
The Autophagy Protein Pacer Positively Regulates the Therapeutic Potential of Mesenchymal Stem Cells in a Mouse Model of DSS-Induced Colitis

 ,
,  , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Mice
2.2. Cells
2.3. Autophagy Assays
2.4. Real-Time PCR
2.5. Immunoblot
2.6. T-Cell Proliferation Assay
2.7. DSS-induced Colitis Mouse Model
2.8. Histological Evaluation and Immunohistochemistry (IHC)
2.9. Statistical Analysis
3. Results
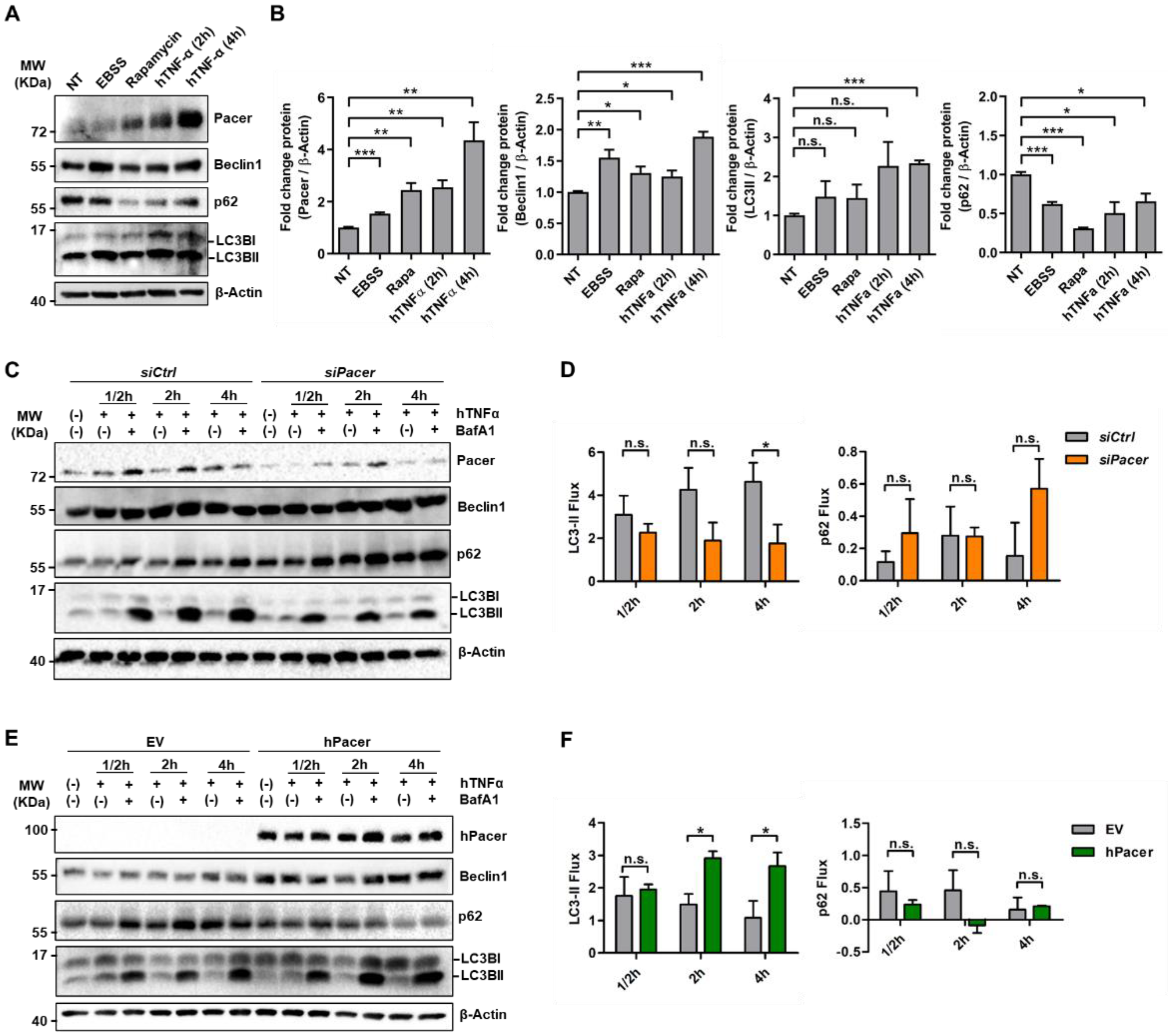
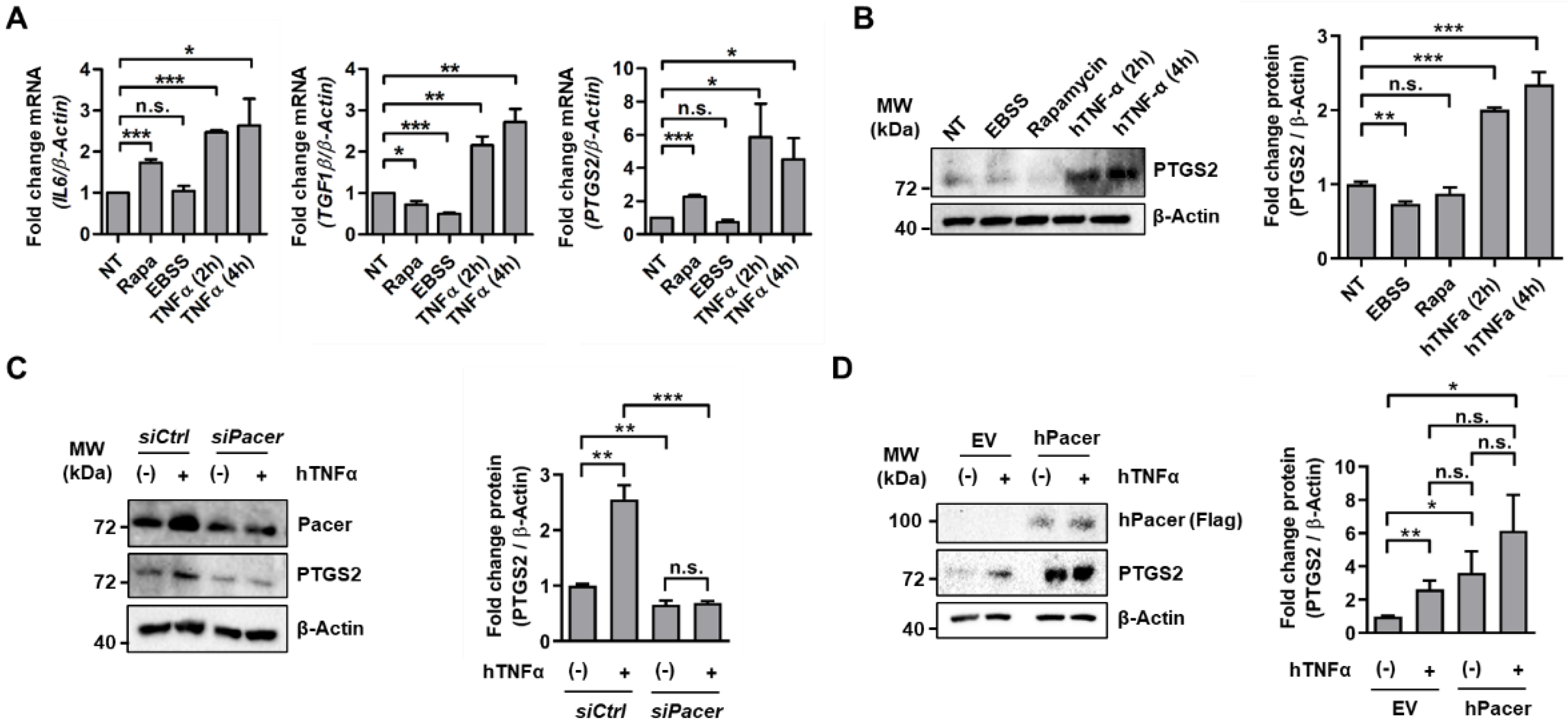
3.1. The Autophagy Enhancer Pacer Is Upregulated in MSC in Response to the Pro-Inflammatory Cytokine TNFα
3.2. Pacer Is Required for the Immunosuppressive Function of MSC
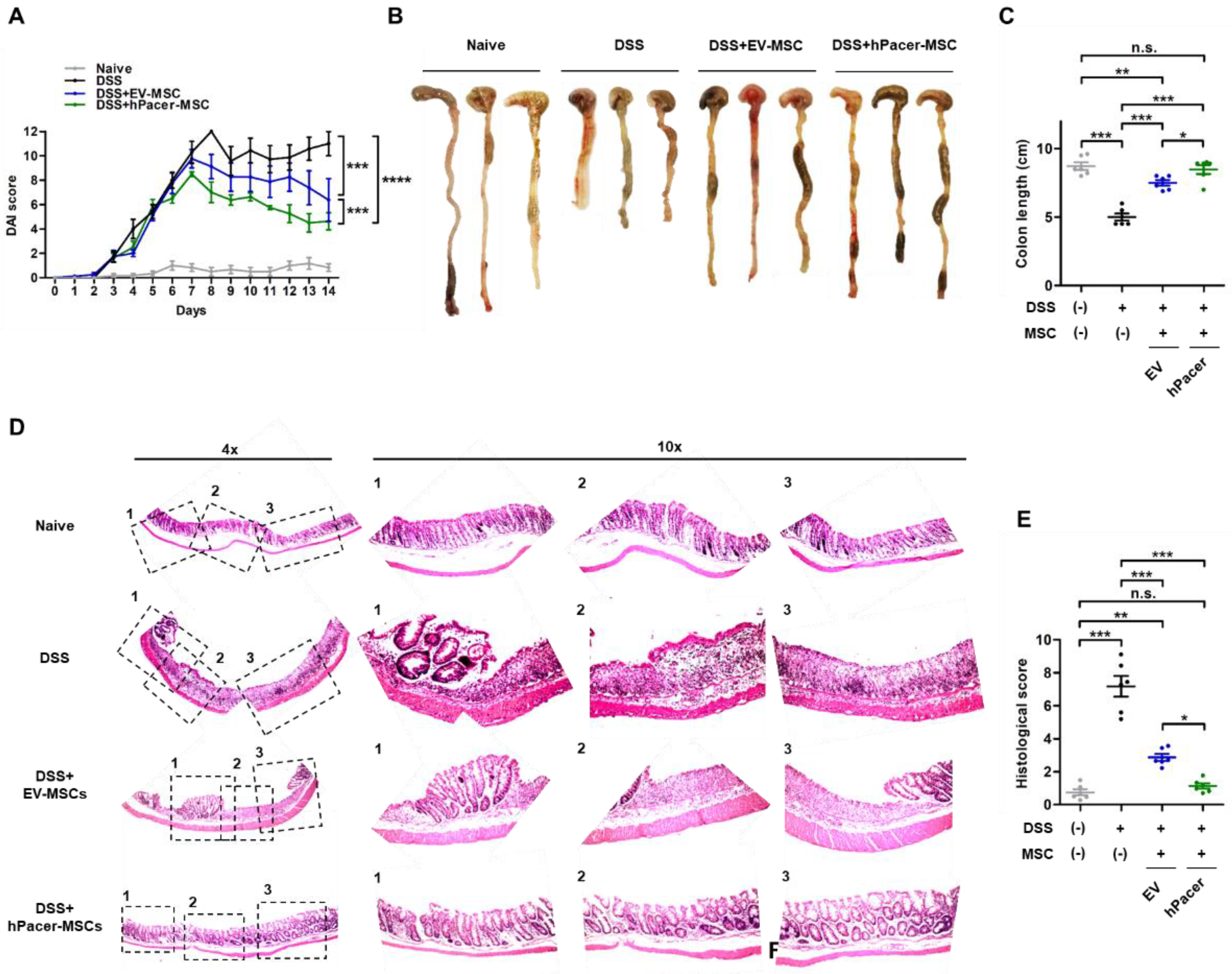
3.3. Increasing Pacer Levels in MSC Improves Their Therapeutic Effect in a Mouse Model of DSS-Induced Colitis
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ashton, B.A.; Allen, T.D.; Howlett, C.R.; Eaglesom, C.C.; Hattori, A.; Owen, M. Formation of bone and cartilage by marrow stromal cells in diffusion chambers in vivo. Clin. Orthop. Relat. Res. 1980, 151, 294–307. [Google Scholar] [CrossRef]
- Bab, I.; Ashton, B.A.; Gazit, D.; Marx, G.; Williamson, M.C.; Owen, M.E. Kinetics and differentiation of marrow stromal cells in diffusion chambers in vivo. J. Cell Sci. 1986, 84, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Castro-Malaspina, H.; Gay, R.E.; Resnick, G.; Kapoor, N.; Meyers, P.; Chiarieri, D.; McKenzie, S.; Broxmeyer, H.E.; Moore, M.A. Characterization of human bone marrow fibroblast colony-forming cells (CFU-F) and their progeny. Blood 1980, 56, 289–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazdic, M.; Volarevic, V.; Arsenijevic, N.; Stojkovic, M. Mesenchymal stem cells: A friend or foe in immune-mediated diseases. Stem Cell Rev. Rep. 2015, 11, 280–287. [Google Scholar] [CrossRef]
- Volarevic, V.; Gazdic, M.; Simovic Markovic, B.; Jovicic, N.; Djonov, V.; Arsenijevic, N. Mesenchymal stem cell-derived factors: Immuno-modulatory effects and therapeutic potential. Biofactors 2017, 43, 633–644. [Google Scholar] [CrossRef]
- Phinney, D.G.; Pittenger, M.F. Concise Review: MSC-Derived Exosomes for Cell-Free Therapy. Stem Cells 2017, 35, 851–858. [Google Scholar] [CrossRef] [Green Version]
- Phinney, D.G.; Prockop, D.J. Concise review: Mesenchymal stem/multipotent stromal cells: The state of transdifferentiation and modes of tissue repair—Current views. Stem Cells 2007, 25, 2896–2902. [Google Scholar] [CrossRef]
- Ankrum, J.A.; Ong, J.F.; Karp, J.M. Mesenchymal stem cells: Immune evasive, not immune privileged. Nat. Biotechnol. 2014, 32, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, X.; Cao, W.; Shi, Y. Plasticity of mesenchymal stem cells in immunomodulation: Pathological and therapeutic implications. Nat. Immunol. 2014, 15, 1009–1016. [Google Scholar] [CrossRef]
- Eichele, D.D.; Kharbanda, K.K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J. Gastroenterol. 2017, 23, 6016–6029. [Google Scholar] [CrossRef]
- Chang, Y.L.; Lo, H.Y.; Cheng, S.P.; Chang, K.T.; Lin, X.F.; Lee, S.P.; Hsieh, M.F.; Chan, C.K. Therapeutic effects of a single injection of human umbilical mesenchymal stem cells on acute and chronic colitis in mice. Sci. Rep. 2019, 9, 5832. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, S.; Popp, V.; Kindermann, M.; Gerlach, K.; Weigmann, B.; Fichtner-Feigl, S.; Neurath, M.F. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 2017, 12, 1295–1309. [Google Scholar] [CrossRef] [PubMed]
- Braegger, C.P.; Nicholls, S.; Murch, S.H.; Stephens, S.; MacDonald, T.T. Tumour necrosis factor alpha in stool as a marker of intestinal inflammation. Lancet 1992, 339, 89–91. [Google Scholar] [CrossRef]
- Breese, E.J.; Michie, C.A.; Nicholls, S.W.; Murch, S.H.; Williams, C.B.; Domizio, P.; Walker-Smith, J.A.; MacDonald, T.T. Tumor necrosis factor alpha-producing cells in the intestinal mucosa of children with inflammatory bowel disease. Gastroenterology 1994, 106, 1455–1466. [Google Scholar] [CrossRef]
- Murch, S.H.; Lamkin, V.A.; Savage, M.O.; Walker-Smith, J.A.; MacDonald, T.T. Serum concentrations of tumour necrosis factor alpha in childhood chronic inflammatory bowel disease. Gut 1991, 32, 913–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, M.; Watanabe, N.; Neda, H.; Yamauchi, N.; Okamoto, T.; Sasaki, H.; Tsuji, Y.; Akiyama, S.; Tsuji, N.; Niitsu, Y. Serum tumor necrosis factor activity in inflammatory bowel disease. Immunopharmacol. Immunotoxicol. 1992, 14, 451–461. [Google Scholar] [CrossRef]
- Shah, B.; Mayer, L. Current status of monoclonal antibody therapy for the treatment of inflammatory bowel disease. Expert Rev. Clin. Immunol. 2010, 6, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Roda, G.; Jharap, B.; Neeraj, N.; Colombel, J.F. Loss of Response to Anti-TNFs: Definition, Epidemiology, and Management. Clin. Transl. Gastroenterol. 2016, 7, e135. [Google Scholar] [CrossRef]
- Singh, U.P.; Singh, N.P.; Singh, B.; Mishra, M.K.; Nagarkatti, M.; Nagarkatti, P.S.; Singh, S.R. Stem cells as potential therapeutic targets for inflammatory bowel disease. Front. Biosci. 2010, 2, 993–1008. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yuan, Q.; Xie, L. Mesenchymal Stem Cell-Based Immunomodulation: Properties and Clinical Application. Stem Cells Int. 2018, 2018, 3057624. [Google Scholar] [CrossRef]
- Kyurkchiev, D.; Bochev, I.; Ivanova-Todorova, E.; Mourdjeva, M.; Oreshkova, T.; Belemezova, K.; Kyurkchiev, S. Secretion of immunoregulatory cytokines by mesenchymal stem cells. World J. Stem Cells 2014, 6, 552–570. [Google Scholar] [CrossRef]
- Yan, L.; Zheng, D.; Xu, R.H. Critical Role of Tumor Necrosis Factor Signaling in Mesenchymal Stem Cell-Based Therapy for Autoimmune and Inflammatory Diseases. Front. Immunol. 2018, 9, 1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, H.; Wang, Y.; Jin, Y.; Zhang, Q.; Zhang, Y.; Wang, L.; Shen, B.; Yin, S.; Liu, W.; Cui, L.; et al. A critical role of IFNgamma in priming MSC-mediated suppression of T cell proliferation through up-regulation of B7-H1. Cell Res. 2008, 18, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Polchert, D.; Sobinsky, J.; Douglas, G.; Kidd, M.; Moadsiri, A.; Reina, E.; Genrich, K.; Mehrotra, S.; Setty, S.; Smith, B.; et al. IFN-gamma activation of mesenchymal stem cells for treatment and prevention of graft versus host disease. Eur. J. Immunol. 2008, 38, 1745–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakovljevic, J.; Harrell, C.R.; Fellabaum, C.; Arsenijevic, A.; Jovicic, N.; Volarevic, V. Modulation of autophagy as new approach in mesenchymal stem cell-based therapy. Biomed. Pharmacother. 2018, 104, 404–410. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Oliver, L.; Hue, E.; Priault, M.; Vallette, F.M. Basal autophagy decreased during the differentiation of human adult mesenchymal stem cells. Stem Cells Dev. 2012, 21, 2779–2788. [Google Scholar] [CrossRef]
- Salemi, S.; Yousefi, S.; Constantinescu, M.A.; Fey, M.F.; Simon, H.U. Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res. 2012, 22, 432–435. [Google Scholar] [CrossRef]
- Gao, L.; Cen, S.; Wang, P.; Xie, Z.; Liu, Z.; Deng, W.; Su, H.; Wu, X.; Wang, S.; Li, J.; et al. Autophagy Improves the Immunosuppression of CD4+ T Cells by Mesenchymal Stem Cells through Transforming Growth Factor-beta1. Stem Cells Transl. Med. 2016, 5, 1496–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, Y.; Liu, W.J.; Xue, P.; Ma, Y.; Zhang, L.Q.; Zhu, B.; Qi, M.; Li, L.Y.; Zhang, Y.J.; Wang, Q.T.; et al. Autophagy promotes MSC-mediated vascularization in cutaneous wound healing via regulation of VEGF secretion. Cell Death Dis. 2018, 9, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, S.; Nassif, M.; Vicencio, E.; Arcos, J.; Labrador, L.; Cortes, B.I.; Cortez, C.; Bergmann, C.A.; Espinoza, S.; Hernandez, M.F.; et al. Network approach identifies Pacer as an autophagy protein involved in ALS pathogenesis. Mol. Neurodegener. 2019, 14, 14. [Google Scholar] [CrossRef]
- Cheng, X.; Ma, X.; Ding, X.; Li, L.; Jiang, X.; Shen, Z.; Chen, S.; Liu, W.; Gong, W.; Sun, Q. Pacer Mediates the Function of Class III PI3K and HOPS Complexes in Autophagosome Maturation by Engaging Stx17. Mol. Cell 2017, 65, 1029–1043.e1025. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Ma, X.; Zhu, Q.; Song, D.; Ding, X.; Li, L.; Jiang, X.; Wang, X.; Tian, R.; Su, H.; et al. Pacer Is a Mediator of mTORC1 and GSK3-TIP60 Signaling in Regulation of Autophagosome Maturation and Lipid Metabolism. Mol. Cell 2019, 73, 788–802.e787. [Google Scholar] [CrossRef] [Green Version]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Roederer, M. Interpretation of cellular proliferation data: Avoid the panglossian. Cytometry A 2011, 79, 95–101. [Google Scholar] [CrossRef]
- Egger, B.; Bajaj-Elliott, M.; MacDonald, T.T.; Inglin, R.; Eysselein, V.E.; Buchler, M.W. Characterisation of acute murine dextran sodium sulphate colitis: Cytokine profile and dose dependency. Digestion 2000, 62, 240–248. [Google Scholar] [CrossRef]
- Gonzalez-Rey, E.; Delgado, M. Therapeutic application of mesenchymal stromal cells in murine models of inflammatory bowel disease. Methods Mol. Biol. 2014, 1213, 331–339. [Google Scholar] [CrossRef]
- Dang, S.; Xu, H.; Xu, C.; Cai, W.; Li, Q.; Cheng, Y.; Jin, M.; Wang, R.X.; Peng, Y.; Zhang, Y.; et al. Autophagy regulates the therapeutic potential of mesenchymal stem cells in experimental autoimmune encephalomyelitis. Autophagy 2014, 10, 1301–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.Y.; Park, H.J.; Kim, H.N.; Oh, S.H.; Bae, J.S.; Ha, H.J.; Lee, P.H. Mesenchymal stem cells enhance autophagy and increase beta-amyloid clearance in Alzheimer disease models. Autophagy 2014, 10, 32–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Wang, Q.; Feng, X.; Zhang, Z.; Geng, L.; Xu, T.; Wang, D.; Sun, L. Umbilical Cord-Derived Mesenchymal Stem Cells Suppress Autophagy of T Cells in Patients with Systemic Lupus Erythematosus via Transfer of Mitochondria. Stem Cells Int. 2016, 2016, 4062789. [Google Scholar] [CrossRef]
- Strober, W.; Fuss, I.J. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology 2011, 140, 1756–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Soria-Juan, B.; Escacena, N.; Capilla-Gonzalez, V.; Aguilera, Y.; Llanos, L.; Tejedo, J.R.; Bedoya, F.J.; Juan, V.; De la Cuesta, A.; Ruiz-Salmeron, R.; et al. Cost-Effective, Safe, and Personalized Cell Therapy for Critical Limb Ischemia in Type 2 Diabetes Mellitus. Front. Immunol. 2019, 10, 1151. [Google Scholar] [CrossRef]
- Squillaro, T.; Peluso, G.; Galderisi, U. Clinical Trials with Mesenchymal Stem Cells: An Update. Cell Transplant. 2016, 25, 829–848. [Google Scholar] [CrossRef] [Green Version]
- Hosseini, S.; Taghiyar, L.; Safari, F.; Baghaban Eslaminejad, M. Regenerative Medicine Applications of Mesenchymal Stem Cells. Adv. Exp. Med. Biol. 2018, 1089, 115–141. [Google Scholar] [CrossRef]
- Spees, J.L.; Lee, R.H.; Gregory, C.A. Mechanisms of mesenchymal stem/stromal cell function. Stem Cell Res. Ther. 2016, 7, 125. [Google Scholar] [CrossRef] [Green Version]
- Prockop, D.J. Repair of tissues by adult stem/progenitor cells (MSCs): Controversies, myths, and changing paradigms. Mol. Ther. 2009, 17, 939–946. [Google Scholar] [CrossRef]
- Ceccariglia, S.; Cargnoni, A.; Silini, A.R.; Parolini, O. Autophagy: A potential key contributor to the therapeutic action of mesenchymal stem cells. Autophagy 2020, 16, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Ouyang, Y.; Li, W.; Wang, P.; Deng, H.; Song, B.; Hou, J.; Chen, Z.; Xie, Z.; Liu, Z.; et al. Autophagy Plays a Protective Role in Tumor Necrosis Factor-alpha-Induced Apoptosis of Bone Marrow-Derived Mesenchymal Stem Cells. Stem Cells Dev. 2016, 25, 788–797. [Google Scholar] [CrossRef] [PubMed]
- English, K.; Barry, F.P.; Field-Corbett, C.P.; Mahon, B.P. IFN-gamma and TNF-alpha differentially regulate immunomodulation by murine mesenchymal stem cells. Immunol. Lett. 2007, 110, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Chinnadurai, R.; Copland, I.B.; Ng, S.; Garcia, M.; Prasad, M.; Arafat, D.; Gibson, G.; Kugathasan, S.; Galipeau, J. Mesenchymal Stromal Cells Derived from Crohn’s Patients Deploy Indoleamine 2,3-dioxygenase-mediated Immune Suppression, Independent of Autophagy. Mol. Ther. 2015, 23, 1248–1261. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.W.; Moon, S.J.; Park, M.J.; Kim, B.M.; Kim, E.K.; Lee, S.H.; Lee, E.J.; Chung, B.H.; Yang, C.W.; Cho, M.L. Optimization of adipose tissue-derived mesenchymal stem cells by rapamycin in a murine model of acute graft-versus-host disease. Stem Cell Res. Ther. 2015, 6, 202. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergmann, C.A.; Beltran, S.; Vega-Letter, A.M.; Murgas, P.; Hernandez, M.F.; Gomez, L.; Labrador, L.; Cortés, B.I.; Poblete, C.; Quijada, C.; et al. The Autophagy Protein Pacer Positively Regulates the Therapeutic Potential of Mesenchymal Stem Cells in a Mouse Model of DSS-Induced Colitis. Cells 2022, 11, 1503. https://doi.org/10.3390/cells11091503
Bergmann CA, Beltran S, Vega-Letter AM, Murgas P, Hernandez MF, Gomez L, Labrador L, Cortés BI, Poblete C, Quijada C, et al. The Autophagy Protein Pacer Positively Regulates the Therapeutic Potential of Mesenchymal Stem Cells in a Mouse Model of DSS-Induced Colitis. Cells. 2022; 11(9):1503. https://doi.org/10.3390/cells11091503
Chicago/Turabian StyleBergmann, Cristian A., Sebastian Beltran, Ana Maria Vega-Letter, Paola Murgas, Maria Fernanda Hernandez, Laura Gomez, Luis Labrador, Bastián I. Cortés, Cristian Poblete, Cristobal Quijada, and et al. 2022. "The Autophagy Protein Pacer Positively Regulates the Therapeutic Potential of Mesenchymal Stem Cells in a Mouse Model of DSS-Induced Colitis" Cells 11, no. 9: 1503. https://doi.org/10.3390/cells11091503
APA StyleBergmann, C. A., Beltran, S., Vega-Letter, A. M., Murgas, P., Hernandez, M. F., Gomez, L., Labrador, L., Cortés, B. I., Poblete, C., Quijada, C., Carrion, F., Woehlbier, U., & Manque, P. A. (2022). The Autophagy Protein Pacer Positively Regulates the Therapeutic Potential of Mesenchymal Stem Cells in a Mouse Model of DSS-Induced Colitis. Cells, 11(9), 1503. https://doi.org/10.3390/cells11091503