version]
- Kasagi, S.; Kawano, S.; Nakazawa, T.; Sugino, H.; Koshiba, M.; Ichinose, K.; Ida, H.; Eguchi, K.; Kumagai, S. A case of periodic-fever-syndrome-like disorder with lipodystrophy, myositis, and autoimmune abnormalities. Mod. Rheumatol. 2008, 18, 203–207. [Google Scholar] [CrossRef][Green Version]
- Torrelo, A.; Patel, S.; Colmenero, I.; Gurbindo, D.; Lendinez, F.; Hernandez, A.; Lopez-Robledillo, J.C.; Dadban, A.; Requena, L.; Paller, A.S. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J. Am. Acad. Dermatol. 2010, 62, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Ramot, Y.; Czarnowicki, T.; Maly, A.; Navon-Elkan, P.; Zlotogorski, A. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome: A case report. Pediatr. Derm. 2011, 28, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Usui, F.; Karasawa, T.; Kawashima, A.; Shirasuna, K.; Inoue, Y.; Komada, T.; Kobayashi, M.; Mizushina, Y.; Kasahara, T.; et al. Immunoproteasome subunit LMP7 Deficiency Improves Obesity and Metabolic Disorders. Sci. Rep. 2015, 5, 15883. [Google Scholar] [CrossRef]
- Martinez, C.A.; Ebstein, F.; Nicholas, S.K.; De Guzman, M.; Forbes, L.R.; Delmonte, O.M.; Bosticardo, M.; Castagnoli, R.; Krance, R.; Notarangelo, L.D.; et al. HSCT corrects primary immunodeficiency and immune dysregulation in patients with POMP-related auto-inflammatory disease. Blood 2021. [Google Scholar] [CrossRef]
- Studencka-Turski, M.; Cetin, G.; Junker, H.; Ebstein, F.; Kruger, E. Molecular Insight Into the IRE1alpha-Mediated Type I Interferon Response Induced by Proteasome Impairment in Myeloid Cells of the Brain. Front. Immunol. 2019, 10, 2900. [Google Scholar] [CrossRef] [PubMed]
- Minton, K. Sensing proteotoxic stress. Nat. Rev. Immunol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Manel, N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat. Rev. Immunol. 2015, 15, 429–440. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Evans, D.; Pandey, A.; Hraha, T.H.; Smith, K.P.; Markham, N.; Rachubinski, A.L.; Wolter-Warmerdam, K.; Hickey, F.; Espinosa, J.M.; et al. Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation. Sci. Rep. 2017, 7, 14818. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Lewis, H.C.; Hill, A.A.; Pandey, A.; Jackson, L.P.; Cabral, J.M.; Smith, K.P.; Liggett, L.A.; Gomez, E.B.; Galbraith, M.D.; et al. Trisomy 21 consistently activates the interferon response. eLife 2016, 5. [Google Scholar] [CrossRef]
- Waugh, K.A.; Araya, P.; Pandey, A.; Jordan, K.R.; Smith, K.P.; Granrath, R.E.; Khanal, S.; Butcher, E.T.; Estrada, B.E.; Rachubinski, A.L.; et al. Mass Cytometry Reveals Global Immune Remodeling with Multi-lineage Hypersensitivity to Type I Interferon in Down Syndrome. Cell Rep. 2019, 29, 1893–1908.e4. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, H.; Schwartz, D.M.; Stoffels, M.; Park, Y.H.; Zhang, Y.; Yang, D.; Demirkaya, E.; Takeuchi, M.; Tsai, W.L.; et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat. Genet. 2016, 48, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, R.B.; Walker, J.A.; Marco-Casanova, P.; Morgan, N.V.; Titheradge, H.L.; Elliott, P.R.; McHale, D.; Maher, E.R.; McKenzie, A.N.J.; Komander, D. The Deubiquitinase OTULIN Is an Essential Negative Regulator of Inflammation and Autoimmunity. Cell 2016, 166, 1215–1230. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, R.B.; Elliott, P.R.; Swatek, K.N.; Maher, E.R.; Stepensky, P.; Elpeleg, O.; Komander, D.; Berkun, Y. OTULIN deficiency in ORAS causes cell type-specific LUBAC degradation, dysregulated TNF signalling and cell death. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, M.E.; Schot, R.; Buta, S.; Oudesluijs, G.; Tinschert, S.; Speer, S.D.; Li, Z.; van Unen, L.; Heijsman, D.; Goldmann, T.; et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J. Exp. Med. 2016, 213, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Boisson, B.; Laplantine, E.; Dobbs, K.; Cobat, A.; Tarantino, N.; Hazen, M.; Lidov, H.G.; Hopkins, G.; Du, L.; Belkadi, A.; et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med. 2015, 212, 939–951. [Google Scholar] [CrossRef]
- Boisson, B.; Laplantine, E.; Prando, C.; Giliani, S.; Israelsson, E.; Xu, Z.; Abhyankar, A.; Israel, L.; Trevejo-Nunez, G.; Bogunovic, D.; et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 2012, 13, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Oda, H.; Beck, D.B.; Kuehn, H.S.; Sampaio Moura, N.; Hoffmann, P.; Ibarra, M.; Stoddard, J.; Tsai, W.L.; Gutierrez-Cruz, G.; Gadina, M.; et al. Second Case of HOIP Deficiency Expands Clinical Features and Defines Inflammatory Transcriptome Regulated by LUBAC. Front. Immunol. 2019, 10, 479. [Google Scholar] [CrossRef]
- Di Donato, G.; d’Angelo, D.M.; Breda, L.; Chiarelli, F. Monogenic Autoinflammatory Diseases: State of the Art and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 6360. [Google Scholar] [CrossRef]
- Zimmermann, A.; Kainz, K.; Andryushkova, A.; Hofer, S.; Madeo, F.; Carmona-Gutierrez, D. Autophagy: One more Nobel Prize for yeast. Microb. Cell 2016, 3, 579–581. [Google Scholar] [CrossRef]
- Tooze, S.A.; Dikic, I. Autophagy Captures the Nobel Prize. Cell 2016, 167, 1433–1435. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Koya, D. Autophagy in metabolic disease and ageing. Nat. Rev. Endocrinol. 2021, 17, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Wong, E. Kinetics of Protein Aggregates Disposal by Aggrephagy. Methods Enzym. 2017, 588, 245–281. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Haack, T.B.; Ignatius, E.; Calvo-Garrido, J.; Iuso, A.; Isohanni, P.; Maffezzini, C.; Lonnqvist, T.; Suomalainen, A.; Gorza, M.; Kremer, L.S.; et al. Absence of the Autophagy Adaptor SQSTM1/p62 Causes Childhood-Onset Neurodegeneration with Ataxia, Dystonia, and Gaze Palsy. Am. J. Hum. Genet. 2016, 99, 735–743. [Google Scholar] [CrossRef]
- Roodman, G.D.; Kurihara, N.; Ohsaki, Y.; Kukita, A.; Hosking, D.; Demulder, A.; Smith, J.F.; Singer, F.R. Interleukin 6. A potential autocrine/paracrine factor in Paget’s disease of bone. J. Clin. Investig. 1992, 89, 46–52. [Google Scholar] [CrossRef]
- Poloni, M.; Facchetti, D.; Mai, R.; Micheli, A.; Agnoletti, L.; Francolini, G.; Mora, G.; Camana, C.; Mazzini, L.; Bachetti, T. Circulating levels of tumour necrosis factor-alpha and its soluble receptors are increased in the blood of patients with amyotrophic lateral sclerosis. Neurosci. Lett. 2000, 287, 211–214. [Google Scholar] [CrossRef]
- Wooten, M.W.; Geetha, T.; Seibenhener, M.L.; Babu, J.R.; Diaz-Meco, M.T.; Moscat, J. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J. Biol. Chem. 2005, 280, 35625–35629. [Google Scholar] [CrossRef]
- Rea, S.L.; Walsh, J.P.; Layfield, R.; Ratajczak, T.; Xu, J. New insights into the role of sequestosome 1/p62 mutant proteins in the pathogenesis of Paget’s disease of bone. Endocr. Rev. 2013, 34, 501–524. [Google Scholar] [CrossRef]
- Kim, M.; Sandford, E.; Gatica, D.; Qiu, Y.; Liu, X.; Zheng, Y.; Schulman, B.A.; Xu, J.; Semple, I.; Ro, S.H.; et al. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Collier, J.J.; Guissart, C.; Olahova, M.; Sasorith, S.; Piron-Prunier, F.; Suomi, F.; Zhang, D.; Martinez-Lopez, N.; Leboucq, N.; Bahr, A.; et al. Developmental Consequences of Defective ATG7-Mediated Autophagy in Humans. N. Engl. J. Med. 2021, 384, 2406–2417. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.C.; Seifert, U.; Urban, S.; Truong, K.T.; Feinstone, S.M.; Rice, C.M.; Kloetzel, P.M.; Rehermann, B. Proteasome activator and antigen-processing aminopeptidases are regulated by virus-induced type I interferon in the hepatitis C virus-infected liver. J. Interferon Cytokine Res. 2007, 27, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Bousoik, E.; Montazeri Aliabadi, H. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 2018, 8, 287. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteasome | Gene | Variant | Genetic Model | Origin | Phenotype | Reference |
|---|---|---|---|---|---|---|
| 20S Complex | PSMB1 | p.Y103H | Homozygous, monogenic | Recessive inheritance | NDD | [206] |
| PSMB4 | 5′ UTR: c.–9G > A | Compound heterozygous, monogenic | Recessive inheritance | PRAAS | [207] | |
| p.D212_V214del | ||||||
| PSMB4 | p.L78Wfs * 31 | Compound heterozygous, monogenic | Recessive inheritance | PRAAS | [208] | |
| c.494 + 17A > G | ||||||
| PSMB4/ PSMB8 | p.Y222 * | Double heterozygous, digenic | Recessive inheritance | PRAAS | [207] | |
| p.K105Q | ||||||
| PSMB8 | p.G179V | Homozygous, monogenic | Recessive inheritance | PRAAS | [209] | |
| PSMB8 | p.G201V | Homozygous, monogenic | Recessive inheritance | PRAAS | [210] | |
| PSMB8 | p.C135 * | Homozygous, monogenic | Recessive inheritance | PRAAS | [211] | |
| PSMB8 | p.T75M | Homozygous, monogenic | Recessive inheritance | PRAAS | [212] | |
| PSMB8 | p.R125C | Compound heterozygous, monogenic | Recessive inheritance | PRAAS | [213] | |
| p.D119N | ||||||
| PSMB8 | p.Q55 * | Compound heterozygous, monogenic | Recessive inheritance | PRAAS | [214] | |
| p.S118P | ||||||
| PSMB8 | p.A92V | Compound heterozygous, monogenic | Recessive inheritance | PRAAS | [215] | |
| p.K105Q | ||||||
| PSMB8 | p.A92T | Homozygous, monogenic | Recessive inheritance | PRAAS | [216] | |
| PSMB8 | - | Homozygous, monogenic | Recessive inheritance | PRAAS | [217] | |
| PSMB8/ PSMA3 | p.T75M | Double heterozygous, digenic | Recessive inheritance | PRAAS | [207] | |
| p.H111Ffs * 10 | ||||||
| PSMB8/ PSMA3 | p.T75M | Double heterozygous, digenic | Recessive inheritance | PRAAS | [207] | |
| p.R233del | ||||||
| PSMB9/ PSMB4 | p.G165D | Double heterozygous, digenic | Recessive inheritance | PRAAS | [207] | |
| p.P16Sfs * 45 | ||||||
| PSMB9 | p.G156D | Heterozygous, monogenic | de novo, dominant | PRAAS | [218] [219] | |
| PSMB10 | p.F14S | Homozygous, monogenic | Recessive inheritance | PRAAS | [220] | |
| Assembly Factors | POMP | p.E115Dfs * 20 | Heterozygous, monogenic | de novo, dominant | PRAAS | [207] |
| POMP | p.F114Lfs * 18 | Heterozygous, monogenic | de novo, dominant | PRAAS | [221] | |
| POMP | p.I112Wfs * 3 | Heterozygous, monogenic | de novo, dominant | PRAAS | [221] | |
| POMP | p.D109Efs * 2 | Heterozygous, monogenic | de novo, dominant | PRAAS | [222] | |
| PSMG4 | p.Y223Sfs * 2 | Compound heterozygous, monogenic | Recessive inheritance | PRAAS | [223] | |
| p.N225K | ||||||
| 19S Complex | PSMD12 | p.R123 * | Heterozygous, monogenic | de novo, dominant | NDD | [224] |
| PSMD12 | p.L425 * | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.R201 * | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | c.909−2A > G | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | Deletion | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.R201 * | Heterozygous, monogenic | de novo, dominant | NDD | [225] | |
| PSMD12 | p.R182 * | Heterozygous, monogenic | de novo, dominant | NDD | [68] | |
| PSMD12 | p.R357fs * 3 | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.T146Kfs * 3 | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.E313 * | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.Q170Gfs * 40 | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.L149 * | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.Q106 * | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.Q345 * | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | c.1083 + 1G > A | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.Q416 * | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.S176Qfs * 15 | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | c.1162−1G > A | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.S434Hfs * 2 | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | c.795 + 1G > A | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.L50Gfs * 26 | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.T146Kfs * 3 | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.R182 * | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.L354Efs * 6 | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.Y302 * | Heterozygous, monogenic | de novo, dominant | NDD | ||
| PSMD12 | p.R289 * | Heterozygous, monogenic | de novo, dominant | NDD | [226] | |
| PSMC3 | p.S376Rfs15 * | Homozygous, monogenic | Recessive inheritance | NDD | [227] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papendorf, J.J.; Krüger, E.; Ebstein, F. Proteostasis Perturbations and Their Roles in Causing Sterile Inflammation and Autoinflammatory Diseases. Cells 2022, 11, 1422. https://doi.org/10.3390/cells11091422
Papendorf JJ, Krüger E, Ebstein F. Proteostasis Perturbations and Their Roles in Causing Sterile Inflammation and Autoinflammatory Diseases. Cells. 2022; 11(9):1422. https://doi.org/10.3390/cells11091422
Chicago/Turabian StylePapendorf, Jonas Johannes, Elke Krüger, and Frédéric Ebstein. 2022. "Proteostasis Perturbations and Their Roles in Causing Sterile Inflammation and Autoinflammatory Diseases" Cells 11, no. 9: 1422. https://doi.org/10.3390/cells11091422
APA StylePapendorf, J. J., Krüger, E., & Ebstein, F. (2022). Proteostasis Perturbations and Their Roles in Causing Sterile Inflammation and Autoinflammatory Diseases. Cells, 11(9), 1422. https://doi.org/10.3390/cells11091422