
Shifting CCR7 towards Its Monomeric Form Augments CCL19 Binding and Uptake


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cloning of Expression Vectors and Plasmids
2.3. Chemokine Production and Site-Specific Labelling
2.4. Cell Lines and Cell Transfection
2.5. Validation of Transfection Efficiency by RT-qPCR
2.6. CCR7 Dimerisation Assessed by a Split-Citrine Complementation Assay
2.7. CCR7 Dimerisation Assessed by a Split-Luciferase Complementation Assay
2.8. Endocytosis and G-Protein Activation Assessment by Bioluminescence Resonance Energy Transfer (BRET)
2.9. CCR7 Surface Expression Determined by Flow Cytometry
2.10. CCL19 Binding and Uptake
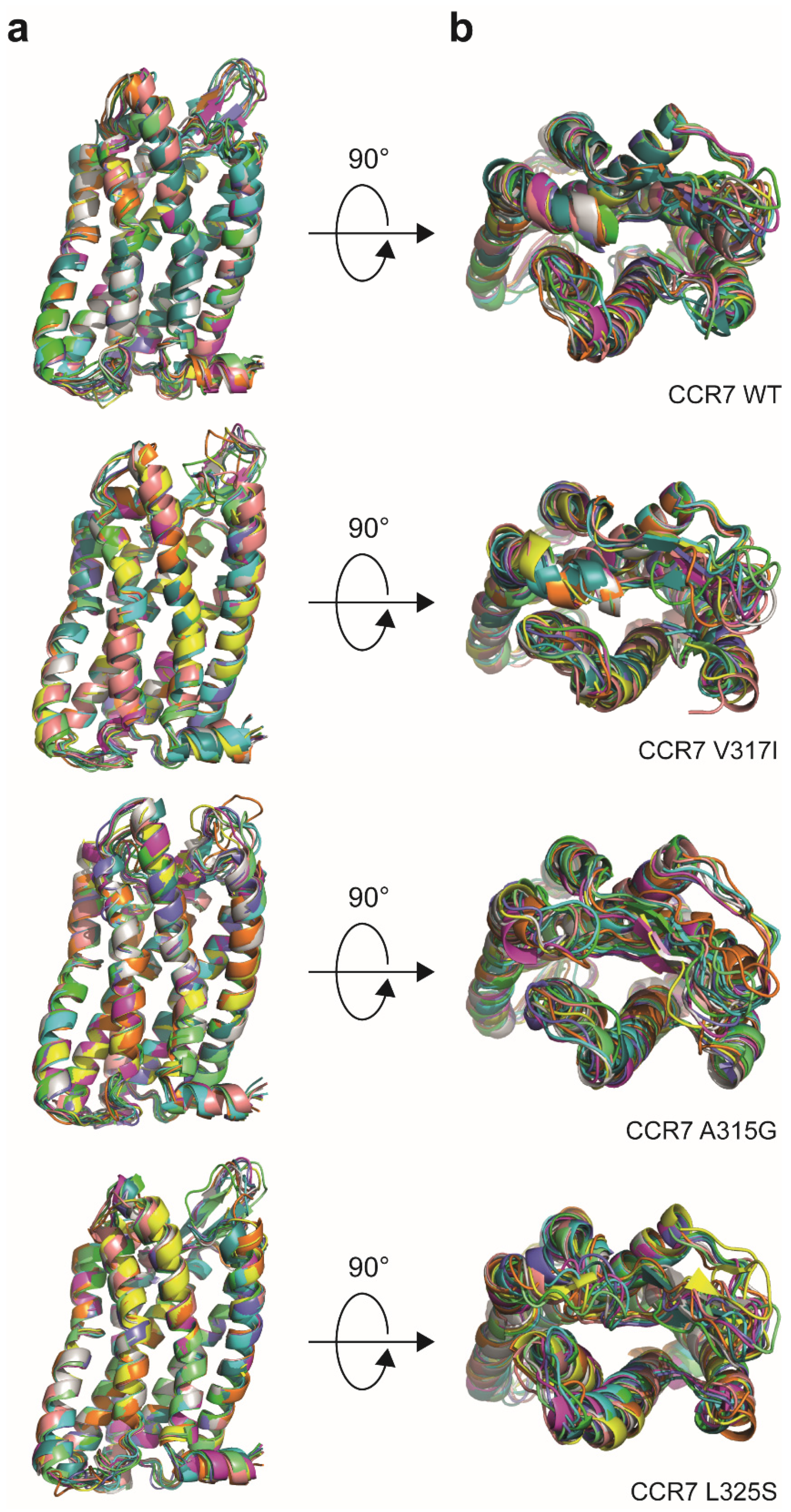
2.11. Receptor Modelling
2.12. Statistical Analysis
3. Results
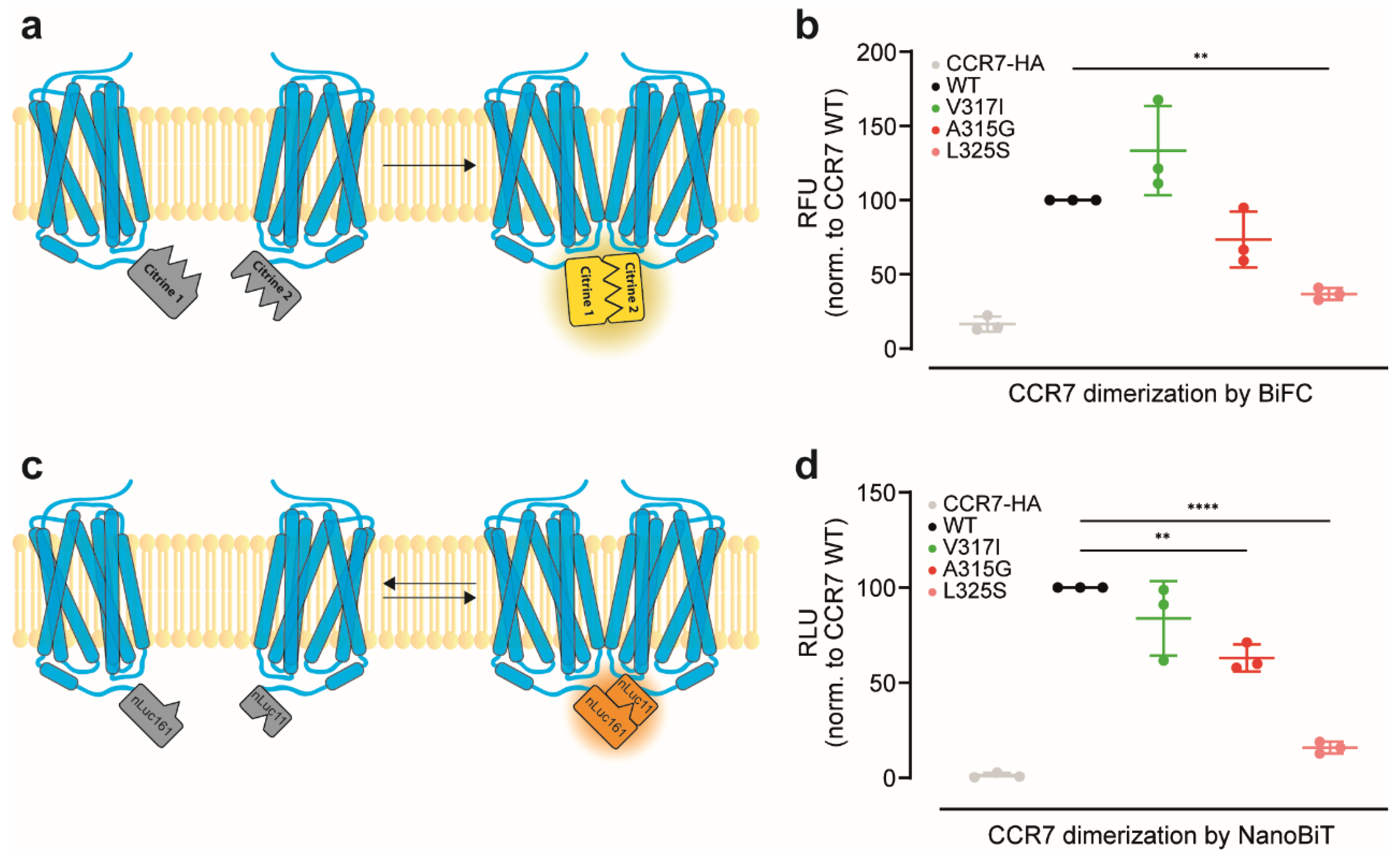
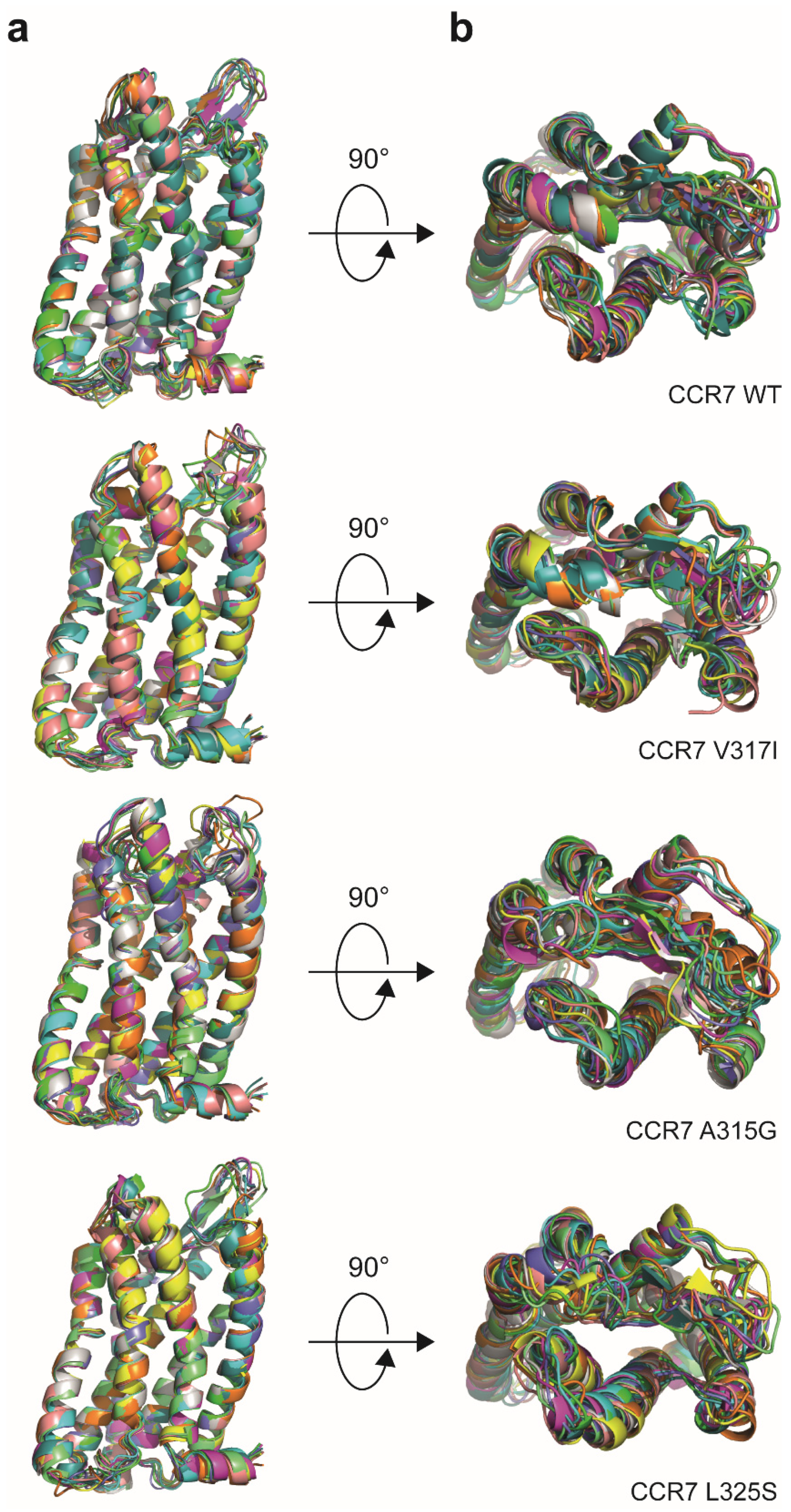
3.1. Reinvestigation of CCR7 Dimerisation: Forced versus More Dynamic Dimerisation
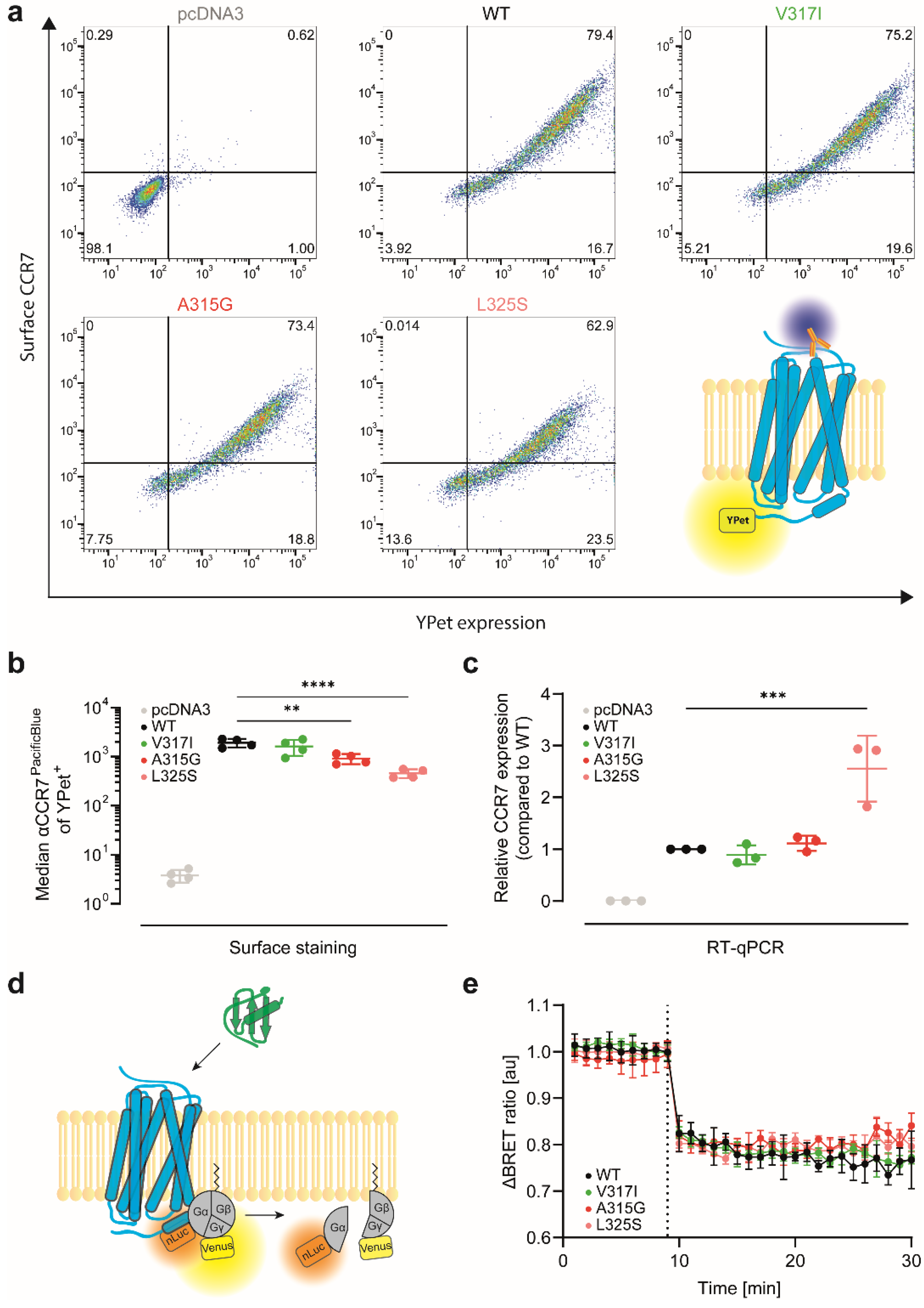
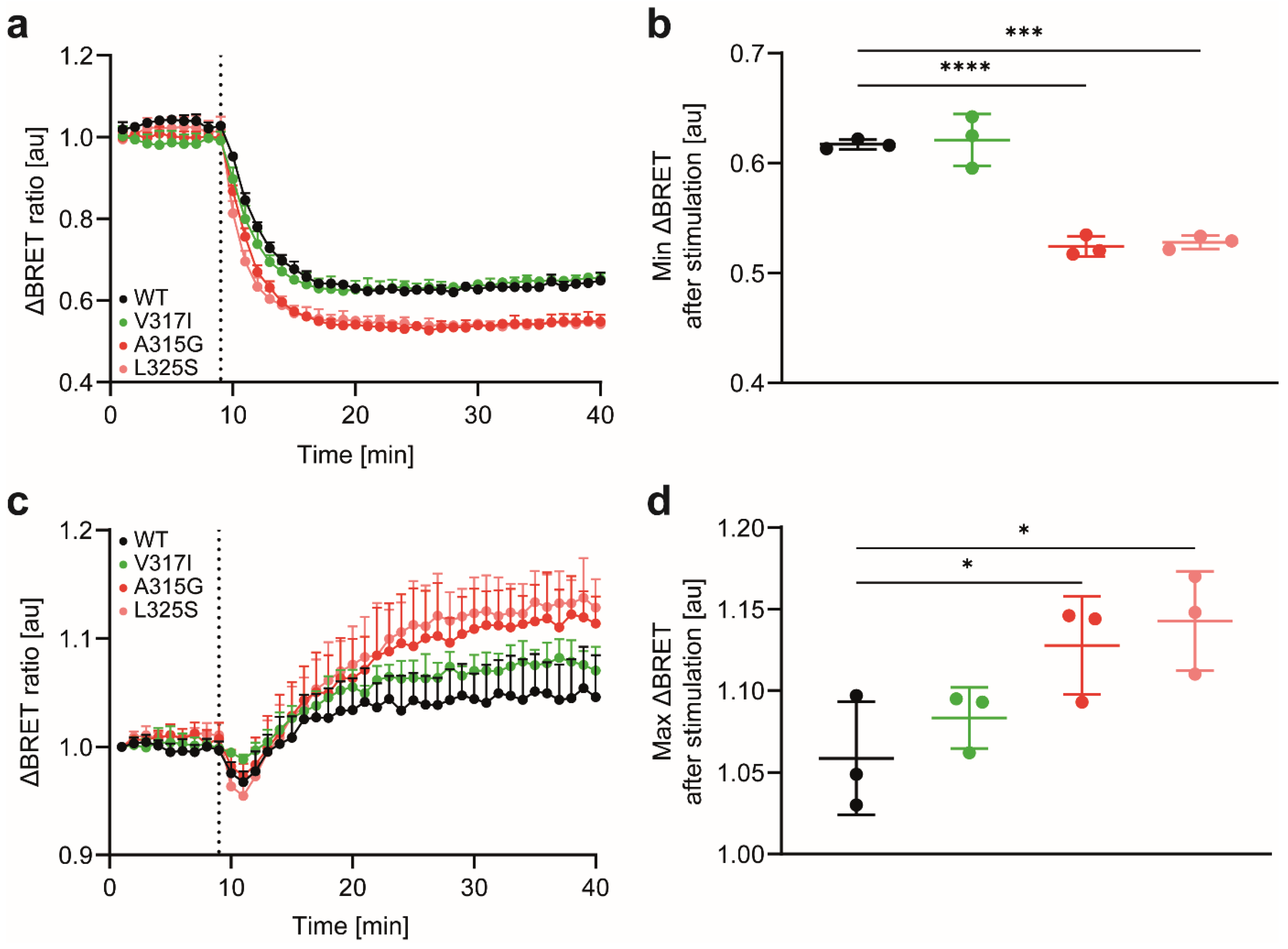
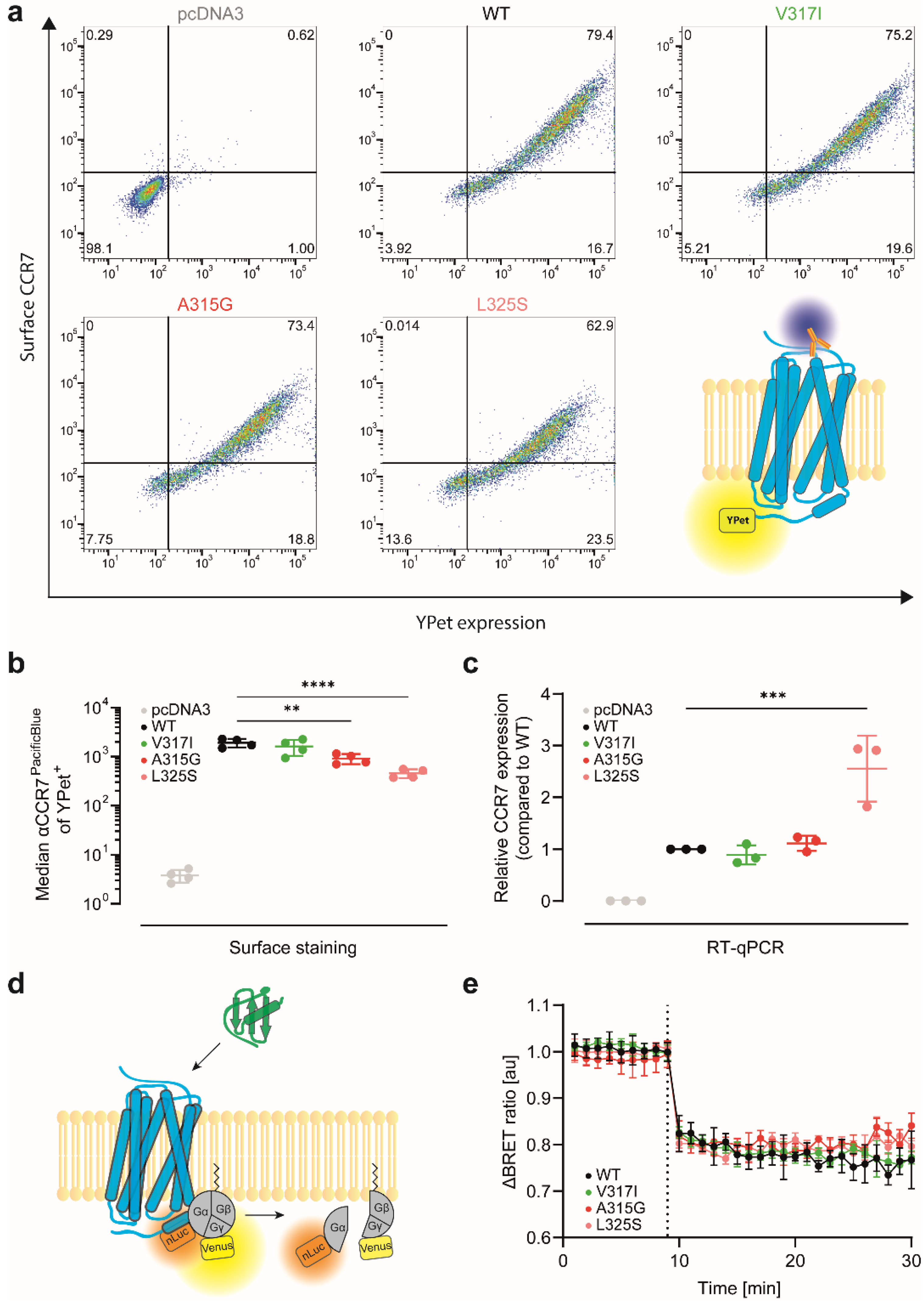
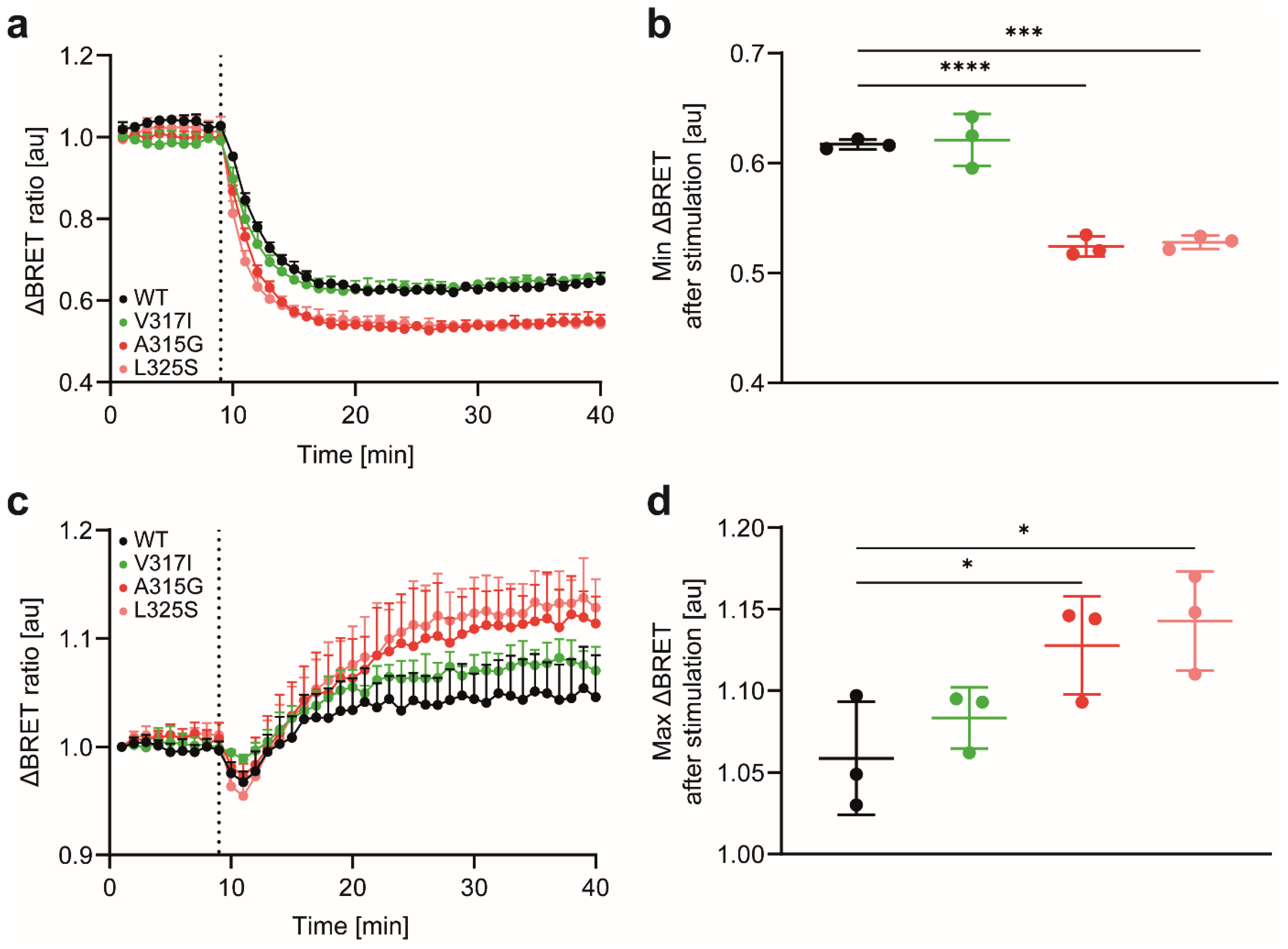
3.2. CCR7 Dimerisation Mutants Are Expressed at the Cell Surface and Activate Gi-Proteins
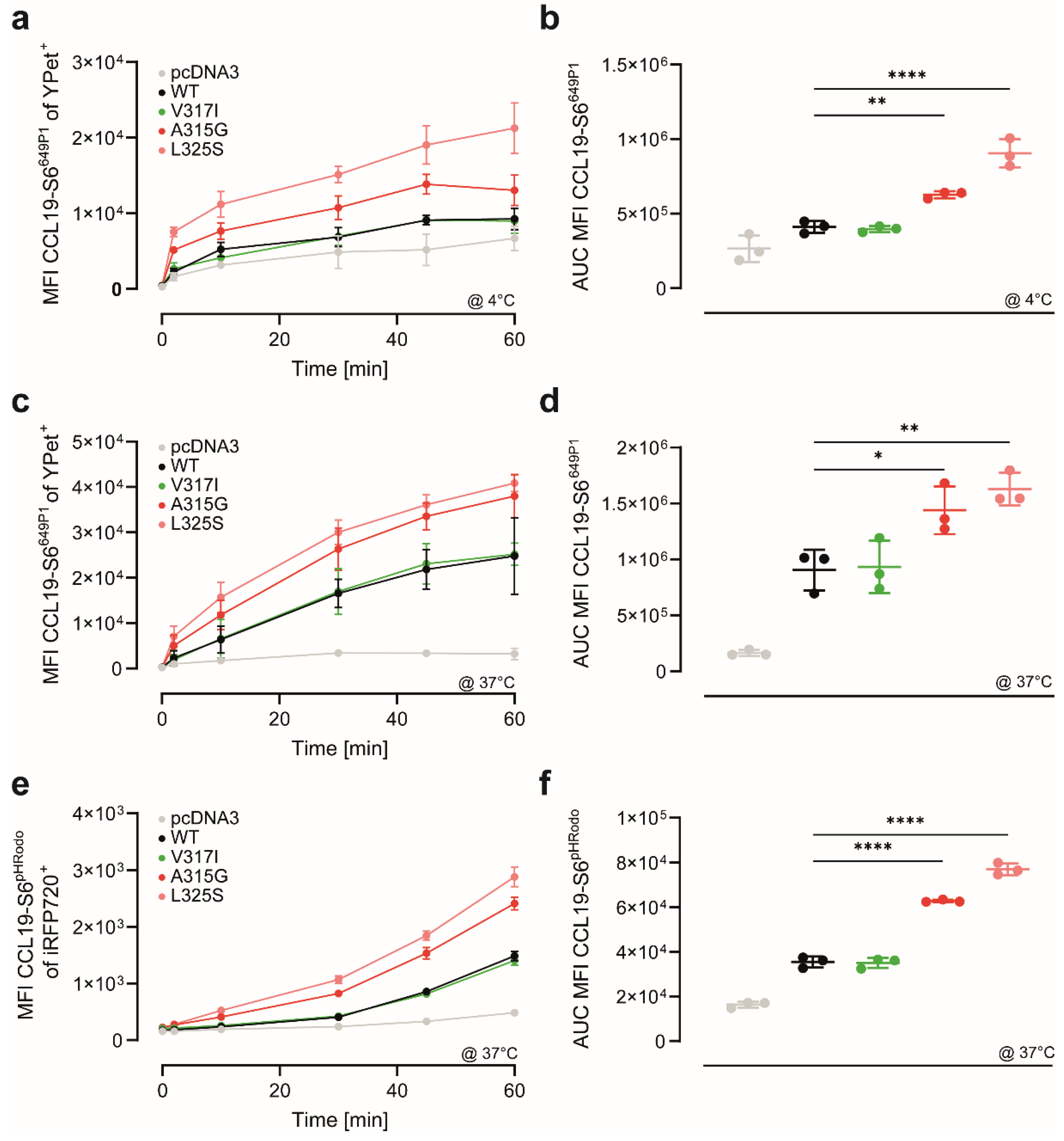
3.3. CCR7 Dimerisation-Defective Mutants Are Superior in CCL19 Binding and Uptake
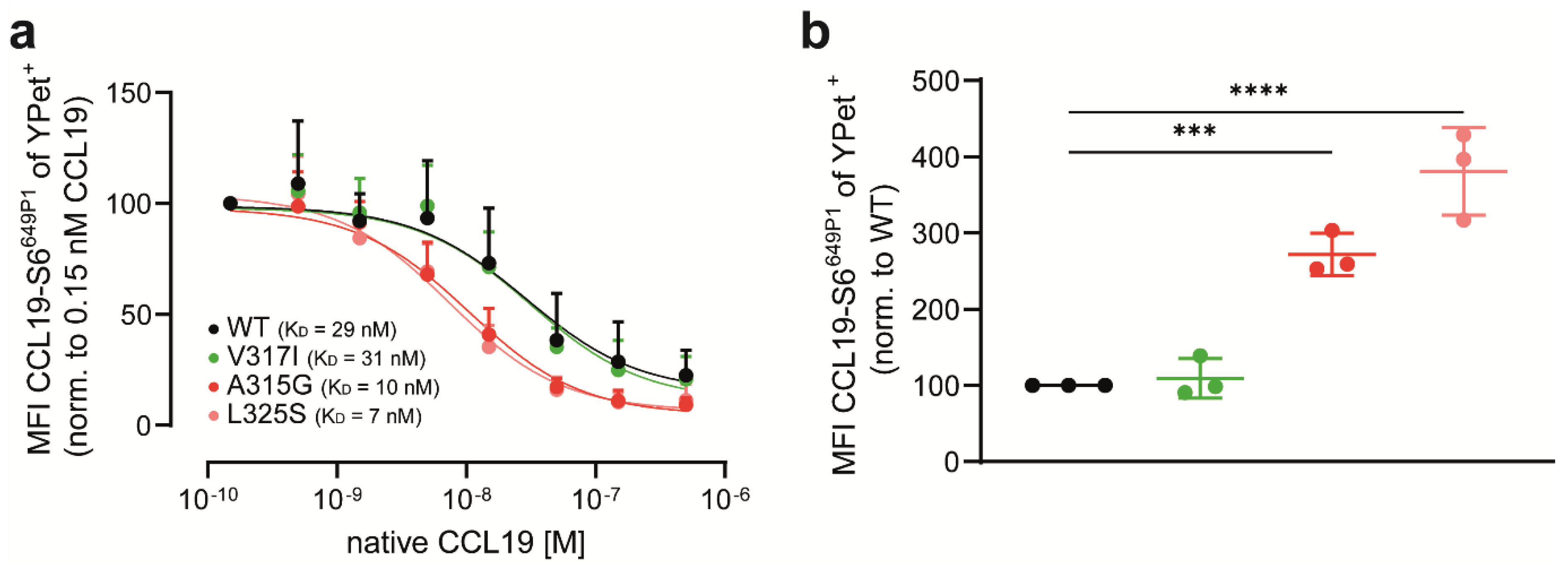
3.4. CCL19 Binds with Higher Affinities to CCR7 Dimerisation-Defective Mutants
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [Green Version]
- Legler, D.F.; Thelen, M. New insights in chemokine signaling. F1000Research 2018, 7, 95. [Google Scholar] [CrossRef]
- Comerford, I.; Harata-Lee, Y.; Bunting, M.D.; Gregor, C.; Kara, E.E.; McColl, S.R. A myriad of functions and complex regulation of the CCR7/CCL19/CCL21 chemokine axis in the adaptive immune system. Cytokine Growth Factor Rev. 2013, 24, 269–283. [Google Scholar] [CrossRef]
- Hauser, M.A.; Legler, D.F. Common and biased signaling pathways of the chemokine receptor CCR7 elicited by its ligands CCL19 and CCL21 in leukocytes. J. Leukoc. Biol. 2016, 99, 869–882. [Google Scholar] [CrossRef] [Green Version]
- Legler, D.F.; Uetz-von Allmen, E.; Hauser, M.A. CCR7: Roles in cancer cell dissemination, migration and metastasis formation. Int. J. Biochem. Cell Biol. 2014, 54C, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Cuesta-Mateos, C.; Brown, J.R.; Terron, F.; Munoz-Calleja, C. Of Lymph Nodes and CLL Cells: Deciphering the Role of CCR7 in the Pathogenesis of CLL and Understanding Its Potential as Therapeutic Target. Front. Immunol. 2021, 12, 662866. [Google Scholar] [CrossRef]
- Hauser, M.A.; Schaeuble, K.; Kindinger, I.; Impellizzieri, D.; Krueger, W.A.; Hauck, C.R.; Boyman, O.; Legler, D.F. Inflammation-Induced CCR7 Oligomers Form Scaffolds to Integrate Distinct Signaling Pathways for Efficient Cell Migration. Immunity 2016, 44, 59–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legler, D.F.; Matti, C.; Laufer, J.M.; Jakobs, B.D.; Purvanov, V.; Uetz-von Allmen, E.; Thelen, M. Modulation of Chemokine Receptor Function by Cholesterol: New Prospects for Pharmacological Intervention. Mol. Pharmacol. 2017, 91, 331–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellado, M.; Rodriguez-Frade, J.M.; Vila-Coro, A.J.; Fernandez, S.; Martin de Ana, A.; Jones, D.R.; Toran, J.L.; Martinez, A.C. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 2001, 20, 2497–2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanpain, C.; Vanderwinden, J.M.; Cihak, J.; Wittamer, V.; Le Poul, E.; Issafras, H.; Stangassinger, M.; Vassart, G.; Marullo, S.; Schlndorff, D.; et al. Multiple active states and oligomerization of CCR5 revealed by functional properties of monoclonal antibodies. Mol. Biol. Cell 2002, 13, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, B.; Handel, T.M. Chemokine receptor oligomerization and allostery. Prog. Mol. Biol. Transl. Sci. 2013, 115, 375–420. [Google Scholar] [PubMed] [Green Version]
- Terrillon, S.; Bouvier, M. Roles of G-protein-coupled receptor dimerization. EMBO Rep. 2004, 5, 30–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Momboisse, F.; Boncompain, G.; Koensgen, F.; Zhou, Z.; Cordeiro, N.; Arenzana-Seisdedos, F.; Perez, F.; Lagane, B.; Kellenberger, E.; et al. CCR5 adopts three homodimeric conformations that control cell surface delivery. Sci. Signal. 2018, 11, 529. [Google Scholar] [CrossRef] [Green Version]
- Laufer, J.M.; Hauser, M.A.; Kindinger, I.; Purvanov, V.; Pauli, A.; Legler, D.F. Chemokine Receptor CCR7 Triggers an Endomembrane Signaling Complex for Spatial Rac Activation. Cell Rep. 2019, 29, 995–1009.e6. [Google Scholar] [CrossRef] [Green Version]
- Uetz-von Allmen, E.; Rippl, A.V.; Farhan, H.; Legler, D.F. A unique signal sequence of the chemokine receptor CCR7 promotes package into COPII vesicles for efficient receptor trafficking. J. Leukoc. Biol. 2018, 104, 375–389. [Google Scholar] [CrossRef] [Green Version]
- Otero, C.; Groettrup, M.; Legler, D.F. Opposite Fate of Endocytosed CCR7 and Its Ligands: Recycling versus Degradation. J. Immunol. 2006, 177, 2314–2323. [Google Scholar] [CrossRef] [Green Version]
- Schaeuble, K.; Hauser, M.A.; Rippl, A.V.; Bruderer, R.; Otero, C.; Groettrup, M.; Legler, D.F. Ubiquitylation of the chemokine receptor CCR7 enables efficient receptor recycling and cell migration. J. Cell Sci. 2012, 125 Pt 19, 4463–4474. [Google Scholar] [CrossRef] [Green Version]
- Matti, C.; D’Uonnolo, G.; Artinger, M.; Melgrati, S.; Salnikov, A.; Thelen, S.; Purvanov, V.; Strobel, T.D.; Spannagel, L.; Thelen, M.; et al. CCL20 is a novel ligand for the scavenging atypical chemokine receptor 4. J. Leukoc. Biol. 2020, 107, 1137–1154. [Google Scholar] [CrossRef]
- Artinger, M.; Matti, C.; Gerken, O.J.; Veldkamp, C.T.; Legler, D.F. A Versatile Toolkit for Semi-Automated Production of Fluorescent Chemokines to Study CCR7 Expression and Functions. Int. J. Mol. Sci. 2021, 22, 4158. [Google Scholar] [CrossRef]
- Matti, C.; Salnikov, A.; Artinger, M.; D’Agostino, G.; Kindinger, I.; Uguccioni, M.; Thelen, M.; Legler, D.F. ACKR4 Recruits GRK3 Prior to β-Arrestins but Can Scavenge Chemokines in the Absence of β-Arrestins. Front. Immunol. 2020, 11, 720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namkung, Y.; Le Gouill, C.; Lukashova, V.; Kobayashi, H.; Hogue, M.; Khoury, E.; Song, M.; Bouvier, M.; Laporte, S.A. Monitoring G protein-coupled receptor and beta-arrestin trafficking in live cells using enhanced bystander BRET. Nat. Commun. 2016, 7, 12178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loening, A.M.; Fenn, T.D.; Wu, A.M.; Gambhir, S.S. Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng. Des. Sel. PEDS 2006, 19, 391–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerppola, T.K. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 2006, 1, 1278–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koker, T.; Fernandez, A.; Pinaud, F. Characterization of Split Fluorescent Protein Variants and Quantitative Analyses of Their Self-Assembly Process. Sci. Rep. 2018, 8, 5344. [Google Scholar] [CrossRef] [Green Version]
- Dixon, A.S.; Schwinn, M.K.; Hall, M.P.; Zimmerman, K.; Otto, P.; Lubben, T.H.; Butler, B.L.; Binkowski, B.F.; Machleidt, T.; Kirkland, T.A.; et al. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem. Biol. 2016, 11, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Stephens, B.S.; Holden, L.G.; Qin, L.; Zhao, C.; Kawamura, T.; Abagyan, R.; Handel, T.M. Stoichiometry and geometry of the CXC chemokine receptor 4 complex with CXC ligand 12: Molecular modeling and experimental validation. Proc. Natl. Acad. Sci. USA 2014, 111, E5363–E5372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldkamp, C.T.; Kiermaier, E.; Gabel-Eissens, S.J.; Gillitzer, M.L.; Lippner, D.R.; DiSilvio, F.A.; Mueller, C.J.; Wantuch, P.L.; Chaffee, G.R.; Famiglietti, M.W.; et al. Solution structure of CCL19 and identification of overlapping CCR7 and PSGL-1 binding sites. Biochemistry 2015, 54, 4163–4166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeger, K.; Bruenle, S.; Weinert, T.; Guba, W.; Muehle, J.; Miyazaki, T.; Weber, M.; Furrer, A.; Haenggi, N.; Tetaz, T.; et al. Structural Basis for Allosteric Ligand Recognition in the Human CC Chemokine Receptor 7. Cell 2019, 178, 1222–1230.e10. [Google Scholar] [CrossRef] [Green Version]
- Kufareva, I.; Gustavsson, M.; Zheng, Y.; Stephens, B.S.; Handel, T.M. What Do Structures Tell Us About Chemokine Receptor Function and Antagonism? Annu. Rev. Biophys. 2017, 46, 175–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerken, O.J.; Artinger, M.; Legler, D.F. Shifting CCR7 towards Its Monomeric Form Augments CCL19 Binding and Uptake. Cells 2022, 11, 1444. https://doi.org/10.3390/cells11091444
Gerken OJ, Artinger M, Legler DF. Shifting CCR7 towards Its Monomeric Form Augments CCL19 Binding and Uptake. Cells. 2022; 11(9):1444. https://doi.org/10.3390/cells11091444
Chicago/Turabian StyleGerken, Oliver J., Marc Artinger, and Daniel F. Legler. 2022. "Shifting CCR7 towards Its Monomeric Form Augments CCL19 Binding and Uptake" Cells 11, no. 9: 1444. https://doi.org/10.3390/cells11091444
APA StyleGerken, O. J., Artinger, M., & Legler, D. F. (2022). Shifting CCR7 towards Its Monomeric Form Augments CCL19 Binding and Uptake. Cells, 11(9), 1444. https://doi.org/10.3390/cells11091444