
How CAR T Cells Breathe

 , and
, and 
Abstract
:1. Introduction
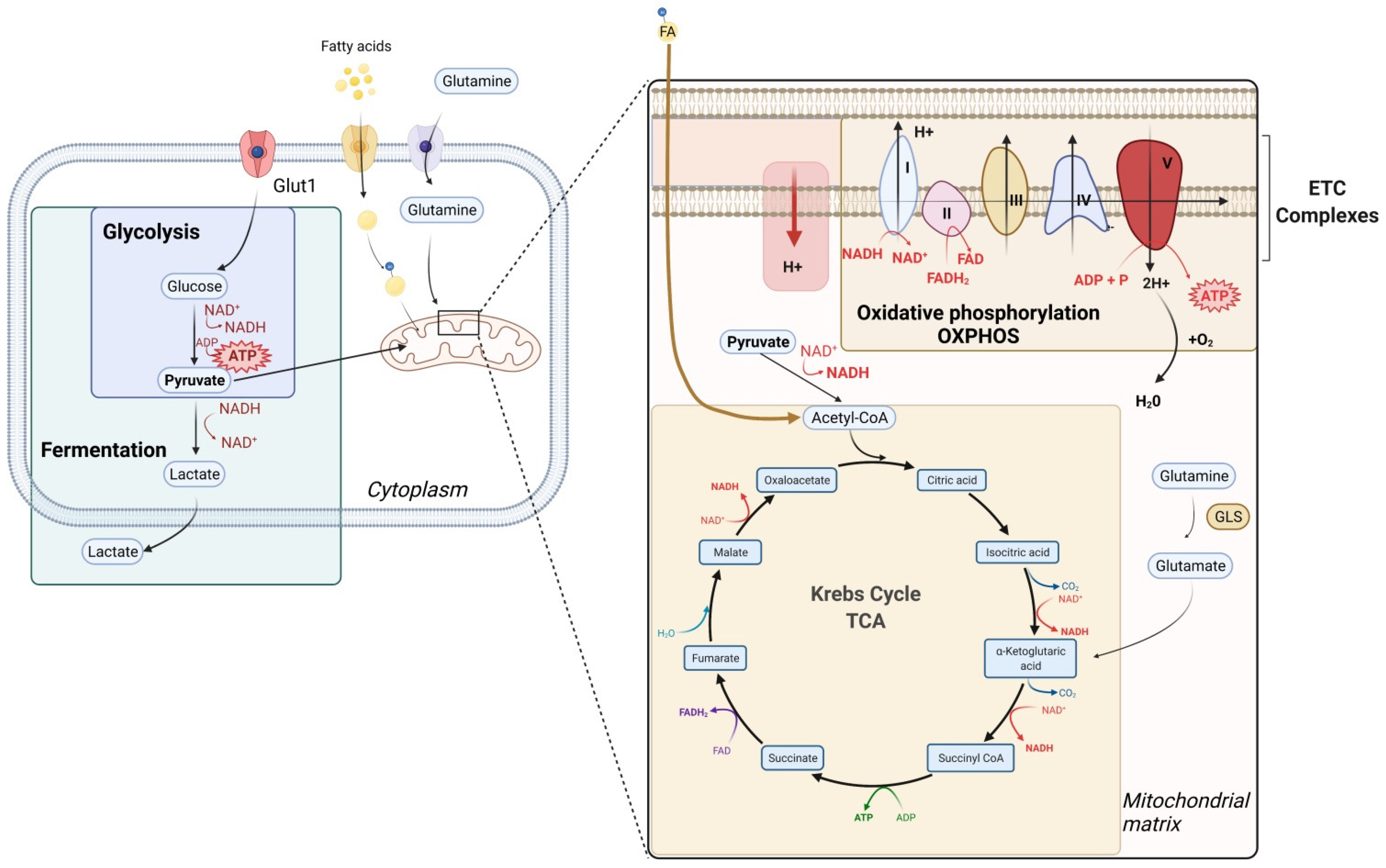
2. General Considerations on Metabolism
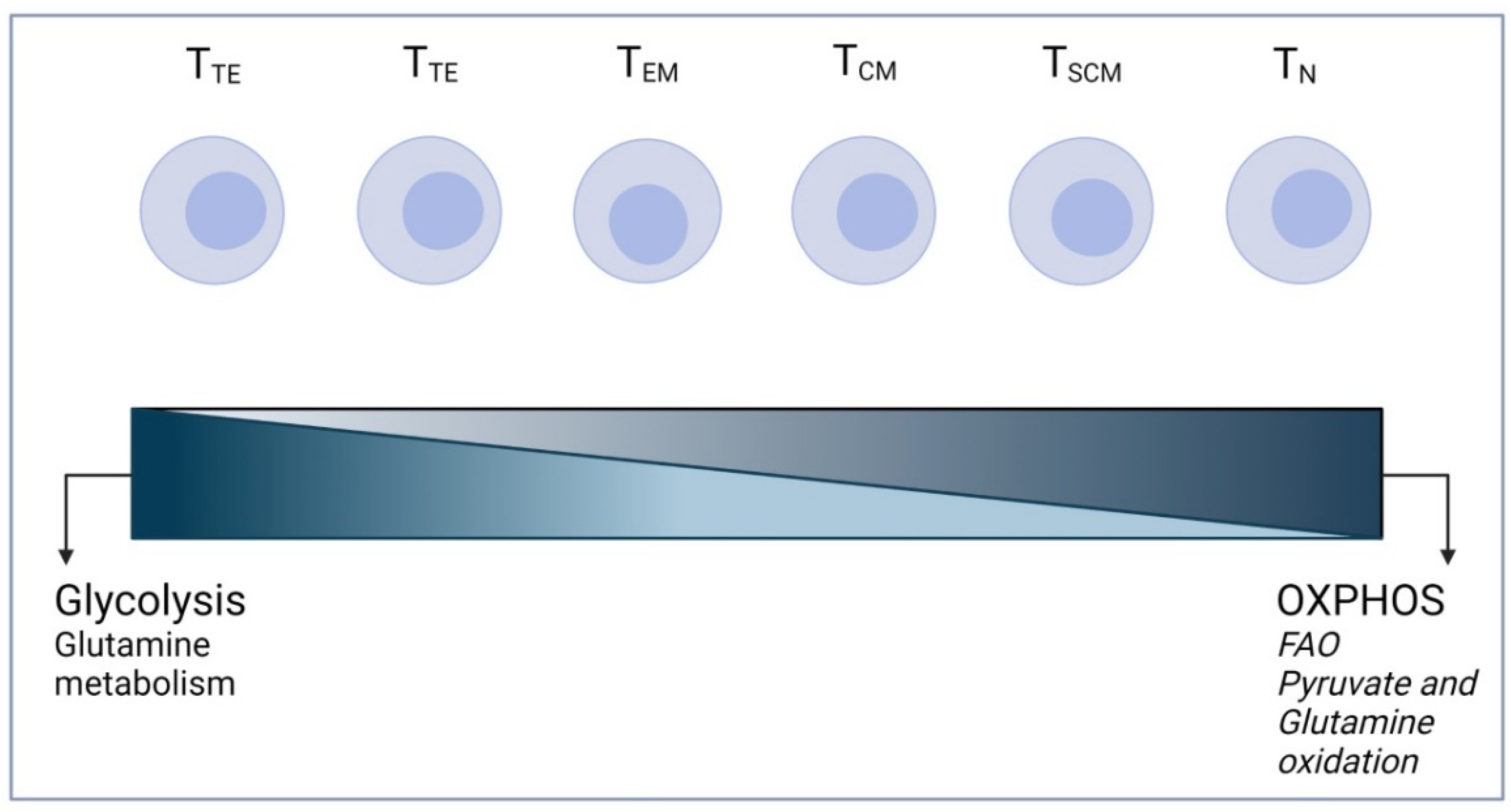
3. T Cell Metabolism
4. Importance of Studying CAR T Cell Metabolism
5. Study of Metabolism in CAR T Cells
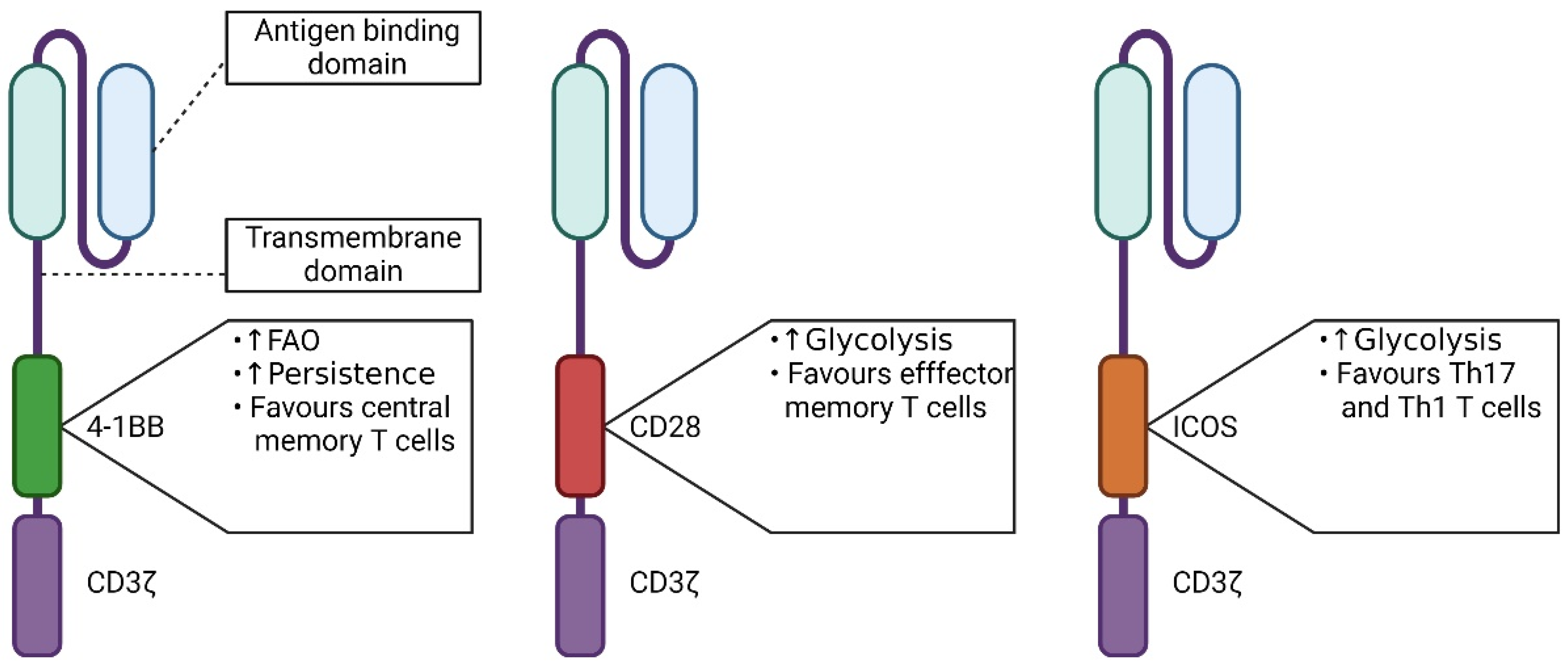
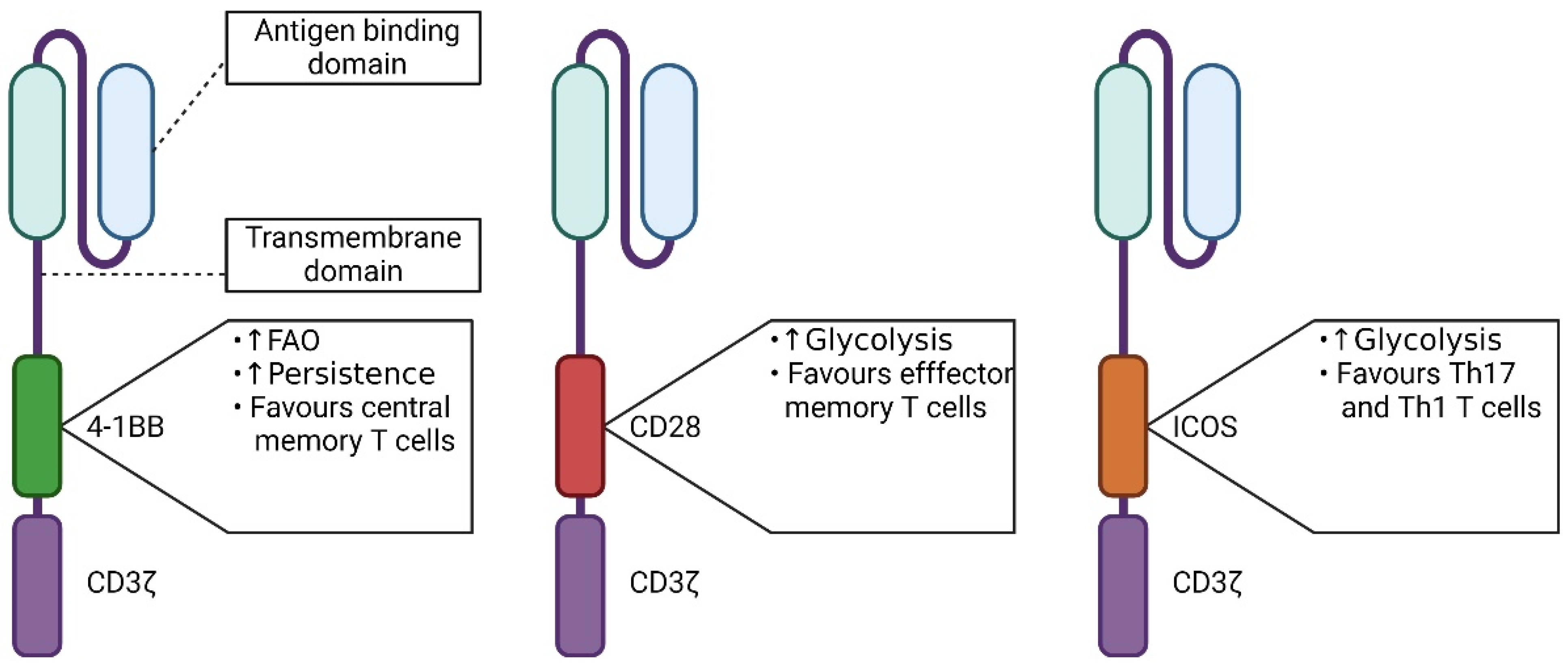
5.1. The Influence of the Signaling Domain
5.2. The Influence of the CAR Design
5.3. Polarization of CAR T Cells
5.4. Effect of the Combination of CAR T Cells and Combinatorial Designs
5.5. CAR Modified to Enhance T Cell Resistance to TME
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clark, L.C.; Wolf, R.; Granger, D.; Taylor, Z. Continuous Recording of Blood Oxygen Tensions by Polarography. J. Appl. Physiol. 1953, 6, 189–193. [Google Scholar] [CrossRef]
- Wang, J. Electrochemical Glucose Biosensors. Chem. Rev. 2008, 108, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.C., Jr.; Lyons, C. Electrode Systems for Continuous Monitoring in Cardiovascular Surgery. Ann. N. Y. Acad. Sci. 1962, 102, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Bittner, C.; Loaiza, A.; Ruminot, I.; Larenas, V.; Sotelo-Hitschfe, T.; Gutiérrez, R.; Córdova, A.; Valdebenito, R.; Frommer, W.; Barros, L.F. High Resolution Measurement of the Glycolytic Rate. Front. Neuroenergetics 2010, 2, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takanaga, H.; Chaudhuri, B.; Frommer, W.B. GLUT1 and GLUT9 as the Major Contributors to Glucose Influx in HEPG2 Cells Identified by a High Sensitivity Intramolecular FRET Glucose Sensor. Biochim. Biophys. Acta 2008, 1778, 1091–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.P.; Ward, N.P.; DeNicola, G.M. Recent Advances in Cancer Metabolism: A Technological Perspective. Exp. Mol. Med. 2018, 50, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumsey, W.; Vanderkooi, J.; Wilson, D. Imaging of Phosphorescence: A Novel Method for Measuring Oxygen Distribution in Perfused Tissue. Science 1988, 241, 1649–1651. [Google Scholar] [CrossRef] [PubMed]
- Dmitriev, R.I.; Papkovsky, D.B. Optical Probes and Techniques for O2 Measurement in Live Cells and Tissue. Cell Mol. Life Sci. 2012, 69, 2025–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argüello, R.J.; Combes, A.J.; Char, R.; Gigan, J.-P.; Baaziz, A.I.; Bousiquot, E.; Camosseto, V.; Samad, B.; Tsui, J.; Yan, P.; et al. SCENITH: A Flow Cytometry Based Method to Functionally Profile Energy Metabolism with Single Cell Resolution. Cell Metab. 2020, 32, 1063–1075.e7. [Google Scholar] [CrossRef]
- Horan, M.P.; Pichaud, N.; Ballard, J.W.O. Review: Quantifying Mitochondrial Dysfunction in Complex Diseases of Aging. J. Gerontol. Ser. A 2012, 67, 1022–1035. [Google Scholar] [CrossRef] [Green Version]
- Bhagavan, N.V. Chapter 13-Carbohydrate Metabolism I: Glycolysis and TCA Cycle. In Medical Biochemistry, 4th ed.; Blanco, A., Blanco, G., Eds.; Academic Press: Washington, DC, USA, 2017; pp. 275–281. ISBN 978-0-12-803550-4. [Google Scholar]
- Almeida, L.; Lochner, M.; Berod, L.; Sparwasser, T. Metabolic Pathways in T Cell Activation and Lineage Differentiation. Semin. Immunol. 2016, 28, 514–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Patergnani, S.; Rimessi, A.; De Marchi, E.; Suski, J.M.; Bononi, A.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. ATP Synthesis and Storage. Purinergic Signal. 2012, 8, 343–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigoulet, M.; Bouchez, C.L.; Paumard, P.; Ransac, S.; Cuvellier, S.; Duvezin-Caubet, S.; Mazat, J.P.; Devin, A. Cell Energy Metabolism: An Update. Biochim. Biophys. Acta (BBA)-Bioenerg. 2020, 1861, 148276. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; Enerbäck, S. Lactate: The Ugly Duckling of Energy Metabolism. Nat. Metab. 2020, 2, 566–571. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.-C.; van der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Jones, W.; Bianchi, K. Aerobic Glycolysis: Beyond Proliferation. Front. Immunol. 2015, 6, 227. [Google Scholar] [CrossRef] [Green Version]
- Mertens, S.; Noll, T.; Spahr, R.; Krutzfeldt, A.; Piper, H.M. Energetic Response of Coronary Endothelial Cells to Hypoxia. Am. J. Physiol.-Heart Circ. Physiol. 1990, 258, H689–H694. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lochner, M.; Berod, L.; Sparwasser, T. Fatty Acid Metabolism in the Regulation of T Cell Function. Trends Immunol. 2015, 36, 81–91. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Yaqoob, P.; Calder, P.C. Glutamine Requirement of Proliferating T Lymphocytes. Nutrition 1997, 13, 646–651. [Google Scholar] [CrossRef]
- Sinclair, L.V.; Rolf, J.; Emslie, E.; Shi, Y.-B.; Taylor, P.M.; Cantrell, D.A. Control of Amino-Acid Transport by Antigen Receptors Coordinates the Metabolic Reprogramming Essential for T Cell Differentiation. Nat. Immunol. 2013, 14, 500–508. [Google Scholar] [CrossRef] [Green Version]
- Hope, H.C.; Salmond, R.J. The Role of Non-Essential Amino Acids in T Cell Function and Anti-Tumour Immunity. Arch. Immunol. Ther. Exp. 2021, 69, 29. [Google Scholar] [CrossRef]
- Mahnke, Y.D.; Brodie, T.M.; Sallusto, F.; Roederer, M.; Lugli, E. The Who’s Who of T-Cell Differentiation: Human Memory T-Cell Subsets. Eur. J. Immunol. 2013, 43, 2797–2809. [Google Scholar] [CrossRef]
- Rangel Rivera, G.O.; Knochelmann, H.M.; Dwyer, C.J.; Smith, A.S.; Wyatt, M.M.; Rivera-Reyes, A.M.; Thaxton, J.E.; Paulos, C.M. Fundamentals of T Cell Metabolism and Strategies to Enhance Cancer Immunotherapy. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Saxena, A.; Dagur, P.K.; Biancotto, A. Multiparametric Flow Cytometry Analysis of Naïve, Memory, and Effector T Cells. In Immunophenotyping: Methods and Protocols; McCoy, J., Philip, J., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 129–140. ISBN 978-1-4939-9650-6. [Google Scholar]
- Jacobs, S.R.; Michalek, R.D.; Rathmell, J.C. IL-7 Is Essential for Homeostatic Control of T Cell Metabolism In Vivo. J. Immunol. 2010, 184, 3461–3469. [Google Scholar] [CrossRef]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic Regulation of T Lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef] [Green Version]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 Signaling Pathway Regulates Glucose Metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Pellegrino, M.; Del Bufalo, F.; De Angelis, B.; Quintarelli, C.; Caruana, I.; de Billy, E. Manipulating the Metabolism to Improve the Efficacy of CAR T-Cell Immunotherapy. Cells 2020, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Shrestha, S.; Zeng, H.; Karmaus, P.W.F.; Neale, G.; Vogel, P.; Guertin, D.A.; Lamb, R.F.; Chi, H. T Cell Exit from Quiescence and Differentiation into Th2 Cells Depend on Raptor-MTORC1-Mediated Metabolic Reprogramming. Immunity 2013, 39, 1043–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shyer, J.A.; Flavell, R.A.; Bailis, W. Metabolic Signaling in T Cells. Cell Res. 2020, 30, 649–659. [Google Scholar] [CrossRef]
- Johnson, M.O.; Wolf, M.M.; Madden, M.Z.; Andrejeva, G.; Sugiura, A.; Contreras, D.C.; Maseda, D.; Liberti, M.V.; Paz, K.; Kishton, R.J.; et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 2018, 175, 1780–1795.e19. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1α–Dependent Glycolytic Pathway Orchestrates a Metabolic Checkpoint for the Differentiation of TH17 and Treg Cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Raynor, J.; Nguyen, T.-L.M.; Chi, H. Nutrient and Metabolic Sensing in T Cell Responses. Front. Immunol. 2017, 8, 247. [Google Scholar] [CrossRef] [Green Version]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The Clinical Role of the TME in Solid Cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef]
- Rostamian, H.; Fallah-Mehrjardi, K.; Khakpoor-Koosheh, M.; Pawelek, J.M.; Hadjati, J.; Brown, C.E.; Mirzaei, H.R. A Metabolic Switch to Memory CAR T Cells: Implications for Cancer Treatment. Cancer Lett. 2021, 500, 107–118. [Google Scholar] [CrossRef]
- Kishton, R.J.; Sukumar, M.; Restifo, N.P. Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metab. 2017, 26, 94–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting Costimulatory Domains for Chimeric Antigen Receptors: Functional and Clinical Considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.; Cohen, S.; Guy, C.; Shrestha, S.; Neale, G.; Brown, S.A.; Cloer, C.; Kishton, R.J.; Gao, X.; Youngblood, B.; et al. MTORC1 and MTORC2 Kinase Signaling and Glucose Metabolism Drive Follicular Helper T Cell Differentiation. Immunity 2016, 45, 540–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedan, S.; Chen, X.; Madar, A.; Carpenito, C.; McGettigan, S.E.; Frigault, M.J.; Lee, J.; Posey, A.D.; Scholler, J.; Scholler, N.; et al. ICOS-Based Chimeric Antigen Receptors Program Bipolar TH17/TH1 Cells. Blood 2014, 124, 1070–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.K.; Lee, D.Y.; Lee, D.G.; Kim, Y.H.; Kim, S.-H.; Oh, H.S.; Han, C.; Kwon, B.S. 4-1BB Signaling Activates Glucose and Fatty Acid Metabolism to Enhance CD8+ T Cell Proliferation. Cell Mol. Immunol. 2017, 14, 748–757. [Google Scholar] [CrossRef]
- Teijeira, A.; Garasa, S.; Etxeberria, I.; Gato-Cañas, M.; Melero, I.; Delgoffe, G.M. Metabolic Consequences of T-Cell Costimulation in Anticancer Immunity. Cancer Immunol Res. 2019, 7, 1564–1569. [Google Scholar] [CrossRef]
- Li, W.; Qiu, S.; Chen, J.; Jiang, S.; Chen, W.; Jiang, J.; Wang, F.; Si, W.; Shu, Y.; Wei, P.; et al. Chimeric Antigen Receptor Designed to Prevent Ubiquitination and Downregulation Showed Durable Antitumor Efficacy. Immunity 2020, 53, 456–470.e6. [Google Scholar] [CrossRef]
- Liu, L.; Bi, E.; Ma, X.; Xiong, W.; Qian, J.; Ye, L.; Su, P.; Wang, Q.; Xiao, L.; Yang, M.; et al. Enhanced CAR-T Activity against Established Tumors by Polarizing Human T Cells to Secrete Interleukin-9. Nat. Commun. 2020, 11, 5920. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; O’Hear, C.E.; Alli, R.; Basham, J.H.; Abdelsamed, H.; Palmer, L.E.; Jones, L.L.; Youngblood, B.; Geiger, T.L. PI3K Orchestration of the in Vivo Persistence of Chimeric Antigen Receptor-Modified T Cells. Leukemia 2018, 32, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Perkins, M.R.; Grande, S.; Hamel, A.; Horton, H.M.; Garrett, T.E.; Miller, S.M.; Latimer, H.J., IV.; Horvath, C.J.; Kuczewski, M.; Friedman, K.M.; et al. Manufacturing an Enhanced CAR T Cell Product by Inhibition of the PI3K/Akt Pathway during T Cell Expansion Results in Improved In Vivo Efficacy of Anti-BCMA CAR T Cells. Blood 2015, 126, 1893. [Google Scholar] [CrossRef]
- Fultang, L.; Booth, S.; Yogev, O.; Martins da Costa, B.; Tubb, V.; Panetti, S.; Stavrou, V.; Scarpa, U.; Jankevics, A.; Lloyd, G.; et al. Metabolic Engineering against the Arginine Microenvironment Enhances CAR-T Cell Proliferation and Therapeutic Activity. Blood 2020, 136, 1155–1160. [Google Scholar] [CrossRef]
- Li, W.; Zhang, L. Rewiring Mitochondrial Metabolism for CD8+ T Cell Memory Formation and Effective Cancer Immunotherapy. Front. Immunol. 2020, 11, 1834. [Google Scholar] [CrossRef]
- Rostamian, H.; Khakpoor-Koosheh, M.; Fallah-Mehrjardi, K.; Mirzaei, H.R.; Brown, C.E. Mitochondria as Playmakers of CAR T-Cell Fate and Longevity. Cancer Immunol. Res. 2021, 9, 856–861. [Google Scholar] [CrossRef]
- Ferrick, D.A.; Neilson, A.; Beeson, C. Advances in Measuring Cellular Bioenergetics Using Extracellular Flux. Drug Discov. Today 2008, 13, 268–274. [Google Scholar] [CrossRef]
- Lardy, H.A.; Johnson, D.; McMurray, W.C. Antibiotics as Tools for Metabolic Studies. I. A Survey of Toxic Antibiotics in Respiratory, Phosphorylative and Glycolytic Systems. Arch. Biochem. Biophys. 1958, 78, 587–597. [Google Scholar] [CrossRef]
- Öberg, K.E. The Site of the Action of Rotenone in the Respiratory Chain. Exp. Cell Res. 1961, 24, 163–164. [Google Scholar] [CrossRef]
- Kim, H.; Esser, L.; Hossain, M.B.; Xia, D.; Yu, C.-A.; Rizo, J.; Van Der Helm, D.; Deisenhofer, J. Structure of Antimycin A1, a Specific Electron Transfer Inhibitor of Ubiquinol−Cytochrome c Oxidoreductase. Available online: https://pubs.acs.org/doi/pdf/10.1021/ja990190h (accessed on 7 September 2021).
- Baur, J.R.; Workman, M. Influence of Carbonyl Cyanide Phenylhydrazone Derivatives on the Respiration Rate of Banana Pulp Tissue. Nature 1964, 201, 612. [Google Scholar] [CrossRef]
- Wu, M.; Neilson, A.; Swift, A.L.; Moran, R.; Tamagnine, J.; Parslow, D.; Armistead, S.; Lemire, K.; Orrell, J.; Teich, J.; et al. Multiparameter Metabolic Analysis Reveals a Close Link between Attenuated Mitochondrial Bioenergetic Function and Enhanced Glycolysis Dependency in Human Tumor Cells. Am. J. Physiol. Cell Physiol. 2007, 292, C125–C136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Liu, W.; Zhou, S.; Yang, C.; Tian, M.; Jia, G.; Wang, H.; Zhu, B.; Feng, M.; Lu, Y.; et al. Identification of FABP5 as an Immunometabolic Marker in Human Hepatocellular Carcinoma. J. Immunother. Cancer 2020, 8, e000501. [Google Scholar] [CrossRef] [PubMed]
- Köksal, H.; Dillard, P.; Juzeniene, A.; Kvalheim, G.; Smeland, E.B.; Myklebust, J.H.; Inderberg, E.M.; Wälchli, S. Combinatorial CAR Design Improves Target Restriction. J. Biol. Chem. 2020, 100116. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, B.; Muthugounder, S.; Jambon, S.; Tibbetts, R.; Hung, L.; Bassiri, H.; Hogarty, M.D.; Barrett, D.M.; Shimada, H.; Asgharzadeh, S. Preclinical Assessment of the Efficacy and Specificity of GD2-B7H3 SynNotch CAR-T in Metastatic Neuroblastoma. Nat. Commun. 2021, 12, 511. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, C.; Novakovsky, G.E.; Qi, C.N.; Weber, E.W.; Mackall, C.L.; Levings, M.K. Repeated Stimulation or Tonic-Signaling Chimeric Antigen Receptors Drive Regulatory T Cell Exhaustion. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hirabayashi, K.; Du, H.; Xu, Y.; Shou, P.; Zhou, X.; Fucá, G.; Landoni, E.; Sun, C.; Chen, Y.; Savoldo, B.; et al. Dual-Targeting CAR-T Cells with Optimal Co-Stimulation and Metabolic Fitness Enhance Antitumor Activity and Prevent Escape in Solid Tumors. Nat. Cancer 2021, 2, 904–918. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, Y.; Wang, S.; Du, J.; Gao, X.; Yuan, Y.; Zhao, L.; Yang, Y.; Xu, L.; Lei, Y.; et al. Secretion of Human Soluble Programmed Cell Death Protein 1 by Chimeric Antigen Receptor-Modified T Cells Enhances Anti-Tumor Efficacy. Cytotherapy 2020, 22, 734–743. [Google Scholar] [CrossRef]
- Serganova, I.; Moroz, E.; Cohen, I.; Moroz, M.; Mane, M.; Zurita, J.; Shenker, L.; Ponomarev, V.; Blasberg, R. Enhancement of PSMA-Directed CAR Adoptive Immunotherapy by PD-1/PD-L1 Blockade. Mol. Ther. Oncolytics 2017, 4, 41–54. [Google Scholar] [CrossRef]
- Sabatino, M.; Hu, J.; Sommariva, M.; Gautam, S.; Fellowes, V.; Hocker, J.D.; Dougherty, S.; Qin, H.; Klebanoff, C.A.; Fry, T.J.; et al. Generation of Clinical-Grade CD19-Specific CAR-Modified CD8+ Memory Stem Cells for the Treatment of Human B-Cell Malignancies. Blood 2016, 128, 519–528. [Google Scholar] [CrossRef]
- Garcia-Canaveras, J.C.; Heo, D.; Trefely, S.; Leferovich, J.; Xu, C.; Philipson, B.I.; Ghassemi, S.; Milone, M.C.; Moon, E.K.; Snyder, N.W.; et al. CAR T-Cells Depend on the Coupling of NADH Oxidation with ATP Production. Cells 2021, 10, 2334. [Google Scholar] [CrossRef]
- Yang, Q.; Hao, J.; Chi, M.; Wang, Y.; Li, J.; Huang, J.; Zhang, J.; Zhang, M.; Lu, J.; Zhou, S.; et al. D2HGDH-Mediated D2HG Catabolism Enhances the Anti-Tumor Activities of CAR-T Cells in an Immunosuppressive Microenvironment. Mol. Ther. 2022. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Wall, S.R.; Olley, P.M.; Davies, N.J. Etomoxir, a Carnitine Palmitoyltransferase I Inhibitor, Protects Hearts from Fatty Acid-Induced Ischemic Injury Independent of Changes in Long Chain Acylcarnitine. Circ. Res. 1988, 63, 1036–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
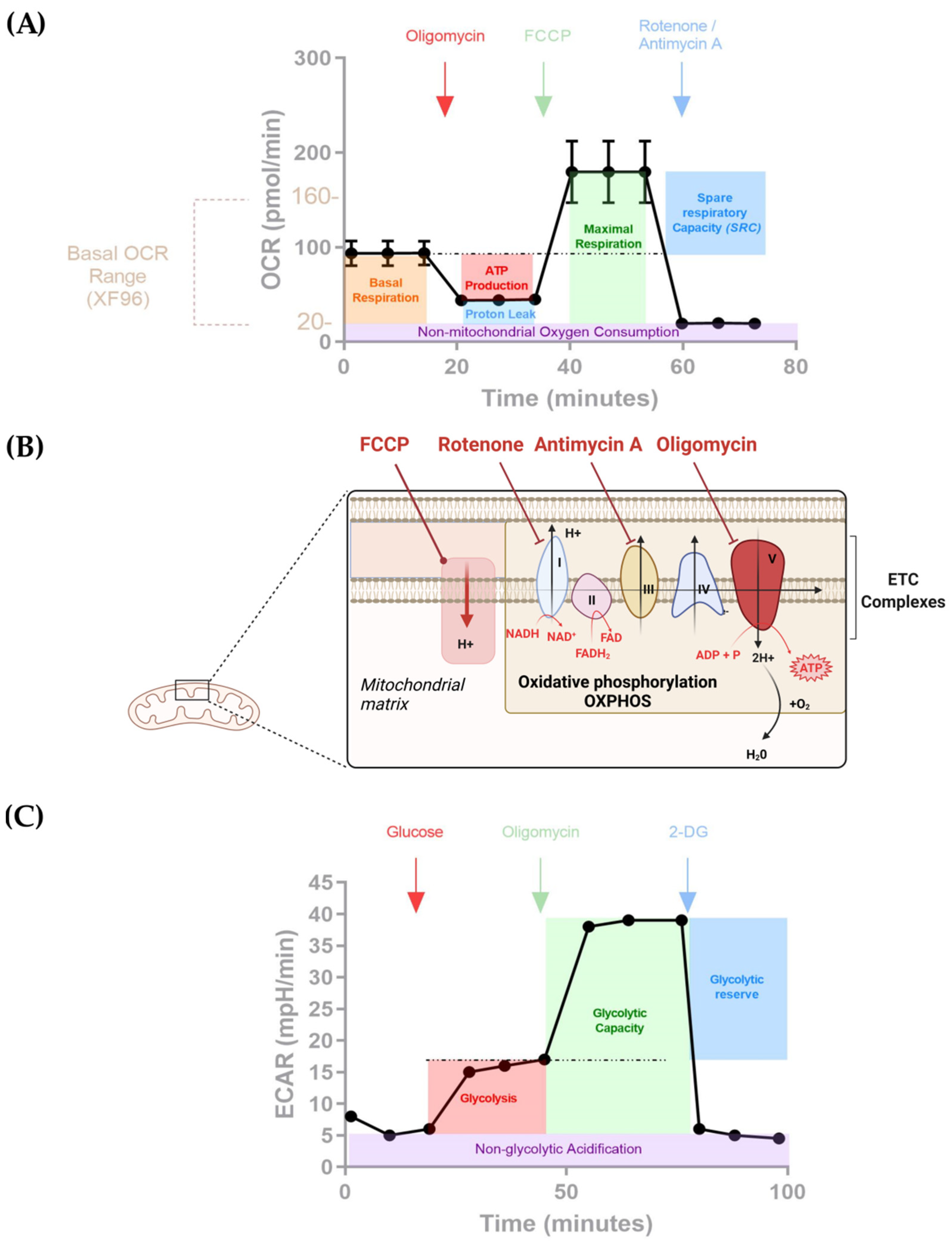{kind=link}
| CAR | Seahorse | Reference | ||||
|---|---|---|---|---|---|---|
| Seahorse XF | Kit | Drugs | Number of Cells/Wells | Cell Stimulation | ||
| CD19 CAR with a CD28 co-stimulatory domain/CD19 CAR with a 4-1BB co-stimulatory domain and Mesothelin CD28-CAR/Mesothelin 4-1BB CAR | Seahorse XF24 and 96 | Cell Mito Stress kit | Oligomycin/FCCP/Rotenone Antimycin A | 1 × 106 | Before/After anti-idiotype stimulation (for 7 days and 21 days) | [49] |
| CD19 CAR with or without TSCM-enrichment | Seahorse XF24 | Cell Mito Stress kit - Glycolysis Stress kit | Oligomycin/FCCP/Rotenone Antimycin A - Oligo/Glucose/2DG | 1 × 106 | Not specified | [72] |
| Anti-hPSMA CAR/Untransduced T cells | Seahorse XF96 | Cell Mito Stress kit - Glycolysis Stress kit | Oligomycin/FCCP/Rotenone Antimycin A - Oligo/Glucose/2DG | 3.5 × 105 | Phytohaemagglutinin (PHA) Stimulation | [71] |
| Mesothelin CD28-CAR/Mesothelin 4-1BB CAR identification | Seahorse XF96 | Cell Mito Stress kit | Oligomycin/FCCP/Rotenone Antimycin A + Etomixir | 1.5 × 105 | Not specified | [65] |
| CD19 CAR/CD19 CAR with ubiquitination blocked by mutating all lysines in the CAR cytoplasmic domain | Seahorse XF24 | Cell Mito Stress kit | Oligomycin/FCCP/Rotenone Antimycin A | 1.5 × 105 | After stimulation with irradiated target cells for 14 days | [52] |
| CD19 CAR/CD19 CAR releasing human soluble PD-1 protein (sPD-1 CAR T) /Untransduced T cells | Seahorse XF96 | Cell Mito Stress kit | Oligomycin/FCCP/Rotenone Antimycin A | Not specified | Before/After 48h in co-culture with target tumor cells | [70] |
| GD2 CAR/CD19 CAR/Untransduced T cells | Seahorse XF96 | Cell Mito Stress kit | Oligomycin/FCCP/Rotenone Antimycin A | 2 × 105 | Not specified | [68] |
| IGK CAR/IGK CD-19 CAR | Seahorse XF96 | Cell Mito Stress kit | Oligomycin/FCCP/Rotenone Antimycin A Anti A | 1 × 105 | After stimulation with surface-coated specific (IgG) and unspecific (anti-CD3) antibodies | [66] |
| GD2 CAR/GD2-B7H3 CAR /Untransduced T cells | Seahorse XF24 | Cell Mito Stress kit | Oligomycin/FCCP/Rotenone Antimycin A | 1 × 106 | After 48h in coculture with target tumor cells | [67] |
| CD19 CAR Th9 or Th1 polarization | Seahorse XF24 | Cell Mito Stress kit | Oligomycin/FCCP/Rotenone Antimycin A | Not specified | Not specified | [53] |
| Mesothelin CD28 CAR with LbNOX or GFP | Seahorse XF96 | Cell Mito Stress Kit | Sodium–L–lactate/Rotenone Antimycin A | 2 × 105 | None | [73] |
| GD2 CAR with a CD28 co-stimulatory domain/GD2 CAR co-expressed with B7H3 with a 4-1BB co-stimulatory domain | Seahorse XF24 | Cell Mito Stress kit - Glycolysis Stress kit | Oligomycin/FCCP/Rotenone Antimycin A - Rotenone/Antimycin A/2DG | 5 × 105 | After stimulation with surface-coated specific immunoglobulin (1A7 mAb) and chimera (4Ig-B7-H3) for CAR activation | [69] |
| CD19 CAR with a 4-1BB co-stimulatory domain in D2HGDH knocked out or overexpressing T cells | Seahorse XF24 | Cell Mito Stress kit | Oligomycin/FCCP/Rotenone Antimycin A | 1 × 106 | None | [74] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forcados, C.; Joaquina, S.; Casey, N.P.; Caulier, B.; Wälchli, S. How CAR T Cells Breathe. Cells 2022, 11, 1454. https://doi.org/10.3390/cells11091454
Forcados C, Joaquina S, Casey NP, Caulier B, Wälchli S. How CAR T Cells Breathe. Cells. 2022; 11(9):1454. https://doi.org/10.3390/cells11091454
Chicago/Turabian StyleForcados, Christopher, Sandy Joaquina, Nicholas Paul Casey, Benjamin Caulier, and Sébastien Wälchli. 2022. "How CAR T Cells Breathe" Cells 11, no. 9: 1454. https://doi.org/10.3390/cells11091454
APA StyleForcados, C., Joaquina, S., Casey, N. P., Caulier, B., & Wälchli, S. (2022). How CAR T Cells Breathe. Cells, 11(9), 1454. https://doi.org/10.3390/cells11091454