
HSP90 Modulates T2R Bitter Taste Receptor Nitric Oxide Production and Innate Immune Responses in Human Airway Epithelial Cells and Macrophages

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Live-Cell Imaging of Ca2+, NO, and cGMP
2.3. Measurement of Ciliary Beat Frequency (CBF)
2.4. Bacteria Culture
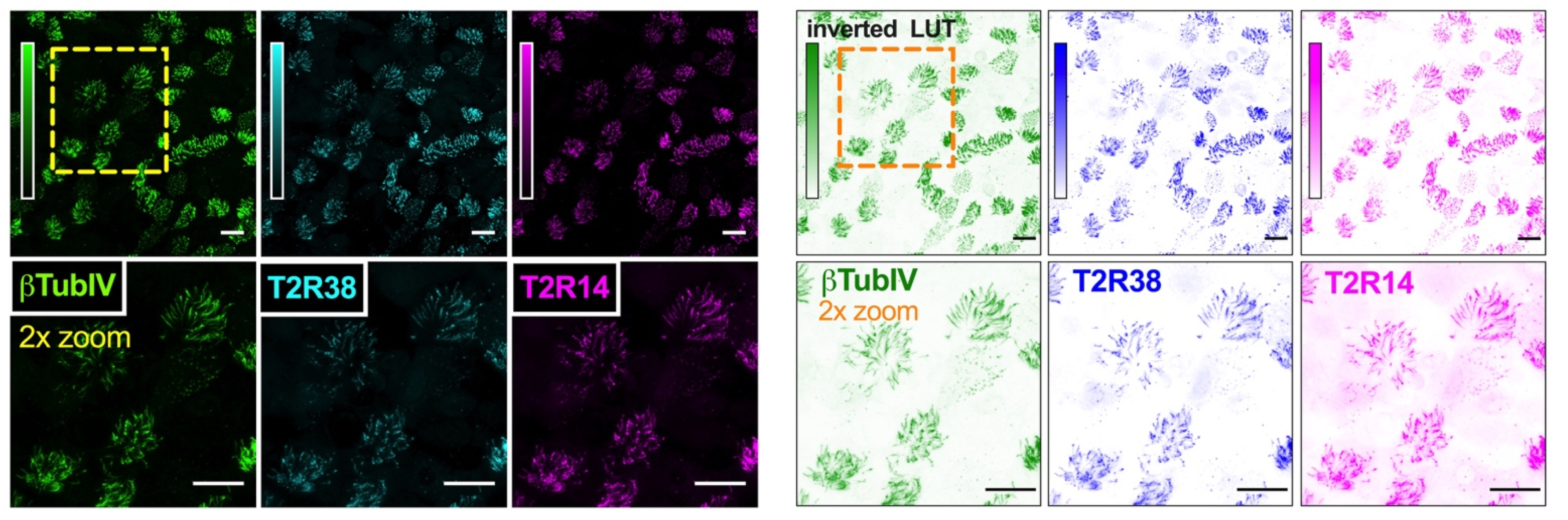
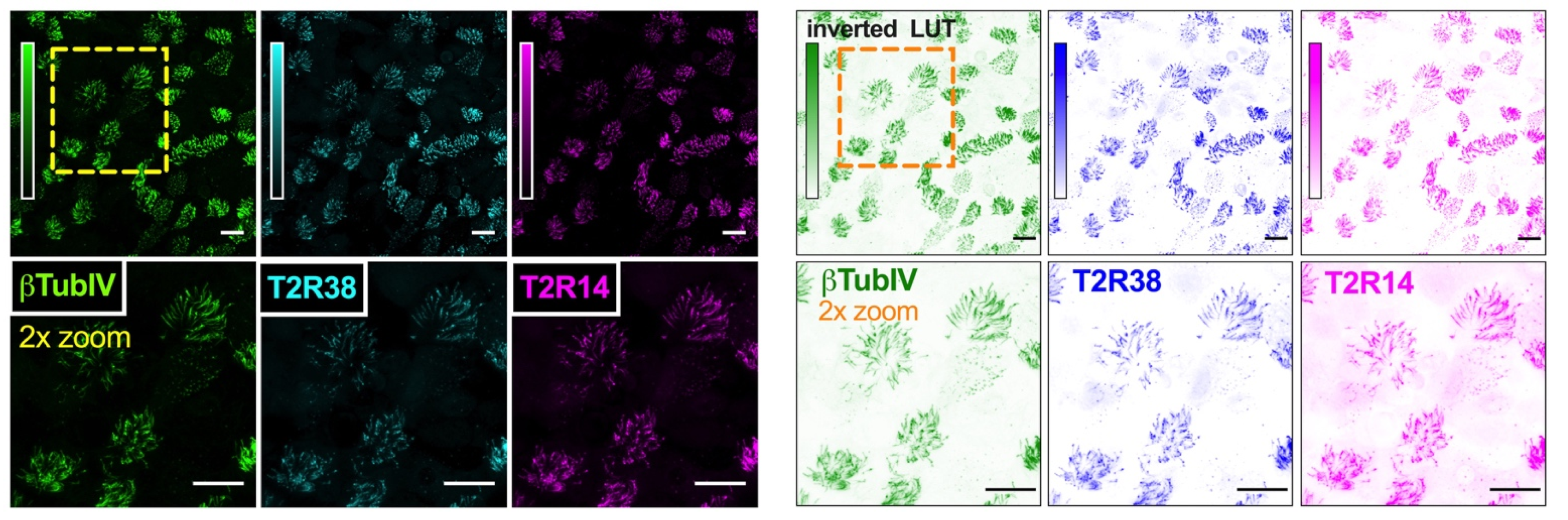
2.5. Immunofluorescence (IF) Microscopy
2.6. Phagocytosis Assays
2.7. Data Analysis and Statistics
3. Results
3.1. HSP90 Inhibition Reduces Heterologously Expressed eNOS Function in HEK293Ts and A549s
3.2. HSP90 Inhibition Reduces Endogenous T2R-Stimulated eNOS Function in Submerged H441 Cells
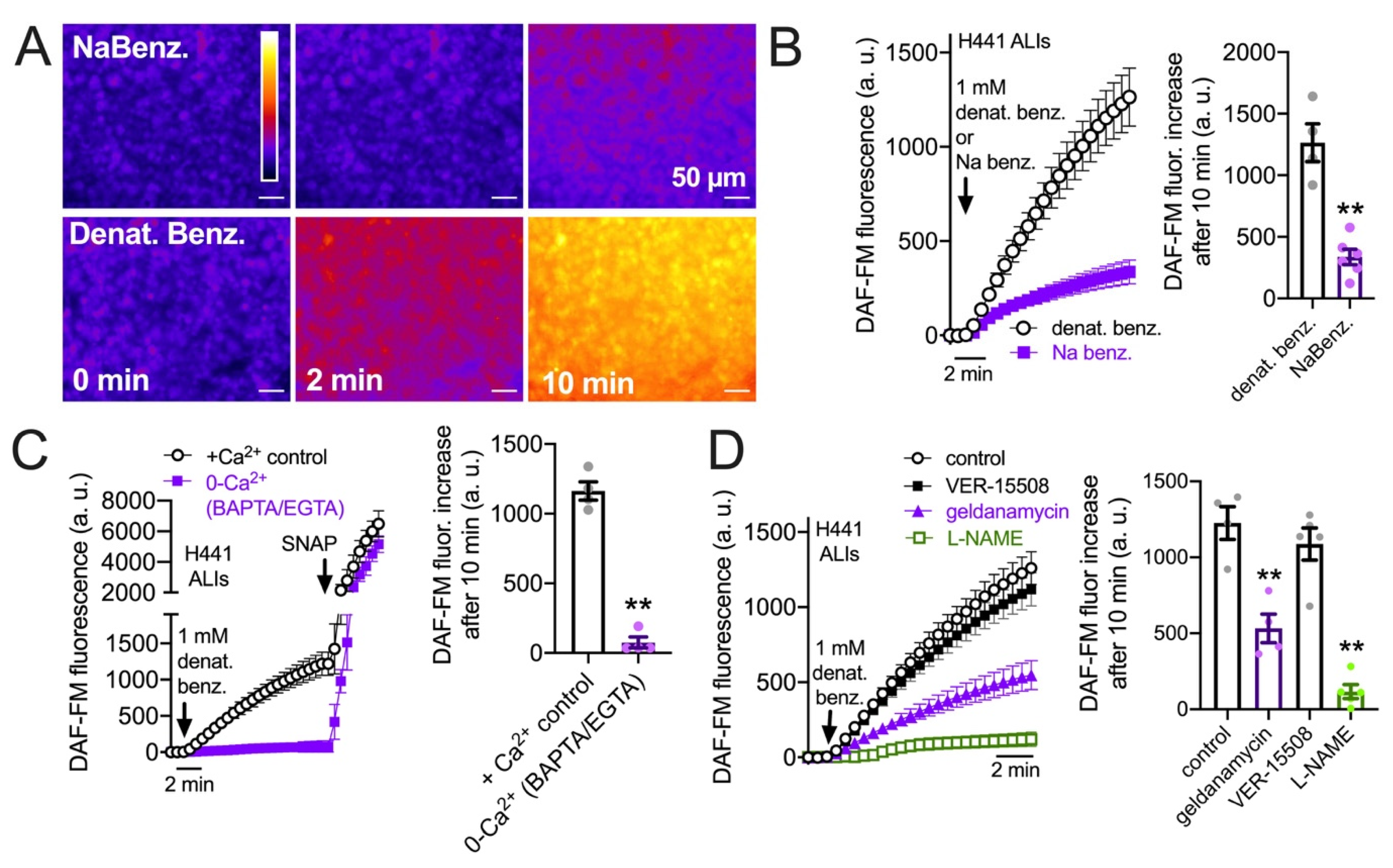
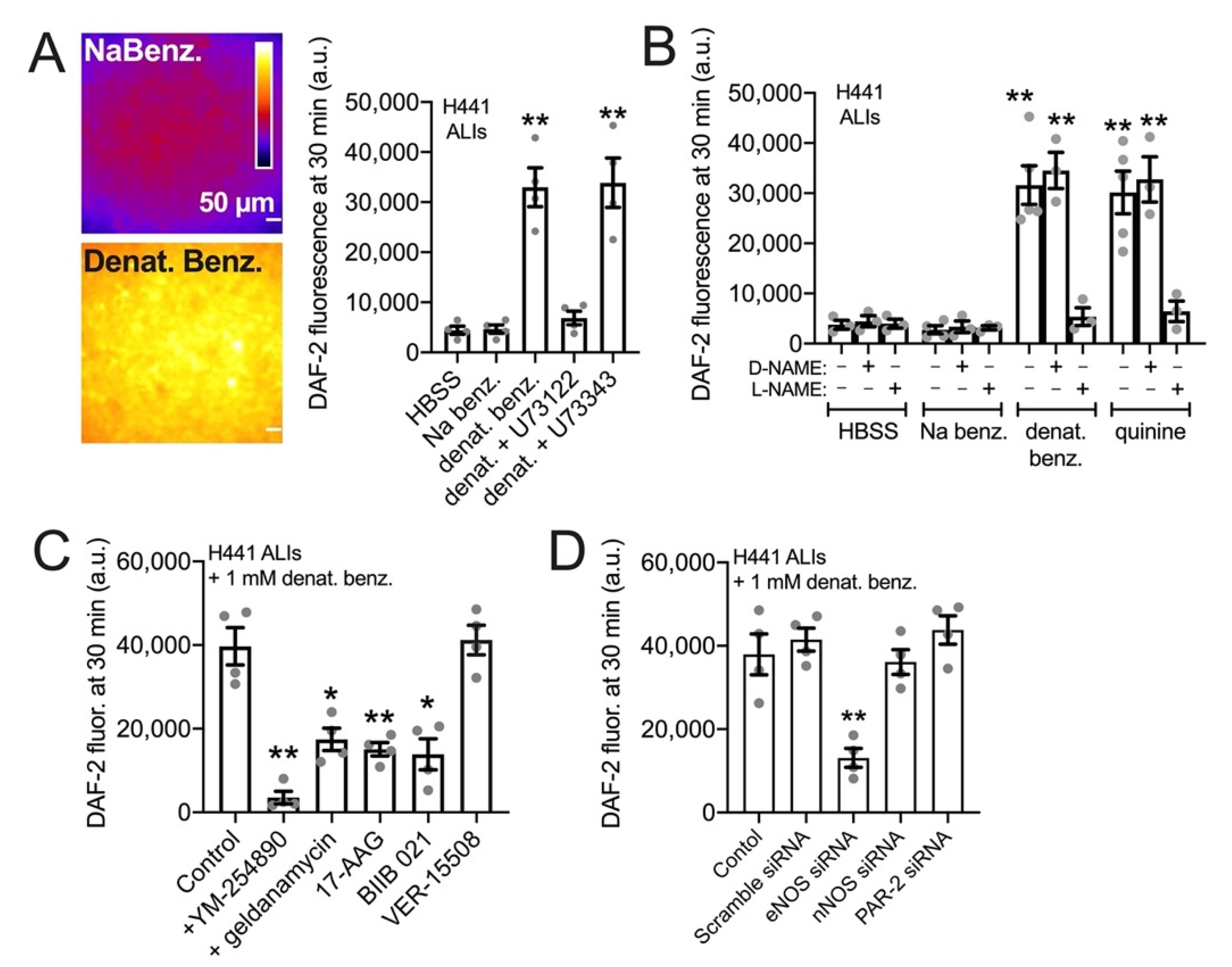
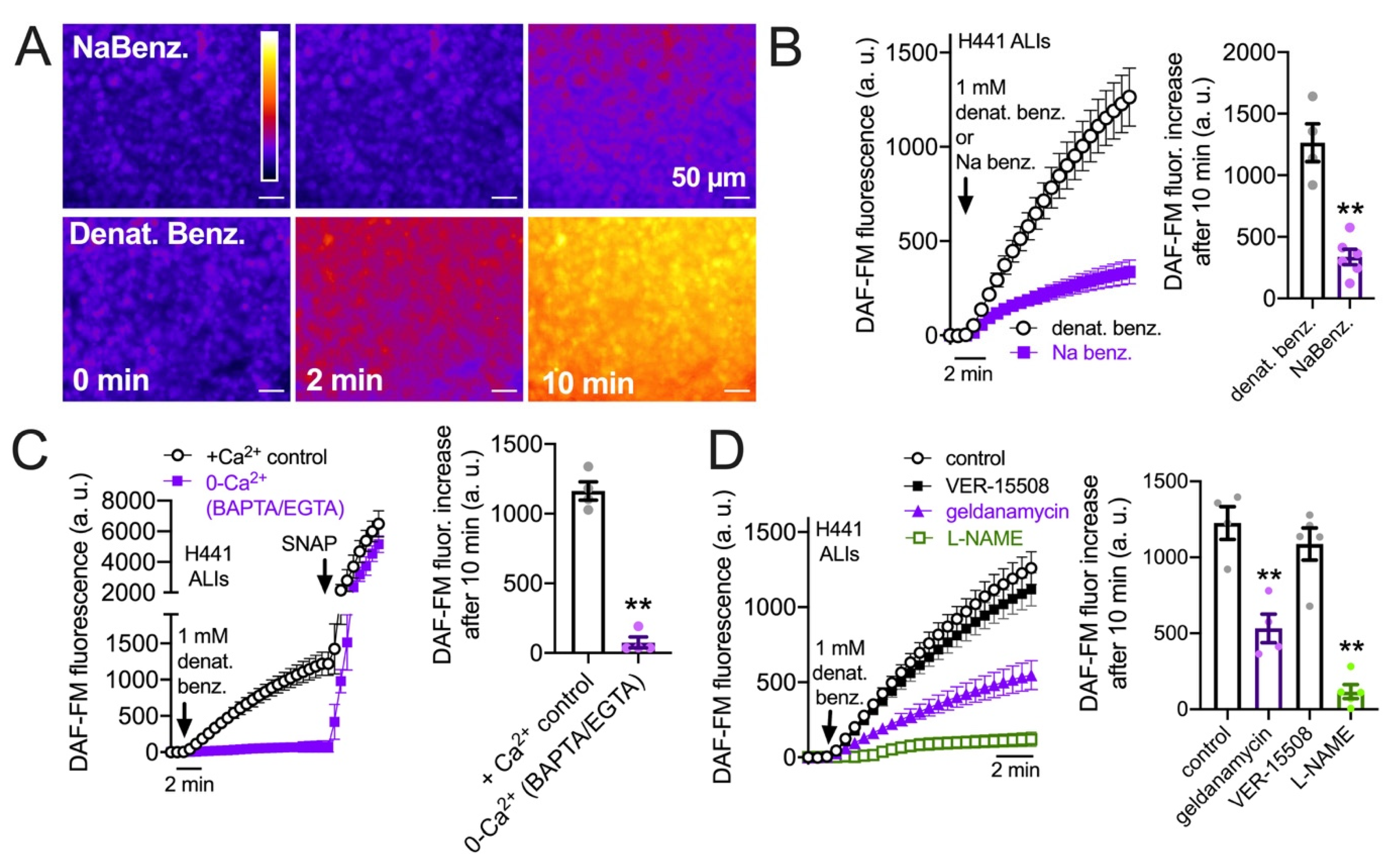
3.3. HSP90 Inhibition Reduces Endogenous T2R-Stimulated eNOS Function in H441 Monolayers Cultured at the Air–Liquid Interface
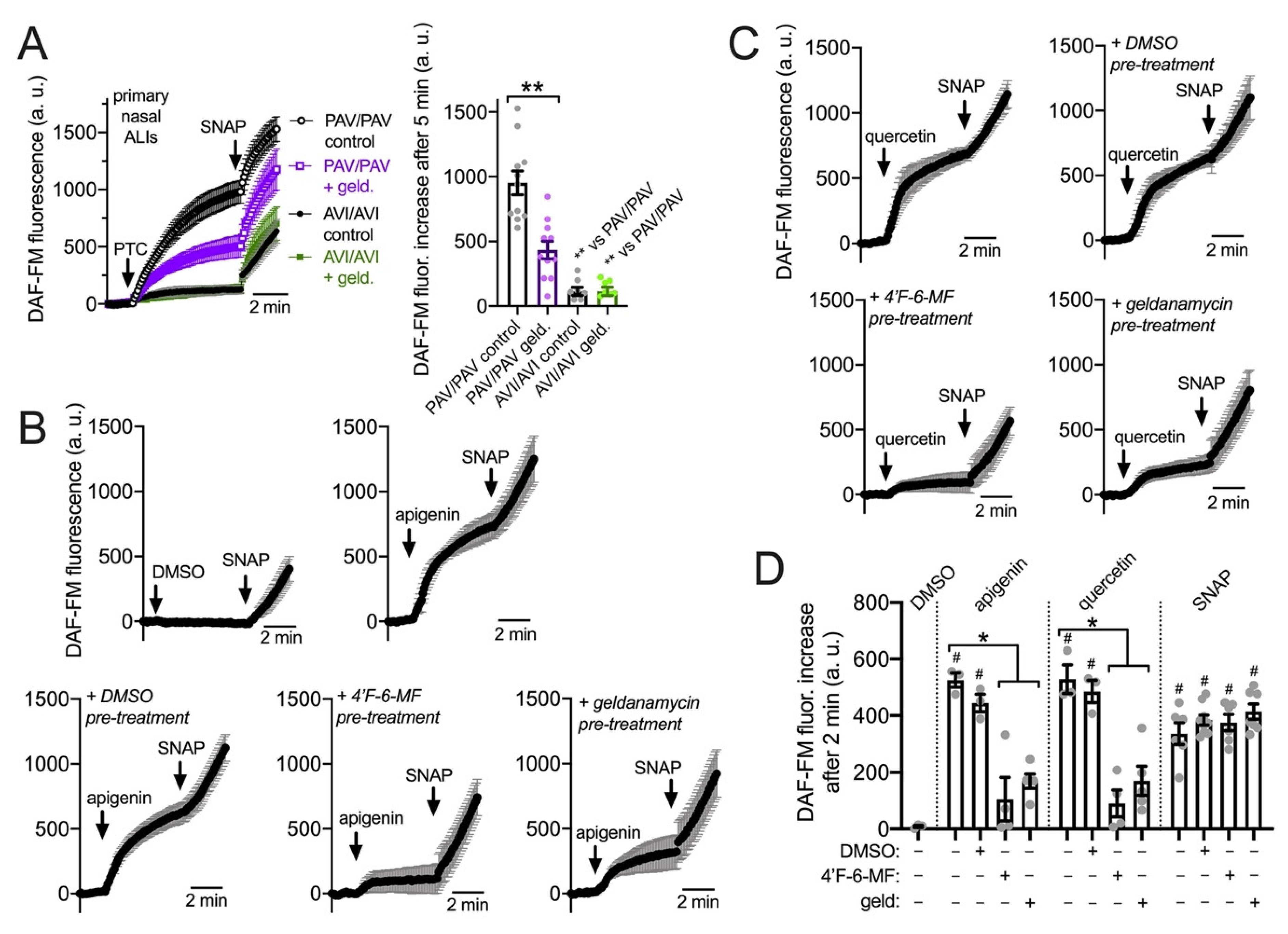
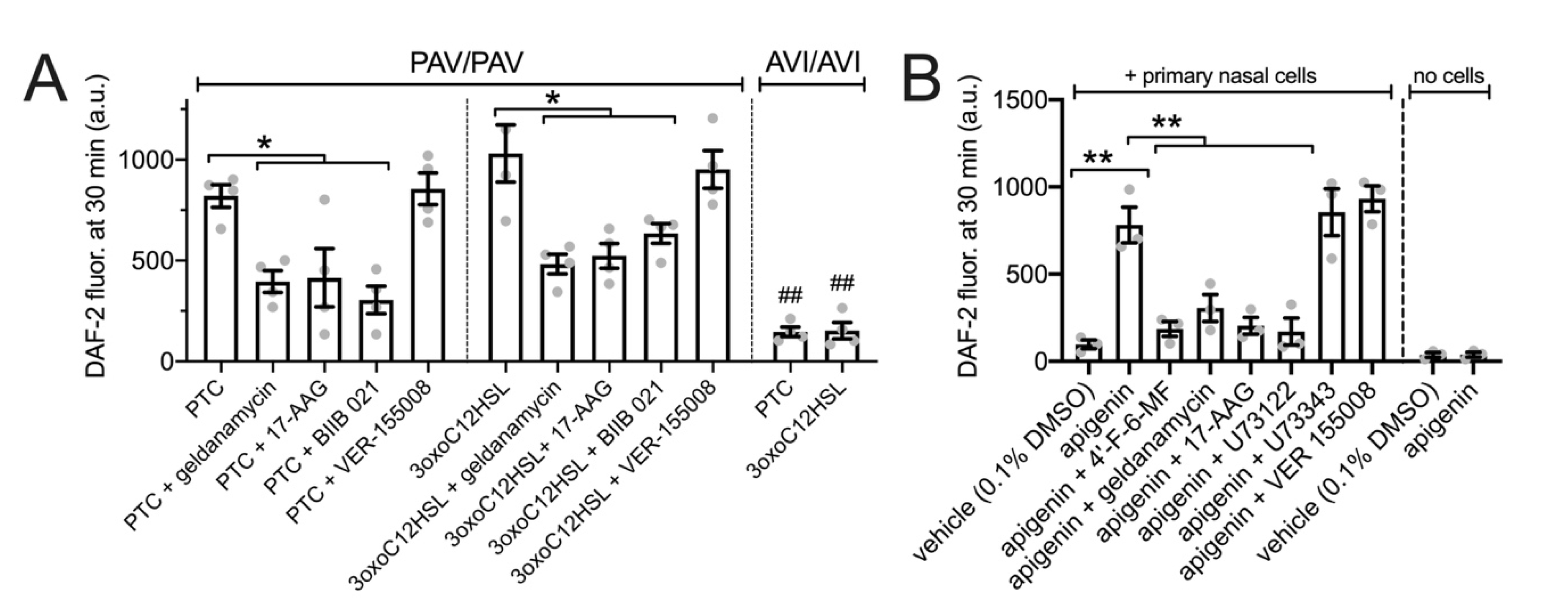
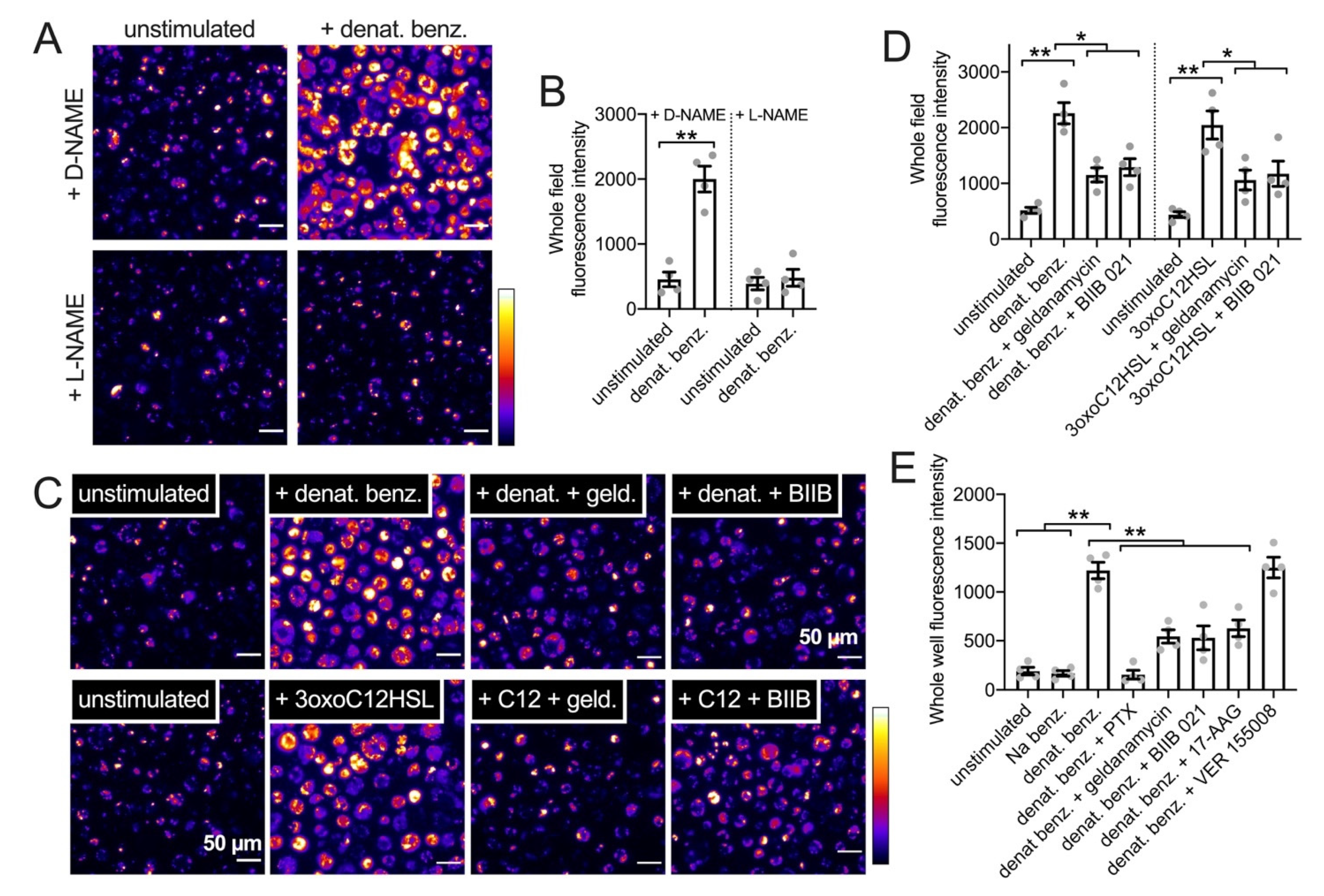
3.4. HSP90 Inhibition Reduces T2R-Driven NO Production in Primary Nasal Epithelial Air–Liquid Interface Cultures
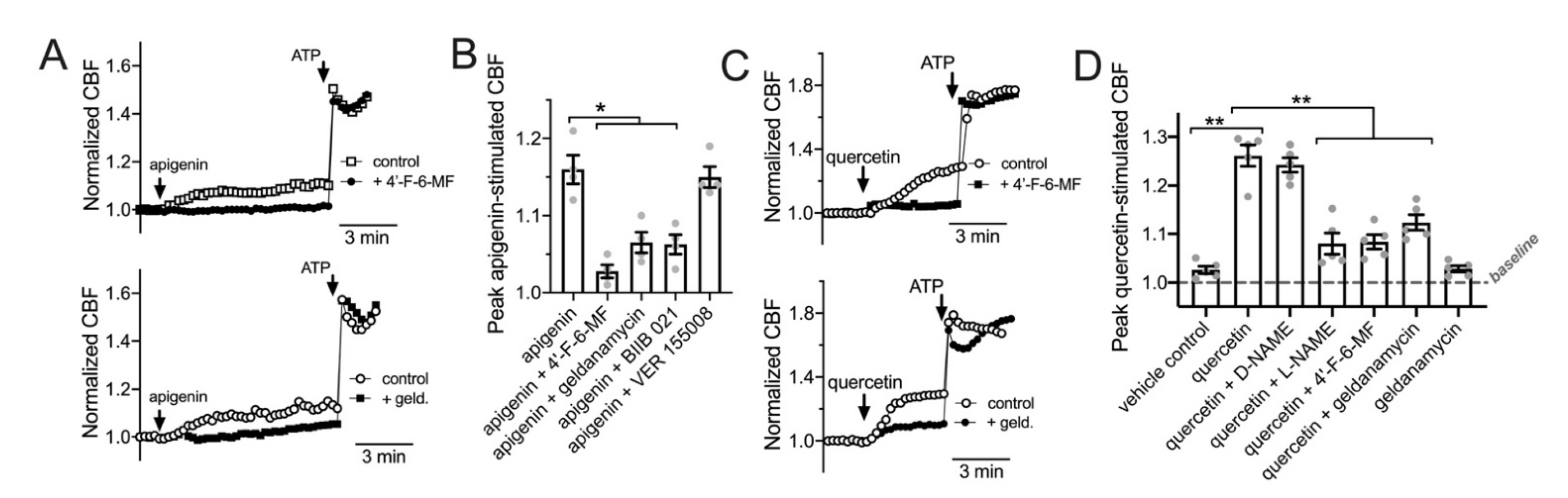
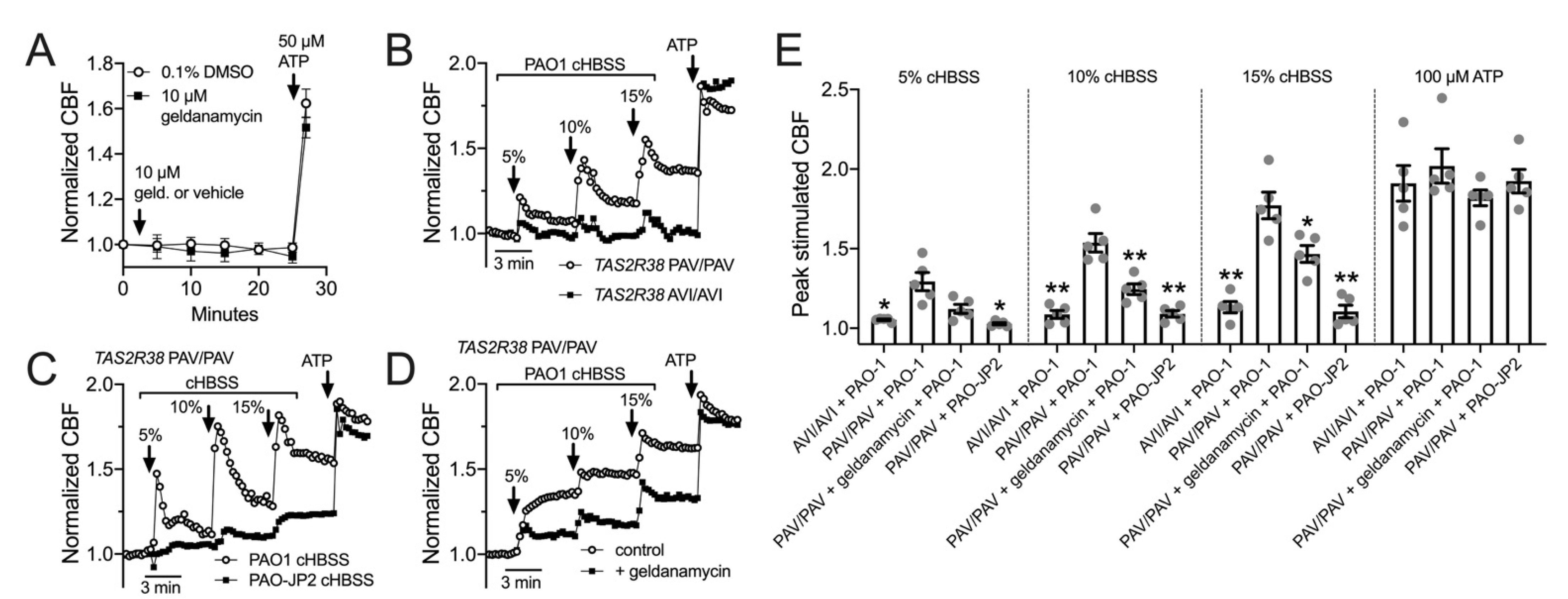
3.5. HSP90 Inhibition Reduces T2R/NO-Driven Nasal Ciliary Beating
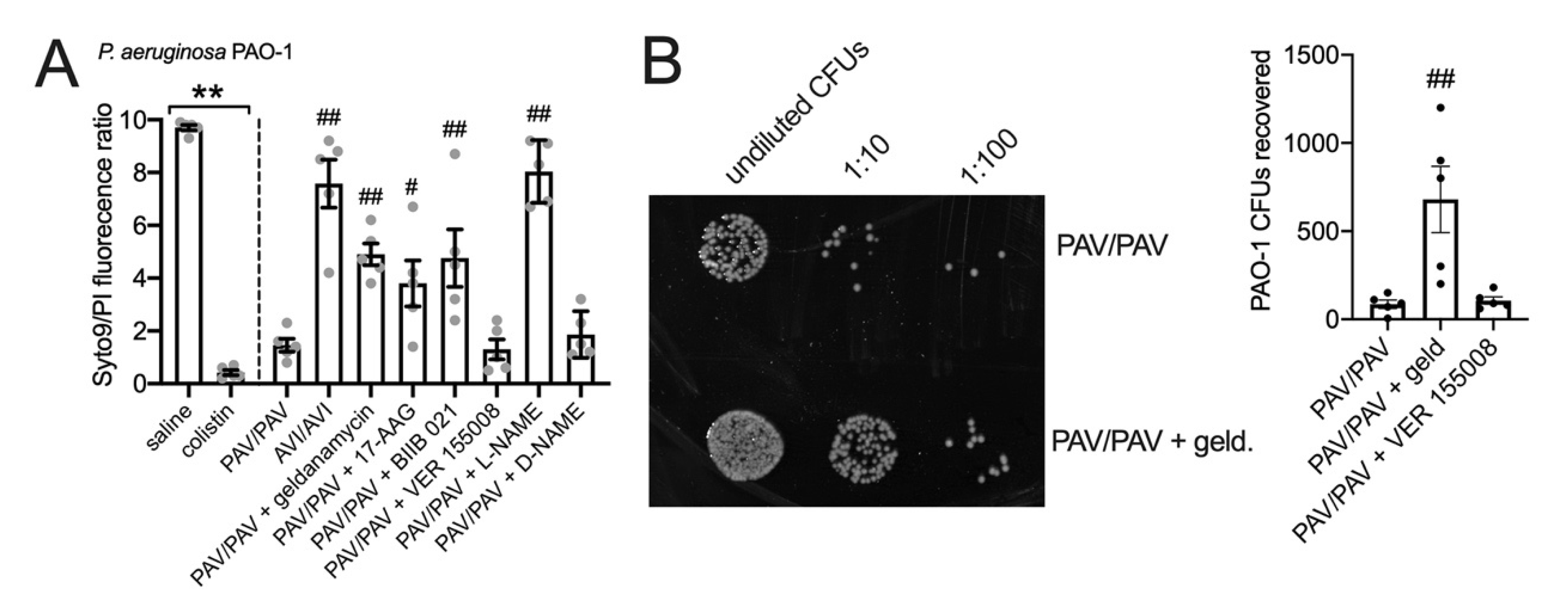
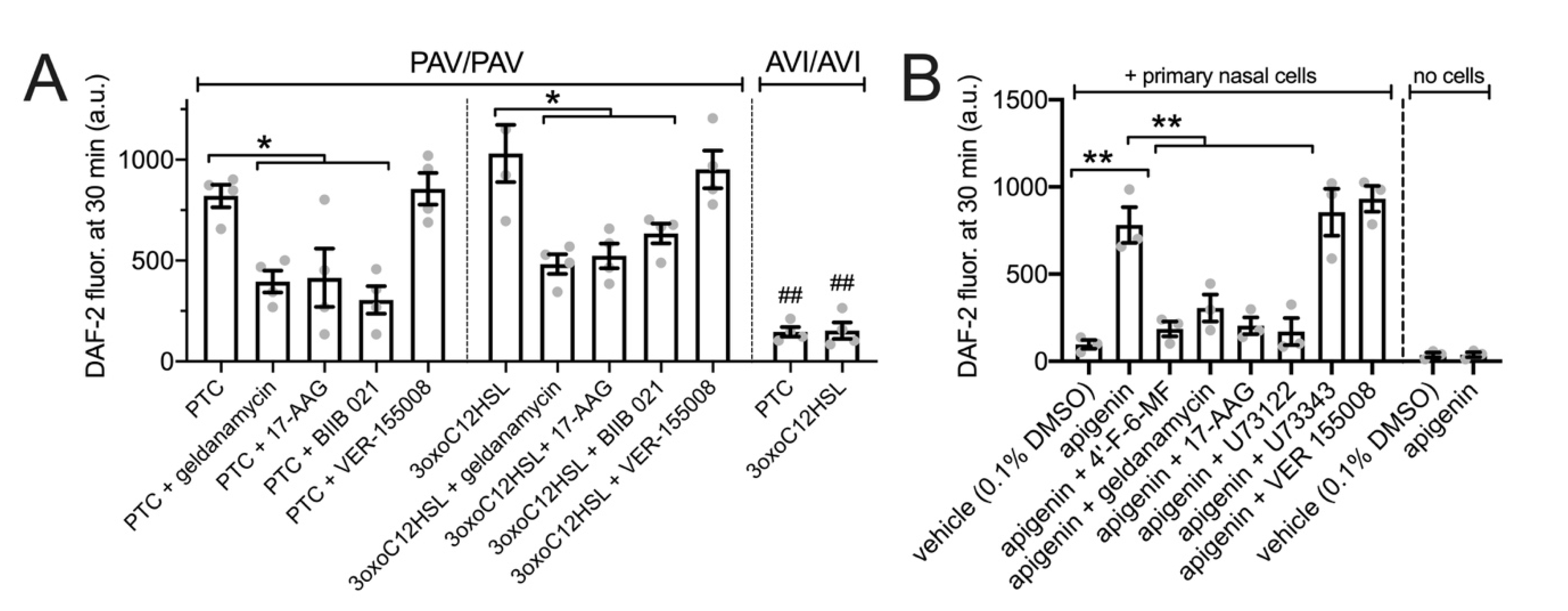
3.6. HSP90 Inhibition Reduces T2R/NO-Driven Bacterial Killing
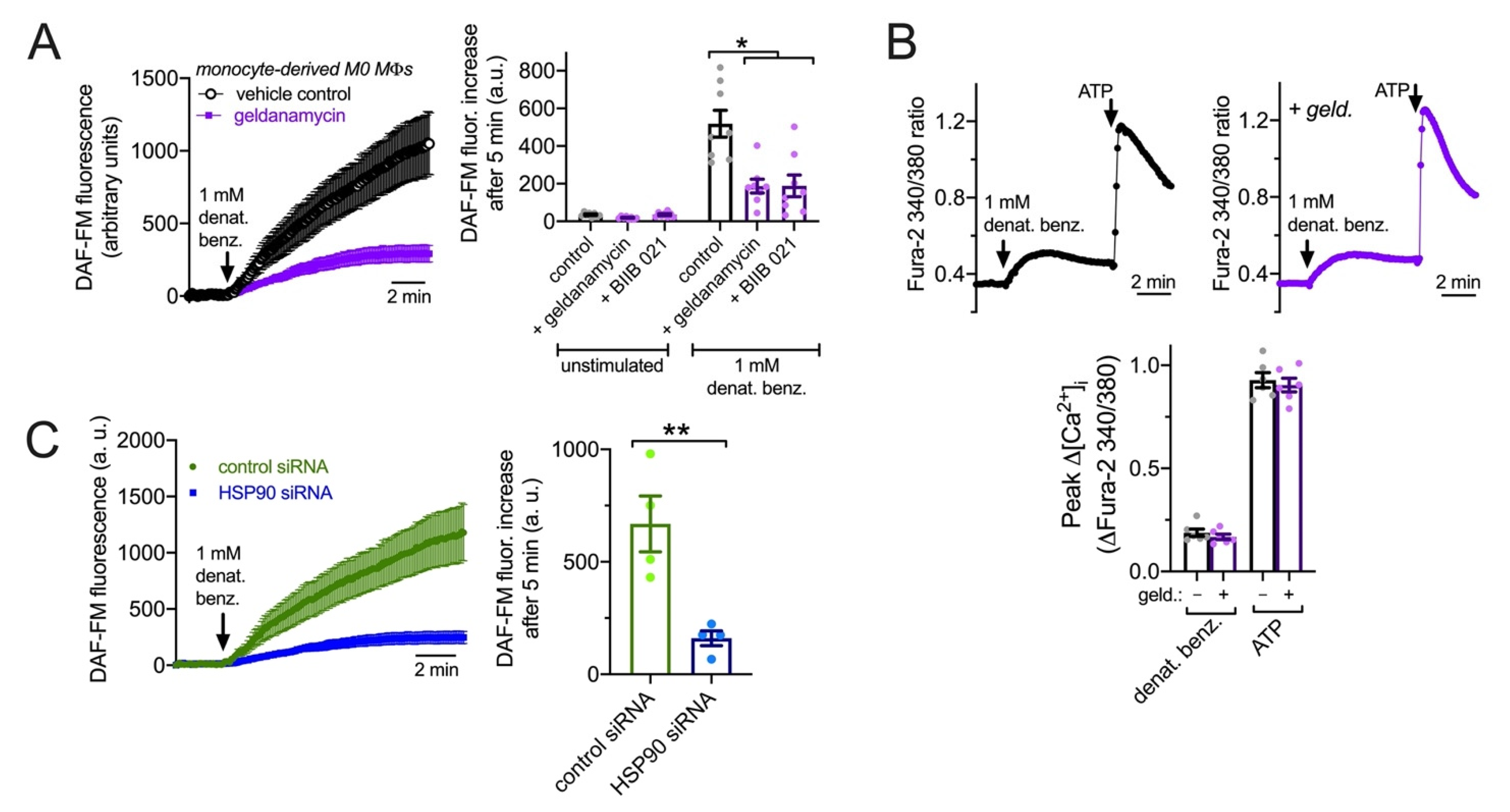
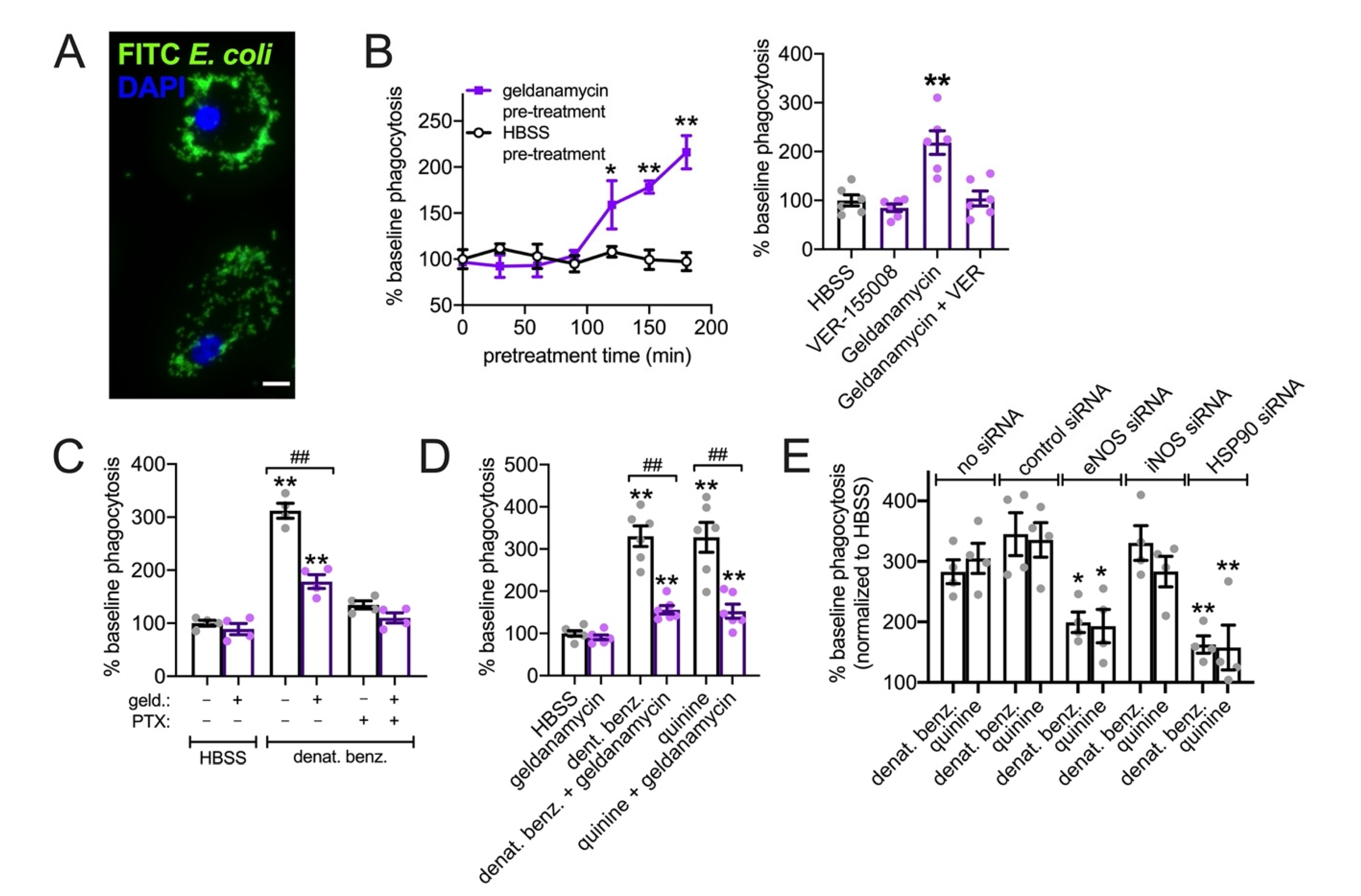
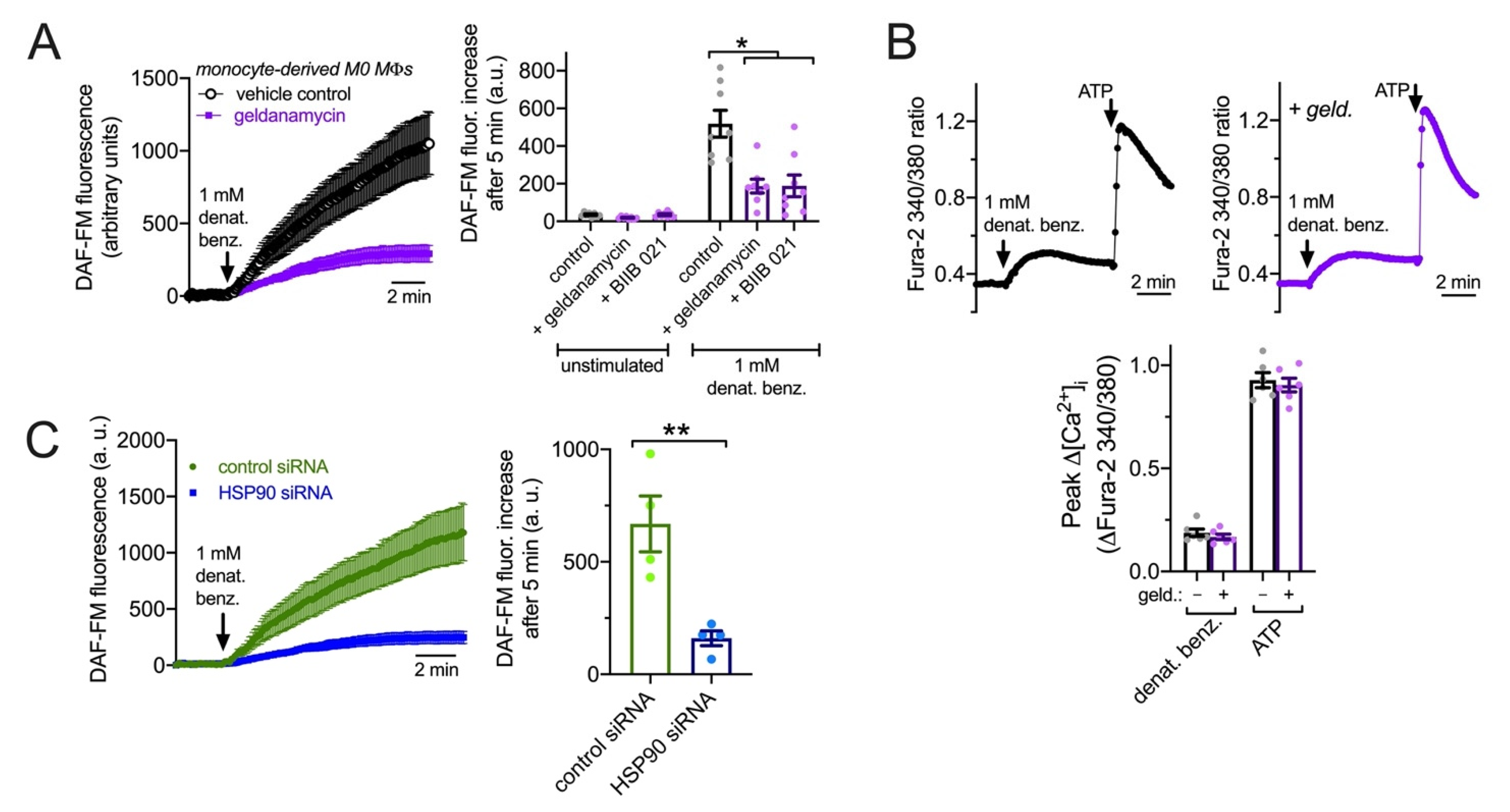
3.7. HSP90 Inhibition Reduces T2R NO Production and Phagocytosis in Primary Human MΦs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, F. Taste perception: From the tongue to the testis. Mol. Hum. Reprod. 2013, 19, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Ishimaru, Y. Oral and extra-oral taste perception. Semin. Cell Dev. Biol. 2013, 24, 240–246. [Google Scholar] [CrossRef] [PubMed]
- An, S.S.; Liggett, S.B. Taste and smell GPCRs in the lung: Evidence for a previously unrecognized widespread chemosensory system. Cell. Signal. 2017, 41, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.S.; Ben-Shahar, Y.; Moninger, T.O.; Kline, J.N.; Welsh, M.J. Motile cilia of human airway epithelia are chemosensory. Science 2009, 325, 1131–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, R.M.; Lee, R.J. Taste Receptors in Upper Airway Innate Immunity. Nutrients 2019, 11, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, D.A.; Wang, W.C.; McIlmoyle, E.L.; Robinett, K.S.; Schillinger, R.M.; An, S.S.; Sham, J.S.; Liggett, S.B. Bitter taste receptors on airway smooth muscle bronchodilate by localized calcium signaling and reverse obstruction. Nat. Med. 2010, 16, 1299–1304. [Google Scholar] [CrossRef] [Green Version]
- An, S.S.; Wang, W.C.; Koziol-White, C.J.; Ahn, K.; Lee, D.Y.; Kurten, R.C.; Panettieri, R.A., Jr.; Liggett, S.B. TAS2R activation promotes airway smooth muscle relaxation despite beta(2)-adrenergic receptor tachyphylaxis. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2012, 303, L304–L311. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.H.; Lifshitz, L.M.; Uy, K.F.; Ikebe, M.; Fogarty, K.E.; ZhuGe, R. The cellular and molecular basis of bitter tastant-induced bronchodilation. PLoS Biol. 2013, 11, e1001501. [Google Scholar] [CrossRef]
- Tan, X.; Sanderson, M.J. Bitter tasting compounds dilate airways by inhibiting airway smooth muscle calcium oscillations and calcium sensitivity. Br. J. Pharmacol. 2014, 171, 646–662. [Google Scholar] [CrossRef] [Green Version]
- Medapati, M.R.; Singh, N.; Bhagirath, A.Y.; Duan, K.; Triggs-Raine, B.; Batista, E.L., Jr.; Chelikani, P. Bitter taste receptor T2R14 detects quorum sensing molecules from cariogenic Streptococcus mutans and mediates innate immune responses in gingival epithelial cells. FASEB J. 2021, 35, e21375. [Google Scholar] [CrossRef]
- Grassin-Delyle, S.; Salvator, H.; Mantov, N.; Abrial, C.; Brollo, M.; Faisy, C.; Naline, E.; Couderc, L.J.; Devillier, P. Bitter Taste Receptors (TAS2Rs) in Human Lung Macrophages: Receptor Expression and Inhibitory Effects of TAS2R Agonists. Front. Physiol. 2019, 10, 1267. [Google Scholar] [CrossRef]
- Tran, H.T.T.; Herz, C.; Ruf, P.; Stetter, R.; Lamy, E. Human T2R38 Bitter Taste Receptor Expression in Resting and Activated Lymphocytes. Front. Immunol. 2018, 9, 2949. [Google Scholar] [CrossRef]
- Malki, A.; Fiedler, J.; Fricke, K.; Ballweg, I.; Pfaffl, M.W.; Krautwurst, D. Class I odorant receptors, TAS1R and TAS2R taste receptors, are markers for subpopulations of circulating leukocytes. J. Leukoc. Biol. 2015, 97, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Kong, I.G.; Kim, D.W. Pathogenesis of Recalcitrant Chronic Rhinosinusitis: The Emerging Role of Innate Immune Cells. Immune Netw. 2018, 18, e6. [Google Scholar] [CrossRef] [Green Version]
- Belchamber, K.B.R.; Donnelly, L.E. Macrophage Dysfunction in Respiratory Disease. Results Probl. Cell Differ. 2017, 62, 299–313. [Google Scholar] [CrossRef]
- Jaggupilli, A.; Singh, N.; Jesus, V.C.; Duan, K.; Chelikani, P. Characterization of the Binding Sites for Bacterial Acyl Homoserine Lactones (AHLs) on Human Bitter Taste Receptors (T2Rs). ACS Infect. Dis. 2018, 4, 1146–1156. [Google Scholar] [CrossRef]
- Lossow, K.; Hubner, S.; Roudnitzky, N.; Slack, J.P.; Pollastro, F.; Behrens, M.; Meyerhof, W. Comprehensive Analysis of Mouse Bitter Taste Receptors Reveals Different Molecular Receptive Ranges for Orthologous Receptors in Mice and Humans. J. Biol. Chem. 2016, 291, 15358–15377. [Google Scholar] [CrossRef] [Green Version]
- Stout, S.L.; Wyatt, T.A.; Adams, J.J.; Sisson, J.H. Nitric oxide-dependent cilia regulatory enzyme localization in bovine bronchial epithelial cells. J. Histochem. Cytochem. 2007, 55, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Sherman, T.S.; Chen, Z.; Yuhanna, I.S.; Lau, K.S.; Margraf, L.R.; Shaul, P.W. Nitric oxide synthase isoform expression in the developing lung epithelium. Am. J. Physiol. 1999, 276, L383–L390. [Google Scholar] [CrossRef] [Green Version]
- German, Z.; Chambliss, K.L.; Pace, M.C.; Arnet, U.A.; Lowenstein, C.J.; Shaul, P.W. Molecular basis of cell-specific endothelial nitric-oxide synthase expression in airway epithelium. J. Biol. Chem. 2000, 275, 8183–8189. [Google Scholar] [CrossRef] [Green Version]
- Salathe, M. Regulation of mammalian ciliary beating. Ann. Rev. Physiol. 2007, 69, 401–422. [Google Scholar] [CrossRef] [PubMed]
- Carey, R.M.; Adappa, N.D.; Palmer, J.N.; Lee, R.J. Neuropeptide Y Reduces Nasal Epithelial T2R Bitter Taste Receptor-Stimulated Nitric Oxide Production. Nutrients 2021, 13, 3392. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Hoffmann, F.W.; Fay, J.D.; Hashimoto, A.C.; Chapagain, M.L.; Kaufusi, P.H.; Hoffmann, P.R. Stimulation of unprimed macrophages with immune complexes triggers a low output of nitric oxide by calcium-dependent neuronal nitric-oxide synthase. J. Biol. Chem. 2012, 287, 4492–4502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcinkiewicz, J. Nitric oxide and antimicrobial activity of reactive oxygen intermediates. Immunopharmacology 1997, 37, 35–41. [Google Scholar] [CrossRef]
- Fang, F.C. Perspectives series: Host/pathogen interactions. Mechanisms of nitric oxide-related antimicrobial activity. J. Clin. Investig. 1997, 99, 2818–2825. [Google Scholar] [CrossRef] [Green Version]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef] [Green Version]
- Akerstrom, S.; Mousavi-Jazi, M.; Klingstrom, J.; Leijon, M.; Lundkvist, A.; Mirazimi, A. Nitric oxide inhibits the replication cycle of severe acute respiratory syndrome coronavirus. J. Virol. 2005, 79, 1966–1969. [Google Scholar] [CrossRef] [Green Version]
- Akerstrom, S.; Gunalan, V.; Keng, C.T.; Tan, Y.J.; Mirazimi, A. Dual effect of nitric oxide on SARS-CoV replication: Viral RNA production and palmitoylation of the S protein are affected. Virology 2009, 395, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Akaberi, D.; Krambrich, J.; Ling, J.; Luni, C.; Hedenstierna, G.; Jarhult, J.D.; Lennerstrand, J.; Lundkvist, A. Mitigation of the replication of SARS-CoV-2 by nitric oxide in vitro. Redox. Biol. 2020, 37, 101734. [Google Scholar] [CrossRef]
- Hedenstierna, G.; Chen, L.; Hedenstierna, M.; Lieberman, R.; Fine, D.H. Nitric oxide dosed in short bursts at high concentrations may protect against COVID-19. Nitric Oxide 2020, 103, 1–3. [Google Scholar] [CrossRef]
- Gopallawa, I.; Freund, J.R.; Lee, R.J. Bitter taste receptors stimulate phagocytosis in human macrophages through calcium, nitric oxide, and cyclic-GMP signaling. Cell. Mol. Life Sci. 2021, 78, 271–286. [Google Scholar] [CrossRef]
- Jeong, J.H.; Yoo, H.S.; Lee, S.H.; Kim, K.R.; Yoon, H.J.; Kim, S.H. Nasal and exhaled nitric oxide in chronic rhinosinusitis with polyps. Am. J. Rhinol. Allergy 2014, 28, e11–e16. [Google Scholar] [CrossRef]
- Eynott, P.R.; Groneberg, D.A.; Caramori, G.; Adcock, I.M.; Donnelly, L.E.; Kharitonov, S.; Barnes, P.J.; Chung, K.F. Role of nitric oxide in allergic inflammation and bronchial hyperresponsiveness. Eur. J. Pharmacol. 2002, 452, 123–133. [Google Scholar] [CrossRef]
- Lee, R.J.; Xiong, G.; Kofonow, J.M.; Chen, B.; Lysenko, A.; Jiang, P.; Abraham, V.; Doghramji, L.; Adappa, N.D.; Palmer, J.N.; et al. T2R38 taste receptor polymorphisms underlie susceptibility to upper respiratory infection. J. Clin. Investig. 2012, 122, 4145–4159. [Google Scholar] [CrossRef] [Green Version]
- Adappa, N.D.; Truesdale, C.M.; Workman, A.D.; Doghramji, L.; Mansfield, C.; Kennedy, D.W.; Palmer, J.N.; Cowart, B.J.; Cohen, N.A. Correlation of T2R38 taste phenotype and in vitro biofilm formation from nonpolypoid chronic rhinosinusitis patients. Int. Forum Allergy Rhinol. 2016, 6, 783–791. [Google Scholar] [CrossRef]
- Rom, D.I.; Christensen, J.M.; Alvarado, R.; Sacks, R.; Harvey, R.J. The impact of bitter taste receptor genetics on culturable bacteria in chronic rhinosinusitis. Rhinology 2017, 55, 90–94. [Google Scholar] [CrossRef]
- Cantone, E.; Negri, R.; Roscetto, E.; Grassia, R.; Catania, M.R.; Capasso, P.; Maffei, M.; Soriano, A.A.; Leone, C.A.; Iengo, M.; et al. In Vivo Biofilm Formation, Gram-Negative Infections and TAS2R38 Polymorphisms in CRSw NP Patients. Laryngoscope 2018, 128, E339–E345. [Google Scholar] [CrossRef]
- Adappa, N.D.; Zhang, Z.; Palmer, J.N.; Kennedy, D.W.; Doghramji, L.; Lysenko, A.; Reed, D.R.; Scott, T.; Zhao, N.W.; Owens, D.; et al. The bitter taste receptor T2R38 is an independent risk factor for chronic rhinosinusitis requiring sinus surgery. Int. Forum Allergy Rhinol. 2014, 4, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Adappa, N.D.; Howland, T.J.; Palmer, J.N.; Kennedy, D.W.; Doghramji, L.; Lysenko, A.; Reed, D.R.; Lee, R.J.; Cohen, N.A. Genetics of the taste receptor T2R38 correlates with chronic rhinosinusitis necessitating surgical intervention. Int. Forum Allergy Rhinol. 2013, 3, 184–187. [Google Scholar] [CrossRef]
- Mfuna Endam, L.; Filali-Mouhim, A.; Boisvert, P.; Boulet, L.P.; Bosse, Y.; Desrosiers, M. Genetic variations in taste receptors are associated with chronic rhinosinusitis: A replication study. Int. Forum Allergy Rhinol. 2014, 4, 200–206. [Google Scholar] [CrossRef]
- Dzaman, K.; Zagor, M.; Sarnowska, E.; Krzeski, A.; Kantor, I. The correlation of TAS2R38 gene variants with higher risk for chronic rhinosinusitis in Polish patients. Otolaryngol. Pol. 2016, 70, 13–18. [Google Scholar] [CrossRef]
- Adappa, N.D.; Farquhar, D.; Palmer, J.N.; Kennedy, D.W.; Doghramji, L.; Morris, S.A.; Owens, D.; Mansfield, C.; Lysenko, A.; Lee, R.J.; et al. TAS2R38 genotype predicts surgical outcome in nonpolypoid chronic rhinosinusitis. Int. Forum Allergy Rhinol. 2015, 6, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Castaldo, A.; Cernera, G.; Iacotucci, P.; Cimbalo, C.; Gelzo, M.; Comegna, M.; Di Lullo, A.M.; Tosco, A.; Carnovale, V.; Raia, V.; et al. TAS2R38 is a novel modifier gene in patients with cystic fibrosis. Sci. Rep. 2020, 10, 5806. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, A.R.; Murphy, R.; Behrends, V.; Lund-Palau, H.; Simbo, A.; Mariveles, M.; Alton, E.; Bush, A.; Shoemark, A.; Davies, J.C. Impact of T2R38 Receptor Polymorphisms on Pseudomonas aeruginosa Infection in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 197, 1635–1638. [Google Scholar] [CrossRef]
- Dalesio, N.M.; Aksit, M.A.; Ahn, K.; Raraigh, K.S.; Collaco, J.M.; McGrath-Morrow, S.; Zeitlin, P.L.; An, S.S.; Cutting, G.R. Cystic fibrosis transmembrane conductance regulator function, not TAS2R38 gene haplotypes, predict sinus surgery in children and young adults with cystic fibrosis. Int. Forum Allergy Rhinol. 2020, 10, 748–754. [Google Scholar] [CrossRef]
- Parsa, S.; Mogharab, V.; Ebrahimi, M.; Ahmadi, S.R.; Shahi, B.; Mehramiz, N.J.; Foroughian, M.; Zarenezhad, M.; Kalani, N.; Abdi, M.H.; et al. COVID-19 as a worldwide selective event and bitter taste receptor polymorphisms: An ecological correlational study. Int. J. Biol. Macromol. 2021, 177, 204–210. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell. Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Simet, S.M.; Pavlik, J.A.; Sisson, J.H. Proteomic analysis of bovine axonemes exposed to acute alcohol: Role of endothelial nitric oxide synthase and heat shock protein 90 in cilia stimulation. Alcohol. Clin. Exp. Res. 2013, 37, 609–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Zhu, D.Y. Neuronal nitric oxide synthase: Structure, subcellular localization, regulation, and clinical implications. Nitric Oxide 2009, 20, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Black, S.M.; Catravas, J.D. Endothelial nitric oxide (NO) and its pathophysiologic regulation. Vascul. Pharmacol. 2008, 49, 134–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.; Mendelsohn, M.E. Synergistic Activation of Endothelial Nitric-oxide Synthase (eNOS) by HSP90 and Akt. J. Biol. Chem. 2003, 278, 30821–30827. [Google Scholar] [CrossRef] [Green Version]
- Fontana, J.; Fulton, D.; Chen, Y.; Fairchild, T.A.; McCabe, T.J.; Fujita, N.; Tsuruo, T.; Sessa, W.C. Domain mapping studies reveal that the M domain of hsp90 serves as a molecular scaffold to regulate Akt-dependent phosphorylation of endothelial nitric oxide synthase and NO release. Circ. Res. 2002, 90, 866–873. [Google Scholar] [CrossRef]
- Garcia-Cardena, G.; Fan, R.; Shah, V.; Sorrentino, R.; Cirino, G.; Papapetropoulos, A.; Sessa, W.C. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature 1998, 392, 821–824. [Google Scholar] [CrossRef]
- Wang, H.; Zou, X.; Wei, Z.; Wu, Y.; Li, R.; Zeng, R.; Chen, Z.; Liao, K. Hsp90alpha forms a stable complex at the cilium neck for the interaction of signalling molecules in IGF-1 receptor signalling. J. Cell Sci. 2015, 128, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Takaki, E.; Fujimoto, M.; Nakahari, T.; Yonemura, S.; Miyata, Y.; Hayashida, N.; Yamamoto, K.; Vallee, R.B.; Mikuriya, T.; Sugahara, K.; et al. Heat shock transcription factor 1 is required for maintenance of ciliary beating in mice. J. Biol. Chem. 2007, 282, 37285–37292. [Google Scholar] [CrossRef] [Green Version]
- Pezzulo, A.A.; Tudas, R.A.; Stewart, C.G.; Buonfiglio, L.G.V.; Lindsay, B.D.; Taft, P.J.; Gansemer, N.D.; Zabner, J. HSP90 inhibitor geldanamycin reverts IL-13- and IL-17-induced airway goblet cell metaplasia. J. Clin. Investig. 2019, 129, 744–758. [Google Scholar] [CrossRef] [Green Version]
- McMahon, D.B.; Kuek, L.E.; Johnson, M.E.; Johnson, P.O.; Horn, R.L.J.; Carey, R.M.; Adappa, N.D.; Palmer, J.N.; Lee, R.J. The bitter end: T2R bitter receptor agonists elevate nuclear calcium and induce apoptosis in non-ciliated airway epithelial cells. Cell Calcium 2022, 101, 102499. [Google Scholar] [CrossRef]
- Hariri, B.M.; McMahon, D.B.; Chen, B.; Adappa, N.D.; Palmer, J.N.; Kennedy, D.W.; Lee, R.J. Plant flavones enhance antimicrobial activity of respiratory epithelial cell secretions against Pseudomonas aeruginosa. PLoS ONE 2017, 12, e0185203. [Google Scholar] [CrossRef] [Green Version]
- Hariri, B.M.; McMahon, D.B.; Chen, B.; Freund, J.R.; Mansfield, C.J.; Doghramji, L.J.; Adappa, N.D.; Palmer, J.N.; Kennedy, D.W.; Reed, D.R.; et al. Flavones modulate respiratory epithelial innate immunity: Anti-inflammatory effects and activation of the T2R14 receptor. J. Biol. Chem. 2017, 292, 8484–8497. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.W.; Reed, D.R. The genetics of phenylthiocarbamide perception. Ann. Hum. Biol. 2001, 28, 111–142. [Google Scholar]
- Bufe, B.; Breslin, P.A.; Kuhn, C.; Reed, D.R.; Tharp, C.D.; Slack, J.P.; Kim, U.K.; Drayna, D.; Meyerhof, W. The molecular basis of individual differences in phenylthiocarbamide and propylthiouracil bitterness perception. Curr. Biol. 2005, 15, 322–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, R.M.; McMahon, D.B.; Miller, Z.A.; Kim, T.; Rajasekaran, K.; Gopallawa, I.; Newman, J.G.; Basu, D.; Nead, K.T.; White, E.A.; et al. T2R bitter taste receptors regulate apoptosis and may be associated with survival in head and neck squamous cell carcinoma. Mol. Oncol. 2021, 16, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, A.; Amodaj, N.; Hoover, K.; Vale, R.; Stuurman, N. Computer control of microscopes using microManager. Curr. Protoc. Mol. Biol. 2010, 14, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisson, J.H.; Stoner, J.A.; Ammons, B.A.; Wyatt, T.A. All-digital image capture and whole-field analysis of ciliary beat frequency. J. Microsc. 2003, 211, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.P.; Pesci, E.C.; Iglewski, B.H. Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of elastase and rhamnolipid biosynthesis genes. J. Bacteriol. 1997, 179, 5756–5767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, J.P.; Passador, L.; Iglewski, B.H.; Greenberg, E.P. A second N-acylhomoserine lactone signal produced by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 1995, 92, 1490–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Leterrier, C. ChrisLUTs. Available online: https://github.com/cleterrier/ChrisLUTs (accessed on 12 December 2021).
- Xu, H.; Shi, Y.; Wang, J.; Jones, D.; Weilrauch, D.; Ying, R.; Wakim, B.; Pritchard, K.A., Jr. A heat shock protein 90 binding domain in endothelial nitric-oxide synthase influences enzyme function. J. Biol. Chem. 2007, 282, 37567–37574. [Google Scholar] [CrossRef] [Green Version]
- Vladic, N.; Ge, Z.D.; Leucker, T.; Brzezinska, A.K.; Du, J.H.; Shi, Y.; Warltier, D.C.; Pratt, P.F., Jr.; Kersten, J.R. Decreased tetrahydrobiopterin and disrupted association of Hsp90 with eNOS by hyperglycemia impair myocardial ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2130–H2139. [Google Scholar] [CrossRef] [Green Version]
- Miao, R.Q.; Fontana, J.; Fulton, D.; Lin, M.I.; Harrison, K.D.; Sessa, W.C. Dominant-negative Hsp90 reduces VEGF-stimulated nitric oxide release and migration in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Sessa, W.C.; Garcia-Cardena, G.; Liu, J.; Keh, A.; Pollock, J.S.; Bradley, J.; Thiru, S.; Braverman, I.M.; Desai, K.M. The Golgi association of endothelial nitric oxide synthase is necessary for the efficient synthesis of nitric oxide. J. Biol. Chem. 1995, 270, 17641–17644. [Google Scholar] [CrossRef] [Green Version]
- Gopallawa, I.; Kuek, L.E.; Adappa, N.D.; Palmer, J.N.; Lee, R.J. Small-molecule Akt-activation in airway cells induces NO production and reduces IL-8 transcription through Nrf-2. Respir. Res. 2021, 22, 267. [Google Scholar] [CrossRef]
- Zhou, B.P.; Hu, M.C.; Miller, S.A.; Yu, Z.; Xia, W.; Lin, S.Y.; Hung, M.C. HER-2/neu blocks tumor necrosis factor-induced apoptosis via the Akt/NF-kappaB pathway. J. Biol. Chem. 2000, 275, 8027–8031. [Google Scholar] [CrossRef] [Green Version]
- Sowa, G.; Liu, J.; Papapetropoulos, A.; Rex-Haffner, M.; Hughes, T.E.; Sessa, W.C. Trafficking of endothelial nitric-oxide synthase in living cells. Quantitative evidence supporting the role of palmitoylation as a kinetic trapping mechanism limiting membrane diffusion. J. Biol. Chem. 1999, 274, 22524–22531. [Google Scholar] [CrossRef] [Green Version]
- Picard, D.; Suslova, E.; Briand, P.A. 2-color photobleaching experiments reveal distinct intracellular dynamics of two components of the Hsp90 complex. Exp. Cell Res. 2006, 312, 3949–3958. [Google Scholar] [CrossRef]
- Shaul, P.W.; North, A.J.; Wu, L.C.; Wells, L.B.; Brannon, T.S.; Lau, K.S.; Michel, T.; Margraf, L.R.; Star, R.A. Endothelial nitric oxide synthase is expressed in cultured human bronchiolar epithelium. J. Clin. Investig. 1994, 94, 2231–2236. [Google Scholar] [CrossRef] [Green Version]
- Salinas, D.; Sparkman, L.; Berhane, K.; Boggaram, V. Nitric oxide inhibits surfactant protein B gene expression in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L1153–L1165. [Google Scholar] [CrossRef] [Green Version]
- Kirsch, E.A.; Yuhanna, I.S.; Chen, Z.; German, Z.; Sherman, T.S.; Shaul, P.W. Estrogen acutely stimulates endothelial nitric oxide synthase in H441 human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 1999, 20, 658–666. [Google Scholar] [CrossRef]
- Boggaram, V.; Chandru, H.; Gottipati, K.R.; Thakur, V.; Das, A.; Berhane, K. Transcriptional regulation of SP-B gene expression by nitric oxide in H441 lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L252–L262. [Google Scholar] [CrossRef] [Green Version]
- Meyerhof, W.; Batram, C.; Kuhn, C.; Brockhoff, A.; Chudoba, E.; Bufe, B.; Appendino, G.; Behrens, M. The molecular receptive ranges of human TAS2R bitter taste receptors. Chem. Sens. 2010, 35, 157–170. [Google Scholar] [CrossRef]
- Wiener, A.; Shudler, M.; Levit, A.; Niv, M.Y. BitterDB: A database of bitter compounds. Nucleic Acids Res. 2012, 40, D413–D419. [Google Scholar] [CrossRef]
- Workman, A.D.; Maina, I.W.; Brooks, S.G.; Kohanski, M.A.; Cowart, B.J.; Mansfield, C.; Kennedy, D.W.; Palmer, J.N.; Adappa, N.D.; Reed, D.R.; et al. The Role of Quinine-Responsive T2Rs in Airway Immune Defense and Chronic Rhinosinusitis. Front. Immunol. 2018, 9, 624. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.; Michel, C.; Braubach, P.; Fauler, M.; Neubauer, D.; Thompson, K.E.; Frick, M.; Mizaikoff, B.; Dietl, P.; Wittekindt, O.H. Water Permeability Adjusts Resorption in Lung Epithelia to Increased Apical Surface Liquid Volumes. Am. J. Respir. Cell Mol. Biol. 2017, 56, 372–382. [Google Scholar] [CrossRef]
- Nickel, S.; Selo, M.A.; Fallack, J.; Clerkin, C.G.; Huwer, H.; Schneider-Daum, N.; Lehr, C.M.; Ehrhardt, C. Expression and Activity of Breast Cancer Resistance Protein (BCRP/ABCG2) in Human Distal Lung Epithelial Cells In Vitro. Pharm. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Korbmacher, J.P.; Michel, C.; Neubauer, D.; Thompson, K.; Mizaikoff, B.; Frick, M.; Dietl, P.; Wittekindt, O.H. Amiloride-sensitive fluid resorption in NCI-H441 lung epithelia depends on an apical Cl(-) conductance. Physiol. Rep. 2014, 2, e00201. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Song, W.; Davis, I.C.; Shrestha, K.; Schwiebert, E.; Sullender, W.M.; Matalon, S. Inhibition of Na+ transport in lung epithelial cells by respiratory syncytial virus infection. Am. J. Respir. Cell Mol. Biol. 2009, 40, 588–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarbiou, J.; Copreni, E.; Buijs-Offerman, R.M.; van der Wegen, P.; Castellani, S.; Carbone, A.; Tilesi, F.; Fradiani, P.; Hiemstra, P.S.; Yueksekdag, G.; et al. Lentiviral small hairpin RNA delivery reduces apical sodium channel activity in differentiated human airway epithelial cells. J. Gene Med. 2012, 14, 733–745. [Google Scholar] [CrossRef]
- Jiang, J.; Malavia, N.; Suresh, V.; George, S.C. Nitric oxide gas phase release in human small airway epithelial cells. Respir. Res. 2009, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Takasaki, J.; Saito, T.; Taniguchi, M.; Kawasaki, T.; Moritani, Y.; Hayashi, K.; Kobori, M. A novel Galphaq/11-selective inhibitor. J. Biol. Chem. 2004, 279, 47438–47445. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Alqahtani, S.; Nasrullah, M.Z.A.; Shen, J. Functional evidence for biased inhibition of G protein signaling by YM-254890 in human coronary artery endothelial cells. Eur. J. Pharmacol. 2021, 891, 173706. [Google Scholar] [CrossRef]
- Nishimura, A.; Kitano, K.; Takasaki, J.; Taniguchi, M.; Mizuno, N.; Tago, K.; Hakoshima, T.; Itoh, H. Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule. Proc. Natl. Acad. Sci. USA 2010, 107, 13666–13671. [Google Scholar] [CrossRef] [Green Version]
- Wolfle, U.; Elsholz, F.A.; Kersten, A.; Haarhaus, B.; Schumacher, U.; Schempp, C.M. Expression and Functional Activity of the Human Bitter Taste Receptor TAS2R38 in Human Placental Tissues and JEG-3 Cells. Molecules 2016, 21, 306. [Google Scholar] [CrossRef] [Green Version]
- Roland, W.S.; van Buren, L.; Gruppen, H.; Driesse, M.; Gouka, R.J.; Smit, G.; Vincken, J.P. Bitter taste receptor activation by flavonoids and isoflavonoids: Modeled structural requirements for activation of hTAS2R14 and hTAS2R39. J. Agric. Food Chem. 2013, 61, 10454–10466. [Google Scholar] [CrossRef]
- Levit, A.; Nowak, S.; Peters, M.; Wiener, A.; Meyerhof, W.; Behrens, M.; Niv, M.Y. The bitter pill: Clinical drugs that activate the human bitter taste receptor TAS2R14. FASEB J. 2014, 28, 1181–1197. [Google Scholar] [CrossRef]
- Zhang, S.; Smith, N.; Schuster, D.; Azbell, C.; Sorscher, E.J.; Rowe, S.M.; Woodworth, B.A. Quercetin increases cystic fibrosis transmembrane conductance regulator-mediated chloride transport and ciliary beat frequency: Therapeutic implications for chronic rhinosinusitis. Am. J. Rhinol. Allergy 2011, 25, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Niisato, N.; Nishio, K.; Hamilton, K.L.; Marunaka, Y. Distinct action of flavonoids, myricetin and quercetin, on epithelial Cl(-) secretion: Useful tools as regulators of Cl(-) secretion. Biomed. Res. Int. 2014, 2014, 902735. [Google Scholar] [CrossRef]
- Kim, D.; Woo, J.A.; Geffken, E.; An, S.S.; Liggett, S.B. Coupling of Airway Smooth Muscle Bitter Taste Receptors to Intracellular Signaling and Relaxation Is via Galphai1,2,3. Am. J. Respir. Cell Mol. Biol. 2017, 56, 762–771. [Google Scholar] [CrossRef]
- Roland, W.S.; Gouka, R.J.; Gruppen, H.; Driesse, M.; van Buren, L.; Smit, G.; Vincken, J.P. 6-methoxyflavanones as bitter taste receptor blockers for hTAS2R39. PLoS ONE 2014, 9, e94451. [Google Scholar] [CrossRef]
- Pearson, J.P.; Gray, K.M.; Passador, L.; Tucker, K.D.; Eberhard, A.; Iglewski, B.H.; Greenberg, E.P. Structure of the autoinducer required for expression of Pseudomonas aeruginosa virulence genes. Proc. Natl. Acad. Sci. USA 1994, 91, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Pesci, E.C.; Milbank, J.B.; Pearson, J.P.; McKnight, S.; Kende, A.S.; Greenberg, E.P.; Iglewski, B.H. Quinolone signaling in the cell-to-cell communication system of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 1999, 96, 11229–11234. [Google Scholar] [CrossRef] [Green Version]
- Liddiard, K.; Rosas, M.; Davies, L.C.; Jones, S.A.; Taylor, P.R. Macrophage heterogeneity and acute inflammation. Eur. J. Immunol. 2011, 41, 2503–2508. [Google Scholar] [CrossRef]
- Davies, L.C.; Rosas, M.; Jenkins, S.J.; Liao, C.T.; Scurr, M.J.; Brombacher, F.; Fraser, D.J.; Allen, J.E.; Jones, S.A.; Taylor, P.R. Distinct bone marrow-derived and tissue-resident macrophage lineages proliferate at key stages during inflammation. Nat. Commun. 2013, 4, 1886. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Assani, K.; Shrestha, C.L.; Robledo-Avila, F.; Rajaram, M.V.; Partida-Sanchez, S.; Schlesinger, L.S.; Kopp, B.T. Human Cystic Fibrosis Macrophages Have Defective Calcium-Dependent Protein Kinase C Activation of the NADPH Oxidase, an Effect Augmented by Burkholderia cenocepacia. J. Immunol. 2017, 198, 1985–1994. [Google Scholar] [CrossRef] [Green Version]
- Bruscia, E.M.; Bonfield, T.L. Cystic Fibrosis Lung Immunity: The Role of the Macrophage. J. Innate Immun. 2016, 8, 550–563. [Google Scholar] [CrossRef]
- Vega, V.L.; De Maio, A. Increase in phagocytosis after geldanamycin treatment or heat shock: Role of heat shock proteins. J. Immunol. 2005, 175, 5280–5287. [Google Scholar] [CrossRef] [Green Version]
- Vega, V.L.; Charles, W.; Crotty Alexander, L.E. Rescuing of deficient killing and phagocytic activities of macrophages derived from non-obese diabetic mice by treatment with geldanamycin or heat shock: Potential clinical implications. Cell Stress Chaperones 2011, 16, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Ozeck, M.; Brust, P.; Xu, H.; Servant, G. Receptors for bitter, sweet and umami taste couple to inhibitory G protein signaling pathways. Eur. J. Pharmacol. 2004, 489, 139–149. [Google Scholar] [CrossRef]
- McLaughlin, S.K.; McKinnon, P.J.; Margolskee, R.F. Gustducin is a taste-cell-specific G protein closely related to the transducins. Nature 1992, 357, 563–569. [Google Scholar] [CrossRef]
- Zininga, T.; Ramatsui, L.; Shonhai, A. Heat Shock Proteins as Immunomodulants. Molecules 2018, 23, 2846. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streicher, J.M. The Role of Heat Shock Proteins in Regulating Receptor Signal Transduction. Mol. Pharmacol. 2019, 95, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Pai, K.S.; Mahajan, V.B.; Lau, A.; Cunningham, D.D. Thrombin receptor signaling to cytoskeleton requires Hsp90. J. Biol. Chem. 2001, 276, 32642–32647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purnell, P.R.; Addicks, B.L.; Zalzal, H.G.; Shapiro, S.; Wen, S.; Ramadan, H.H.; Setola, V.; Siderovski, D.P. Single Nucleotide Polymorphisms in Chemosensory Pathway Genes GNB3, TAS2R19, and TAS2R38 Are Associated with Chronic Rhinosinusitis. Int. Arch. Allergy Immunol. 2019, 180, 72–78. [Google Scholar] [CrossRef]
- Lin, C.; Civantos, A.M.; Arnold, M.; Stevens, E.M.; Cowart, B.J.; Colquitt, L.R.; Mansfield, C.; Kennedy, D.W.; Brooks, S.G.; Workman, A.D.; et al. Divergent bitter and sweet taste perception intensity in chronic rhinosinusitis patients. Int. Forum Allergy Rhinol. 2020, 11, 857–865. [Google Scholar] [CrossRef]
- Adappa, N.D.; Workman, A.D.; Hadjiliadis, D.; Dorgan, D.J.; Frame, D.; Brooks, S.; Doghramji, L.; Palmer, J.N.; Mansfield, C.; Reed, D.R.; et al. T2R38 genotype is correlated with sinonasal quality of life in homozygous DeltaF508 cystic fibrosis patients. Int. Forum Allergy Rhinol. 2016, 6, 356–361. [Google Scholar] [CrossRef] [Green Version]
- Prodromou, C. Mechanisms of Hsp90 regulation. Biochem. J. 2016, 473, 2439–2452. [Google Scholar] [CrossRef] [Green Version]
- Mollapour, M.; Neckers, L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta. 2012, 1823, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Kupatt, C.; Dessy, C.; Hinkel, R.; Raake, P.; Daneau, G.; Bouzin, C.; Boekstegers, P.; Feron, O. Heat shock protein 90 transfection reduces ischemia-reperfusion-induced myocardial dysfunction via reciprocal endothelial NO synthase serine 1177 phosphorylation and threonine 495 dephosphorylation. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1435–1441. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Xu, W.; Bose, S.; Banerjee, A.K.; Haque, S.J.; Erzurum, S.C. Impaired nitric oxide synthase-2 signaling pathway in cystic fibrosis airway epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L374–L381. [Google Scholar] [CrossRef]
- Thomas, S.R.; Kharitonov, S.A.; Scott, S.F.; Hodson, M.E.; Barnes, P.J. Nasal and exhaled nitric oxide is reduced in adult patients with cystic fibrosis and does not correlate with cystic fibrosis genotype. Chest 2000, 117, 1085–1089. [Google Scholar] [CrossRef] [Green Version]
- Texereau, J.; Marullo, S.; Hubert, D.; Coste, J.; Dusser, D.J.; Dall’Ava-Santucci, J.; Dinh-Xuan, A.T. Nitric oxide synthase 1 as a potential modifier gene of decline in lung function in patients with cystic fibrosis. Thorax 2004, 59, 156–158. [Google Scholar] [CrossRef] [Green Version]
- Texereau, J.; Fajac, I.; Hubert, D.; Coste, J.; Dusser, D.J.; Bienvenu, T.; Dall’Ava-Santucci, J.; Dinh-Xuan, A.T. Reduced exhaled NO is related to impaired nasal potential difference in patients with cystic fibrosis. Vascul. Pharmacol. 2005, 43, 385–389. [Google Scholar] [CrossRef]
- Steagall, W.K.; Elmer, H.L.; Brady, K.G.; Kelley, T.J. Cystic fibrosis transmembrane conductance regulator-dependent regulation of epithelial inducible nitric oxide synthase expression. Am. J. Respir. Cell Mol. Biol. 2000, 22, 45–50. [Google Scholar] [CrossRef]
- Mhanna, M.J.; Ferkol, T.; Martin, R.J.; Dreshaj, I.A.; van Heeckeren, A.M.; Kelley, T.J.; Haxhiu, M.A. Nitric oxide deficiency contributes to impairment of airway relaxation in cystic fibrosis mice. Am. J. Respir. Cell Mol. Biol. 2001, 24, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Keen, C.; Olin, A.C.; Edentoft, A.; Gronowitz, E.; Strandvik, B. Airway nitric oxide in patients with cystic fibrosis is associated with pancreatic function, Pseudomonas infection, and polyunsaturated fatty acids. Chest 2007, 131, 1857–1864. [Google Scholar] [CrossRef]
- Grasemann, H.; Ratjen, F. Nitric oxide and L-arginine deficiency in cystic fibrosis. Curr. Pharm. Des. 2012, 18, 726–736. [Google Scholar] [CrossRef]
- Grasemann, H.; Knauer, N.; Buscher, R.; Hubner, K.; Drazen, J.M.; Ratjen, F. Airway nitric oxide levels in cystic fibrosis patients are related to a polymorphism in the neuronal nitric oxide synthase gene. Am. J. Respir. Crit. Care Med. 2000, 162, 2172–2176. [Google Scholar] [CrossRef] [Green Version]
- Pifferi, M.; Bush, A.; Maggi, F.; Michelucci, A.; Ricci, V.; Conidi, M.E.; Cangiotti, A.M.; Bodini, A.; Simi, P.; Macchia, P.; et al. Nasal nitric oxide and nitric oxide synthase expression in primary ciliary dyskinesia. Eur. Respir. J. 2011, 37, 572–577. [Google Scholar] [CrossRef] [Green Version]
- Noone, P.G.; Leigh, M.W.; Sannuti, A.; Minnix, S.L.; Carson, J.L.; Hazucha, M.; Zariwala, M.A.; Knowles, M.R. Primary ciliary dyskinesia: Diagnostic and phenotypic features. Am. J. Respir. Crit. Care Med. 2004, 169, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Degano, B.; Valmary, S.; Serrano, E.; Brousset, P.; Arnal, J.F. Expression of nitric oxide synthases in primary ciliary dyskinesia. Hum. Pathol. 2011, 42, 1855–1861. [Google Scholar] [CrossRef]
- Kuek, L.E.; Lee, R.J. First contact: The role of respiratory cilia in host-pathogen interactions in the airways. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 319, L603–L619. [Google Scholar] [CrossRef]
- Sisson, J.H.; Pavlik, J.A.; Wyatt, T.A. Alcohol stimulates ciliary motility of isolated airway axonemes through a nitric oxide, cyclase, and cyclic nucleotide-dependent kinase mechanism. Alcohol. Clin. Exp. Res. 2009, 33, 610–616. [Google Scholar] [CrossRef] [Green Version]
- McMahon, D.B.; Workman, A.D.; Kohanski, M.A.; Carey, R.M.; Freund, J.R.; Hariri, B.M.; Chen, B.; Doghramji, L.J.; Adappa, N.D.; Palmer, J.N.; et al. Protease-activated receptor 2 activates airway apical membrane chloride permeability and increases ciliary beating. FASEB J. 2018, 32, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, P.A.; Garvin, J.L. Trafficking and activation of eNOS in epithelial cells. Acta Physiol. Scand. 2003, 179, 107–114. [Google Scholar] [CrossRef]
- Dudzinski, D.M.; Michel, T. Life history of eNOS: Partners and pathways. Cardiovasc. Res. 2007, 75, 247–260. [Google Scholar] [CrossRef]
- Davis, B.J.; Xie, Z.; Viollet, B.; Zou, M.H. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes 2006, 55, 496–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.P.; Mitchelhill, K.I.; Michell, B.J.; Stapleton, D.; Rodriguez-Crespo, I.; Witters, L.A.; Power, D.A.; Ortiz de Montellano, P.R.; Kemp, B.E. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999, 443, 285–289. [Google Scholar] [CrossRef] [Green Version]
- Thors, B.; Halldorsson, H.; Thorgeirsson, G. eNOS activation mediated by AMPK after stimulation of endothelial cells with histamine or thrombin is dependent on LKB1. Biochim. Biophys. Acta 2011, 1813, 322–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, B.; Chhipa, R.R. Evolving Lessons on the Complex Role of AMPK in Normal Physiology and Cancer. Trends Pharmacol. Sci. 2016, 37, 192–206. [Google Scholar] [CrossRef] [Green Version]
- Murphy, B.A.; Fakira, K.A.; Song, Z.; Beuve, A.; Routh, V.H. AMP-activated protein kinase and nitric oxide regulate the glucose sensitivity of ventromedial hypothalamic glucose-inhibited neurons. Am. J. Physiol. Cell Physiol. 2009, 297, C750–C758. [Google Scholar] [CrossRef] [Green Version]
- Morrow, V.A.; Foufelle, F.; Connell, J.M.; Petrie, J.R.; Gould, G.W.; Salt, I.P. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J. Biol. Chem. 2003, 278, 31629–31639. [Google Scholar] [CrossRef] [Green Version]
- El-Mas, M.M.; Fan, M.; Abdel-Rahman, A.A. Facilitation of myocardial PI3K/Akt/nNOS signaling contributes to ethanol-evoked hypotension in female rats. Alcohol. Clin. Exp. Res. 2009, 33, 1158–1168. [Google Scholar] [CrossRef]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef]
- Michell, B.J.; Chen, Z.; Tiganis, T.; Stapleton, D.; Katsis, F.; Power, D.A.; Sim, A.T.; Kemp, B.E. Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J. Biol. Chem. 2001, 276, 17625–17628. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carey, R.M.; Hariri, B.M.; Adappa, N.D.; Palmer, J.N.; Lee, R.J. HSP90 Modulates T2R Bitter Taste Receptor Nitric Oxide Production and Innate Immune Responses in Human Airway Epithelial Cells and Macrophages. Cells 2022, 11, 1478. https://doi.org/10.3390/cells11091478
Carey RM, Hariri BM, Adappa ND, Palmer JN, Lee RJ. HSP90 Modulates T2R Bitter Taste Receptor Nitric Oxide Production and Innate Immune Responses in Human Airway Epithelial Cells and Macrophages. Cells. 2022; 11(9):1478. https://doi.org/10.3390/cells11091478
Chicago/Turabian StyleCarey, Ryan M., Benjamin M. Hariri, Nithin D. Adappa, James N. Palmer, and Robert J. Lee. 2022. "HSP90 Modulates T2R Bitter Taste Receptor Nitric Oxide Production and Innate Immune Responses in Human Airway Epithelial Cells and Macrophages" Cells 11, no. 9: 1478. https://doi.org/10.3390/cells11091478
APA StyleCarey, R. M., Hariri, B. M., Adappa, N. D., Palmer, J. N., & Lee, R. J. (2022). HSP90 Modulates T2R Bitter Taste Receptor Nitric Oxide Production and Innate Immune Responses in Human Airway Epithelial Cells and Macrophages. Cells, 11(9), 1478. https://doi.org/10.3390/cells11091478