Abstract
BCR::ABL1-negative myeloproliferative neoplasms (MPNs) include three major subgroups—polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF)—which are characterized by aberrant hematopoietic proliferation with an increased risk of leukemic transformation. Besides the driver mutations, which are JAK2, CALR, and MPL, more than twenty additional mutations have been identified through the use of next-generation sequencing (NGS), which can be involved with pathways that regulate epigenetic modifications, RNA splicing, or DNA repair. The aim of this short review is to highlight the impact of molecular biology on the diagnosis, prognosis, and therapeutic management of patients with PV, ET, and PMF.
1. Introduction
BCR::ABL1-negative myeloproliferative neoplasms (MPNs) are a group of stem cell disorders characterized by aberrant hematopoietic proliferation with an increased risk of leukemic transformation. BCR::ABL1-negative MPNs include three major subgroups: polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) [1]. A major advancement in our understanding of the genetic landscape of MPNs has been made following the discovery of the JAK2 V617F somatic mutation in 2005. Most of the MPNs have one or more somatic mutations that activate a signaling pathway, conferring a proliferative advantage to the neoplastic cells. Regarding PV, ET, and PMF, the main “driver” mutations affect the JAK2, CALR, and MPL genes, activate the JAK–STAT signaling pathway, and induce a proliferation and antiapoptotic effect in cells. Besides these driver mutations, additional mutations may coexist in pathways that regulate epigenetic modifications, RNA splicing, or DNA repair [2,3,4]. In 2020 and 2021, Loscocco et al. and Ross Dm et al., respectively, listed twenty additional mutations (ASXL1, CBL, DNMT3A, EZH2, IDH1/2, RUNX1, SETBP1, SF3B1, SRSF2, TET2, ZRSR2, LNK/SH2B3, NRAS/KRAS, PTPN11, TP53, and U2AF1) that they identified using next-generation sequencing (NGS) analyses. These mutations occur between 0.5 and 10% in patients with MPN [5,6]. Table 1 summarizes all additional mutations described in patients with PV, ET, and PMF with their respective types and frequencies. The aim of this short review is to highlight the impact of NGS on the diagnostic, prognostic, and therapeutic management of patients with PV, ET, and PMF in 2022. The SF3B1 mutation (associated with myelodysplastic syndrome) is not within the scope of this review.

Table 1.
Recurrent additional mutations in patients with MPN.
2. Impact of Mutational Profile on PV, ET, and PMF Diagnosis
MPNs are characterized by a constitutive activation of the JAK–STAT pathway. Mutations in JAK2, CALR, and MPL are referred to as “driver mutations” because they lead to the determination of the MPN phenotype. In addition, based on the WHO’s 2016 revision to the classification of myeloid neoplasms and acute leukemia, these mutations, together with the clinical, biological, and histological features, allow for a determination of the diagnosis [1]. Even though these three mutations cannot usually coexist, a few cases have been reported where patients turned out to have rare, mutated variations of JAK2 and MPL, particularly in patients initially ranked “without driver mutations” or with a low allelic burden for driver mutations in ET [5,34,35,36]. However, a subset (10–15%) of patients with MPN, called triple-negative (TN) patients, do not present any of these driver mutations but may harbor others that might be associated with a shorter overall survival (OS) and/or leukemia-free survival (LFS) [7,37,38].
2.1. Somatic Myeloid NGS Panel and Erythrocytosis Exploration
As described in the RESPONSE clinical trial, almost 99% of PV cases are associated with somatic mutations in JAK2 (97.3% in JAK2 V617F with a mean variant allele frequency (VAF) of approximately 84% and 1.3% in exon 12) and are hypercellular with trilineage hyperplasia (panmyelosis) with evidence of an increased red cell mass [39]. Moreover, many clinical situations can occur and should be considered. We suppose in these clinical cases that a secondary cause of polyglobulia is not present:
- Clinical situation No. 1: The patient has an absolute erythrocytosis according to the WHO 2016 PV diagnostic criteria (hemoglobin levels > 18.5 g/dL in men (hematocrit > 55.5%) or >16.5 g/dL in women (hematocrit > 49.5%)), and the JAK2 mutation with a high VAF is present. If the EPO level is suppressed (as in 80% of PV), a bone marrow (BM) biopsy is not required [1,40]. A PV diagnosis is confirmed.
- Clinical situation No. 2: The patient has an absolute erythrocytosis according to the WHO 2016 PV diagnostic criteria without the JAK2 mutation (or with a low VAF). Even if the EPO level is suppressed, a BM biopsy is required to highlight BM hypercellularity (present in 90% of PV) and to confirm a PV diagnosis [41].
- Clinical situation No. 3: The patient has an erythrocytosis according to the WHO 2016 classification (HGB > 16.5 g/dL (men) and >16 g/dL (women), HCT > 49% (men) and >48% (women) or increased red cell mass) without meeting the absolute erythrocytosis criteria. The JAK2 mutation is present at a high ratio, and a BM biopsy highlights BM hypercellularity. A PV diagnosis is confirmed.
In these three clinical situations outlined above, a PV diagnosis is not a clinical issue. However, as described above, 1% of patients with PV are JAK2 negative, as the EPO level is not suppressed in 20% of cases, and the BM does not show panmyelosis in 10% of JAK2-positive erythrocytosis. In these rare cases, in which PV is suspected and/or the WHO diagnostic criteria are met despite the absence of a detectable JAK2 mutation, a somatic myeloid NGS panel can help with PV diagnosis in addition to research on noncanonical nucleotide changes leading to V617F or compound mutations in exon 14 [42,43]. In association with concordant BM features, NGS can provide evidence of clonal hematopoiesis, notably by detecting alternative activating JAK–STAT pathway driver mutations, such as LNK/SH2B3 or nondriver mutations such as TET2 or DNMT3A [44].
When the myeloid NGS panel and JAK2 are both negative, the detection of mutations in other genes via germline NGS panels may be useful to diagnose hereditary erythrocytosis [44]. In the majority of laboratories, these genes are typically not included in myeloid NGS panels, but there is some overlap between the germline and somatic candidate gene lists [6]. In the case of JAK2-negative erythrocytosis without panmyelosis in the BM and the presence of a nondriver mutation found through NGS, clonal hematopoiesis of indeterminate potential (CHIP) must be discussed, especially when genes such as DNMT3A, ASXL1, or TET2 are involved [45]. If the somatic and germline myeloid NGS panel are also negative, idiopathic or secondary erythrocytosis should be considered, especially if the EPO level is not suppressed (normal or increased). The different erythrocytosis and PV diagnostic approaches are summarized in Figure 1.
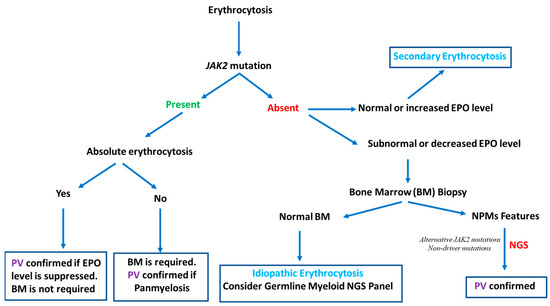
Figure 1.
Erythrocytosis and PV diagnostic approaches: NGS contribution to PV diagnosis.
2.2. Impact of Mutational Status on ET and PMF Diagnosis
ET is characterized by thrombocytosis and megakaryocytic hyperplasia. Because there are many causes of reactive thrombocytosis, the use of driver or nondriver mutations to provide the proof of clonality is essential to make an ET diagnosis. Driver mutations in JAK2, CALR, and MPL are detected in 60–65%, 20–25%, and 5% of patients with ET, respectively [2,30]. Therefore, between 10 and 20% of patients with ET are TN. Outside of some notable cases reported, JAK2, CALR, and MPL driver mutations are mutually exclusive in ET. Regarding the JAK2 V617F mutation in ET, the median VAF is lower than in PV or PMF. In ET, the median VAF is approximately 10 to 20% and rarely exceeds 50% compared to a median VAF frequency above 50% in PV and PMF [46,47]. With regard to CALR, more than 50 mutations have been described for which CALR type 1 (52 base-pair deletions) and type 2 (5 base-pair insertions) represent 80% of cases [48]. In the case of thrombocytosis (with a platelet count > 450.109/L) with no identified driver mutation, no evidence for reactive thrombocytosis, and a strong suspicion of ET, a BM biopsy should be the first intention exam before the myeloid NGS panel. According to the WHO 2016 ET diagnostic criteria, a BM biopsy showing proliferation mainly in the megakaryocytic lineage with increased numbers of enlarged, mature megakaryocytes with hyperlobulated nuclei is a major diagnostic criterion, whereas the presence of a clonal marker found through NGS, for example, is a minor criterion [1]. In the absence of any MPN features in the BM, an ET diagnosis cannot be retained. On the contrary, in the presence of concordant BM, a noncanonical mutation in CALR, JAK2, or MPL and/or nondriver additional mutations detected through NGS can help to make a definitive diagnosis of ET and can help provide information on the patient’s prognosis (see below).
Regarding PMF, the distribution of driver mutations is similar to that in ET, as it is also 10% of TN patients [49]. As with patients with ET, NGS can detect noncanonical mutations in CALR, JAK2, or MPL and/or nondriver additional mutations. Contrary to ET, if the BM biopsy shows the presence of megakaryocytic proliferation and atypia accompanied by either reticulin and/or collagen fibrosis grade 2 or 3 and if reactive causes can be excluded, then a clonality marker is not strictly necessary to make a PMF diagnosis. The WHO 2016 MPN diagnostic criteria revision identified a fourth and independent entity: prefibrotic myelofibrosis (pre-PMF) [1]. The distinction between ET and pre-PMF is not always simple and is theoretically based on morphological criteria with variable reproducibility [50,51]. There are also more robust, minor pre-PMF diagnostic criteria, such as anemia, palpable splenomegaly, leukocytosis, or a raised lactate dehydrogenase level [1]. These minor criteria are classically absent in ET, which instead has criteria such as a high VAF of JAK2V617F above 50% and/or the presence of additional nondriver mutations found through NGS, such as NRAS, ZRSR2, U2AF1, and SRSF2 (see Table 1) [46]. The different ET and PMF diagnostic approaches are summarized in Figure 2.
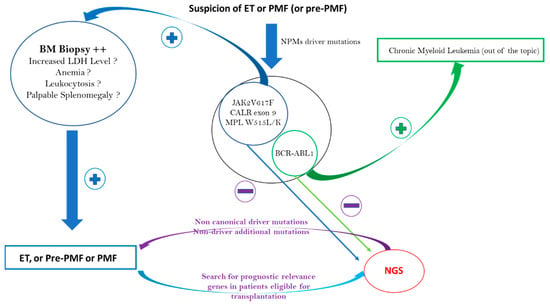
Figure 2.
ET, pre-PMF, and PMF diagnostic approaches: NGS contribution to diagnosis.
3. Impact of Somatic Myeloid NGS Panel in MPN Prognosis and Treatment Management
In 2016, Tefferi et al. performed deep NGS in 133 patients with PV and 183 patients with ET. A little more than half of the patients with PV and patients with ET presented additional nondriver mutations. The most frequent were TET2 and ASXL1. The TET2 mutation occurs in 15 and 10% of patients with PV and patients with ET, respectively (Table 1). In patients with both JAK2 V617F and TET2 mutations, the order in which the JAK2 and TET2 mutations were acquired influenced the clinical features, the response to targeted therapy, the biology of stem and progenitor cells, and the clonal evolution. Indeed, as compared with patients in whom the TET2 mutation was acquired first, patients in whom the JAK2 mutation was acquired first showed a larger probability of presenting with PV than ET, an increased risk of thrombosis, and an increased in vitro sensitivity of JAK2-mutant progenitors to ruxolitinib. Mutation order influenced the proliferative response to JAK2 V617F and the capacity of double-mutant hematopoietic cells and progenitor cells to generate colony-forming cells [8]. High-risk mutations (HRM = adverse prognostic) included ASXL1, SRSF2, and IDH2 in patients with PV, and SH2B3, SF3B1, U2AF1, TP53, IDH2, and EZH2 in patients with ET. These HRMs may be combined in 15% of patients with PV and/or ET. HRMs were associated with inferior OS in patients with PV (median OS, 7.7 vs. 16.9 years) and patients with ET (median OS, 9 vs. 22 years) [9].
The implementation of NGS techniques allows for the identification of additional mutations in patients that influence survival and the risk of transformation into myelofibrosis. A prognostic score called the mutation-enhanced international prognostic model in PV and ET (MIPSS-PV and MIPSS-ET), based on an age > 67 years, leukocytosis (≥15 × 109/L), an abnormal karyotype, and the presence of additional poor prognostic mutations (SRSF2 in PV, SRSF2, SF3B1, U2AF1, and TP53 in ET) allowed for the establishment of a classification of the survival of patients. The classification of low risk, intermediate risk, and high risk with corresponding median survivals of 24, 13.1, and 3.2 years, respectively, can help modulate the management, prognosis, and therapy for the patient [52].
As for patients with PV and patients with ET, 182 patients with PMF were screened with a somatic myeloid NGS panel. Additional mutations were detected in 81% of patients with PMF. The most frequent mutations were ASXL1 (36%), TET2 (18%), SRSF2 (18%), and U2AF1 (16%) (Table 1); furthermore, 35%, 26%, 10%, and 9% of the patients harbored 1, 2, 3, and 4 or more such variants/mutations, respectively. HRMs in patients with PMF, with a pejorative impact on OS or LFS, included ASXL1, SRSF2, CBL, KIT, RUNX1, SH2B3, and CEBPA; their combined prevalence was 56%. HRMs were associated with an inferior OS (median, 3.6 vs. 8.5 years; p < 0.001) and LFS (7-year risk, 25% vs. 4%; p < 0.001), and the effect on survival was independent of both the Dynamic International Prognostic Scoring System Plus (DIPSS+) and JAK2/CALR/MPL mutational status, especially for ASXL1 [7,13,18].
In 2014, 570 patients with PMF were recruited to build a molecular prognostic model based on CALR and ASXL1 mutations. OS was the longest in CALR (+) ASXL1 (-) patients (median OS of 10.4 years) and shortest in CALR (-) ASXL1 (+) patients (median OS of 2.3 years). CALR (+) ASXL1 (+) and CALR (-) ASXL1 (-) patients had similar survival rates and were grouped together in an intermediate-risk category (median OS of 5.8 years). The CALR/ASXL1-mutation-based prognostic model was independent of the DIPSS+ (p < 0.0001) and effective at identifying DIPSS+ low/intermediate-1-risk patients with a shorter (median OS of 4 years) or longer (median OS of 20 years) survival and high/intermediate-2-risk patients with a shorter (median OS of 2.3 years) survival. A multivariable analysis distinguished the CALR (-) ASXL1 (+) mutational status as the most significant risk factor for survival in older patients (age > 65 years) and/or those with an unfavorable karyotype [14]. More recently, a risk stratification based on ASXL1 and/or SRSF2 was proposed for patients with PMF and a low or intermediate-1 risk in the DIPSS+. In this specific subgroup, patients with PMF and ASXL1 and/or SRSF2 have a significantly lower OS than patients with PMF without these two mutations (median OS of approximately 18 years vs. 5 years, p < 0.0001) [15].
In 2017, Newberry et al., based on patients with MF enrolled in a ruxolitinib discontinuation phase 1/2 study, reported that patients with three or more additional mutations (not only HRMs) are less likely to respond to ruxolitinib. After a median follow-up of 79 months, 86 patients had discontinued ruxolitinib (30 of whom died while undergoing therapy). The median follow-up after ruxolitinib discontinuation for the remaining 56 patients was 32 months with a median survival after discontinuation of 14 months. Platelets <260 × 109/L at the start of therapy or <100 × 109/L at the time of discontinuation were associated with a shorter survival after discontinuation. Of the 62 patients with molecular data at baseline and follow-up, 22 (35%) acquired a new mutation while receiving ruxolitinib (14 [61%] in ASXL1). Patients showing clonal evolution had a significantly shorter survival after discontinuation (6 vs. 16 months). Transfusion dependency was the only clinical variable associated with clonal evolution [53].
In 2018, Grinfeld et al. used NGS to sequence 69 myeloid cancer genes from a panel of 1887 patients to develop a genomic classification and prognostic models of MPNs [30]. They highlighted:
- Forty-five truncating mutations on the terminal exon of PPM1D, which is the eighth most frequent mutation;
- The mutation of the MLL3 gene (nonsense or frameshift), which was detected in 20 patients and has already been described in patients with AML;
- Some noncanonical variants in 16 triple-negative patients with ET.
They, therefore, defined eight subgroups:
- (1)
- MPNs with TP53 mutation (often associated with Ch17p aberration and 5q deletion);
- (2)
- MPNs with chromatin or spliceosome regulator mutations;
- (3)
- MPNs associated with CALR mutation;
- (4)
- MPNs associated with MPL mutation;
- (5)
- MPNs associated with JAK2 or homozygous NFE2 mutation;
- (6)
- MPNs associated with heterozygous JAK2 mutation;
- (7)
- MPNs with unknown driver mutation;
- (8)
- MPNs with another driver mutation.
According to the diagnostic groups mentioned above, Grinfeld et al. suggested a prognostic model based on the statistical results established by using a comparison with the JAK2 heterozygous subgroup (sixth subgroup in diagnostic classification). The most significant results are listed below [2]:
- -
- The first subgroup (MPNs with TP53 mutation) was found to be associated with a higher risk of acute myeloid leukemia transformation and earlier death (HR 15.5; 95% CI, 7.5 to 31.4; p < 0.001).
- -
- The second subgroup (MPNs with chromatin or spliceosome regulator mutations) was exposed to a higher risk of transformation to myelofibrosis (HR 5.4; 95% CI, 2.7 to 11; p < 0.001) and had a shorter event-free survival (HR 2.6; 95% CI, 2.1 to 3.2; p < 0.001).
- -
- The patients in the fourth subgroup (MPNs associated with MPL mutation) with MF developed an increased risk of transformation to myeloid leukemia (HR 8.6; 95% CI, 1.4 to 49.1; p = 0.02).
- -
- The fifth group (MPNs associated with JAK2 or homozygous NFE2 mutation) showed a higher risk for myelofibrosis transformation (HR 3.0; 95% CI, 1.3 to 6.6; p = 0.007).
- -
- The eighth subgroup was associated with a good prognosis and only 0.5% and 1% of myelofibrosis and acute leukemia transformation, respectively (HR 0.56; 95% CI, 0.38 to 0.78; p = 0.005).
In 2020, Dunbar et al. worked on the leukemias secondary to myeloproliferative neo-plasms. They found some differences between post-MPN AML and de novo leukemia, which suggests different mechanisms in leukemogenesis [54]. For example, a higher rate of erythroblastic and megakaryoblastic leukemias are classically secondary to MPNs compared to the de novo leukemia. In addition, FLT3, NPM1, and DNMT3A are frequently absent in post-MPN AML (in contrast with de novo leukemia), whereas IDH1/2, TET2, ASXL1, EZH 2, and SRSF2 are frequently present.
Furthermore, the “triple-negative” MPNs have an increased tendency to transform themselves into leukemia. Therefore, the study highlights some strong risk factors for the leukemic transformation of MPNs through:
- -
- Cytogenetic abnormalities, such as a monosomal karyotype, complex karyotype, or two sole abnormalities that include +8, -7/7q, i(17q), -5/5q-, and a chromosome 17p deletion.
- -
- Molecular abnormalities, such as TP53, TET2, ASXL1, EZH2, SRSF2, IDH1/2, RUNX1, and U2AF1Q157.
On the other hand, clonality studies have shown that the order of acquisition of mutations can change. Some earlier mutations in epigenetic genes, such as ASXL1, TET2, or DNMT3A, may lead to a driver mutation the second time, whereas mutations in IDH1/2 generally occur after JAKV617F. When a TET2 mutation occurs before a JAK2 mutation, there is a higher risk of leukemic transformation.
At present, the mechanisms of how mutational order influences stem cells remain unclear. It would appear that some genetic or epigenetic mutations are required in addition to a driver mutation and/or other specific mutation to engender leukemic transformation. For instance, patients with JAK2V617F/IDH1/2 mutations present a higher risk for leukemic transformation, and sometimes, this would involve the loss of the JAK2V617F mutation. This hypothesis is shared by Grinfeld et al. [2], who found that mutations in epigenetic regulators, splicing factors, and RAS signaling pathways are associated with a higher risk of progression to MF or AML. Further progress is still needed to identify patients at risk of progression.
The clinical and biological features of a PMF diagnosis, such as karyotypes and driver and/or nondriver mutations of PMF, alter a patient’s outcome. At diagnosis, 80% of patients with PMF harbor at least one HMR mutation, and NGS is the best way to detect these genetic changes [7]. In 2018, Tefferi et al. proposed a mutation- and karyotype-enhanced international prognostic scoring system for PMF (MIPSS70+) to better stratify the patient’s risk profile at diagnosis. The MIPSS70+ is based, notably, on the PMF HRMs previously described (ASXL1, SRSF2, EZH2, IDH1, IDH2, and U2AF1) [55]. Five risk groups were determined, very low, low, intermediate, high, and very high risk, for which the 10-year OS was 92%, 56%, 37%, 13%, and <5%, respectively. In the same way, Tefferi et al. proposed a genetically inspired prognostic scoring system exclusively based on genetic markers (GIPSS). Based on the analysis of 641 patients with PMF, a multivariable analysis identified a very high risk and unfavorable karyotype, the absence of the type 1/like CALR mutation, and the presence of the ASXL1, SRSF2, or U2AF1Q157 mutation as inter-independent predictors of inferior survival. Four risk groups were determined: high, intermediate-2, intermediate-1, and low risk. The median OS determined with the GIPSS was 2 years for high risk, 4.2 years for intermediate-2 risk, 8 years for intermediate-1 risk, and 26.4 years for low risk. This score is a useful and simple tool, which requires only one karyotype and a few types of mutations. [49].
Allo-HSCT is the only curative therapy for patients with PMF when they are eligible. The median OS of any patient with PMF is approximately 5 years [56]. An optimal risk profile assessment at diagnosis is essential to select patients with PMF in whom allo-HSCT may be justified, especially for younger patients [57]. In 2016, Kroger et al. studied how molecular genetics may influence the outcome for patients with myelofibrosis after allo-HSCT. They screened 169 patients with PMF (n = 110), post-ET/PV MF (n = 46), and transformed MF (n = 13) for mutations on 16 frequently mutated genes. The most frequent mutation reported dealt with JAK2V617F (n = 101) followed by ASXL1 (n = 49), CALR (n = 34), SRSF2 (n = 16), TET2 (n = 10), U2AF1 (n = 11), EZH2 (n = 7), MPL (n = 6), IDH2 (n = 5), IDH1 (n = 4), and CBL (n = 1). The cumulative incidence of non-relapse mortality (NRM) was 21% at 1 year and 25% at 5 years. The 5-year rates of PFS and OS were 48% and 56%, respectively. In a multivariate analysis, the CALR mutation was an independent factor for lower NRMs (p = 0.05), improved PFS (p = 0.01), and OS (p = 0.03). ASXL1 and IDH2 mutations were independent risk factors for a lower PFS (p = 0.008) and OS (p = 0.002), whereas no impact was observed for “triple negative” patients [16]. Table 2 summarizes the additional mutations described in patients with PMF and their potential pejorative prognostic impact on OS and/or AML transformation.
Table 2.
Prognostic impact of additional mutations described in PMF.
Besides conventional diagnostic and/or prognostic approaches, it is quite difficult to propose a therapeutic strategy based on only one or two genes. For instance, in a multi-institutional collaborative project, 1473 patients with MPNs were screened for only IDH1 and IDH2 mutations: 594 with ET, 421 with PV, 312 with PV, 95 with post-PV/ET MF, and 51 with blast-phase MPN. A total of 38 IDH mutations were detected in 5 (0.8%) patients with ET, 8 (1.9%) patients with PV, 13 (4.2%) patients with PMF, 1 (1%) patient with post-PV/ET MF, and 11 (21.6%) patients with blast-phase MPN (p < 0.01). Mutant IDH was documented in the presence or absence of MPN driver mutations with similar mutational frequencies (1% in patients with PV and patients with ET and 4% in patients with PMF). We can note a larger frequency of the IDH mutation occurrence in patients that are nullizygous for the JAK2 haplotype, especially in PMF, and at a lower risk for a present complex karyotype when secondary leukemia occurs. In chronic-phase PMF, JAK2 haplotype nullizygosity but not IDH mutational status had an adverse effect on OS. In contrast, in both patients with blast-phase PMF and patients with blast-phase PV/ET, the IDH mutation predicted a worse survival [11]. Although interesting and rigorous, it is not possible to make a therapeutic decision (in particular, allo-HSCT) based only on these results without data on the other HRMs and the patients’ clinical histories. In the same way for blast-phase MPNs as described by Venton et al. in 2017, conventional clinical factors (age, karyotype, ELN2017 prognostic classification, treatments received, treatment response, and allo-SCT) failed to predict the patients’ outcomes. Only the mutational status accessed via NGS appeared relevant to predict the patients’ prognoses at the blast phase. Three genes, TP53 (p = 0.001), SRSF2 (p = 0.018), and TET2 (p = 0.012), impacted AML prognosis pejoratively at the transformation time. In 33% of cases, the mutation profile was already present for MPNs in the chronic phase. However, only the patients with a SRSF2 mutation presented a lower LFS (52 months) vs. unmutated patients (134 months) (p < 0.001) [65]. Based on these results, it seems, nevertheless, difficult to propose allo-HSCT as a first-line treatment for young/fit patients with chronic-phase MPNs and with the SRSF2 mutation.
4. Conclusions
The use of somatic myeloid NGS panels in routine biology plays an increased role in MPN management, especially as a precious diagnostic aid for triple-negative MPNs, but also, and more importantly, as a method to stratify the risk profile of young/fit patients with PMF. However, first-line allo-HSCT is not systematically proposed for young/fit patients with PMF and HRMs at diagnosis because of the high risk for NRMs of this therapy. Further comparative and prospective studies are needed to better access the impact of a more aggressive treatment on PFS and OS in patients with PMF and HRMs at diagnosis. In contrast, performing NGS in patients with ET/PV and a JAK2/CALR/MPL driver mutation or patients with PMF who are ineligible for allo-HSCT does not present optimal cost-efficiency, since these data do not have a significant impact on our clinical practice.
Author Contributions
G.V., P.P., N.A., V.E., J.G. and R.C. collected the bibliographic data and wrote the manuscript. R.A., A.-L.C., A.T., P.R., L.F., J.C., L.O. and P.M. proofread and corrected this review. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- Magor, G.W.; Tallack, M.R.; Klose, N.M.; Taylor, D.; Korbie, D.; Mollee, P.; Trau, M.; Perkins, A.C. Rapid Molecular Profiling of Myeloproliferative Neoplasms Using Targeted Exon Resequencing of 86 Genes Involved in JAK-STAT Signaling and Epigenetic Regulation. J. Mol. Diagn. 2016, 18, 707–718. [Google Scholar] [CrossRef] [PubMed]
- McClure, R.F.; Ewalt, M.D.; Crow, J.; Temple-Smolkin, R.L.; Pullambhatla, M.; Sargent, R.; Kim, A.S. Clinical Significance of DNA Variants in Chronic Myeloid Neoplasms: A Report of the Association for Molecular Pathology. J. Mol. Diagn. 2018, 20, 717–737. [Google Scholar] [CrossRef] [PubMed]
- Loscocco, G.G.; Guglielmelli, P.; Vannucchi, A.M. Impact of Mutational Profile on the Management of Myeloproliferative Neoplasms: A Short Review of the Emerging Data. OncoTargets Ther. 2020, 13, 12367–12382. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.M.; Thomson, C.; Hamad, N.; Lane, S.W.; Manos, K.; Grigg, A.P.; Guo, B.; Erber, W.N.; Scott, A.; Viiala, N.; et al. Myeloid somatic mutation panel testing in myeloproliferative neoplasms. Pathology 2021, 53, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of Mutation Order on Myeloproliferative Neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef]
- Lasho, T.L.; Mudireddy, M.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Szuber, N.; Begna, K.H.; Patnaik, M.M.; Gangat, N.; Pardanani, A.; et al. Targeted next-generation sequencing in blast phase myeloproliferative neoplasms. Blood Adv. 2018, 2, 370–380. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Abdel-Wahab, O.; Guglielmelli, P.; Patel, J.; Caramazza, D.; Pieri, L.; Finke, C.M.; Kilpivaara, O.; Wadleigh, M.; et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia 2010, 24, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Score, J.; Mannarelli, C.; Pancrazzi, A.; Biamonte, F.; Pardanani, A.; Zoi, K.; Reiter, A.; et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: An international study of 797 patients. Leukemia 2014, 28, 1804–1810. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Finke, C.; Mannarelli, C.; Belachew, A.A.; Pancrazzi, A.; Wassie, E.A.; Ketterling, R.; et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: An international study of 570 patients. Leukemia 2014, 28, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Screening for ASXL1 and SRSF2 mutations is imperative for treatment decision-making in otherwise low or intermediate-1 risk patients with myelofibrosis. Br. J. Haematol. 2017, 183, 678–681. [Google Scholar] [CrossRef] [PubMed]
- Kröger, N.; Panagiota, V.; Badbaran, A.; Zabelina, T.; Triviai, I.; Cruz, M.M.A.; Shahswar, R.; Ayuk, F.; Gehlhaar, M.; Wolschke, C.; et al. Impact of Molecular Genetics on Outcome in Myelofibrosis Patients after Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2017, 23, 1095–1101. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.; Gangat, N.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A. Prognostic significance of ASXL1 mutation types and allele burden in myelofibrosis. Leukemia 2017, 32, 837–839. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Biamonte, F.; Score, J.; Hidalgo-Curtis, C.; Cervantes, F.; Maffioli, M.; Fanelli, T.; Ernst, T.; Winkelman, N.; Jones, A.V.; et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood 2011, 118, 5227–5234. [Google Scholar] [CrossRef]
- Shimizu, T.; Kubovcakova, L.; Nienhold, R.; Zmajkovic, J.; Meyer, S.C.; Hao-Shen, H.; Geier, F.; Dirnhofer, S.; Guglielmelli, P.; Vannucchi, A.M.; et al. Loss of Ezh2 synergizes with JAK2-V617F in initiating myeloproliferative neoplasms and promoting myelofibrosis. J. Exp. Med. 2016, 213, 1479–1496. [Google Scholar] [CrossRef]
- Ok, C.Y.; Trowell, K.T.; Parker, K.G.; Moser, K.; Weinberg, O.K.; Rogers, H.J.; Reichard, K.K.; George, T.I.; Hsi, E.D.; Bueso-Ramos, C.E.; et al. Chronic myeloid neoplasms harboring concomitant mutations in myeloproliferative neoplasm driver genes (JAK2/MPL/CALR) and SF3B1. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 2020, 34, 20–31. [Google Scholar] [CrossRef]
- Zhang, S.-J.; Rampal, R.; Manshouri, T.; Patel, J.; Mensah, N.; Kayserian, A.; Hricik, T.; Heguy, A.; Hedvat, C.; Gönen, M.; et al. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood 2012, 119, 4480–4485. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Nicolosi, M.; Mudireddy, M.; Szuber, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Gangat, N.; et al. Driver mutations and prognosis in primary myelofibrosis: Mayo-Careggi MPN alliance study of 1095 patients. Am. J. Hematol. 2017, 93, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, O.; O’Sullivan, J.; Barkas, N.; Wang, G.; Buck, G.; Hamblin, A.; Tefferi, A.; Al-Ali, H.K.; Barosi, G.; Devos, T.; et al. Spliceosome mutations are common in persons with myeloproliferative neoplasm-associated myelofibrosis with RBC-transfusion-dependence and correlate with response to pomalidomide. Leukemia 2020, 35, 1197–1202. [Google Scholar] [CrossRef] [PubMed]
- Maslah, N.; Cassinat, B.; Verger, E.; Kiladjian, J.-J.; Velazquez, L. The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 2017, 31, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Loscocco, G.G.; Mannarelli, C.; Pacilli, A.; Fanelli, T.; Rotunno, G.; Gesullo, F.; Corbizi-Fattori, G.; Vannucchi, A.M.; Guglielmelli, P. Germline transmission of LNKE208Q variant in a family with myeloproliferative neoplasms. Am. J. Hematol. 2016, 91, E356. [Google Scholar] [CrossRef]
- Rumi, E.; Harutyunyan, A.S.; Pietra, D.; Feenstra, J.D.M.; Cavalloni, C.; Roncoroni, E.; Casetti, I.; Bellini, M.; Milanesi, C.; Renna, M.C.; et al. LNK mutations in familial myeloproliferative neoplasms. Blood 2016, 128, 144–145. [Google Scholar] [CrossRef]
- Sanada, M.; Suzuki, T.; Shih, L.-Y.; Otsu, M.; Kato, M.; Yamazaki, S.; Tamura, A.; Honda, H.; Sakata-Yanagimoto, M.; Kumano, K.; et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature 2009, 460, 904–908. [Google Scholar] [CrossRef]
- Coltro, G.; Rotunno, G.; Mannelli, L.; Mannarelli, C.; Fiaccabrino, S.; Romagnoli, S.; Bartalucci, N.; Ravenda, E.; Gelli, E.; Sant’Antonio, E.; et al. RAS/CBL mutations predict resistance to JAK inhibitors in myelofibrosis and are associated with poor prognostic features. Blood Adv. 2020, 4, 3677–3687. [Google Scholar] [CrossRef]
- Braun, B.S.; Shannon, K.; Masuda, T.; Wada, K.; Nakajima, A.; Okura, M.; Kudo, C.; Kadowaki, T.; Kogo, M.; Kamisaki, Y. Targeting Ras in Myeloid Leukemias. Clin. Cancer Res. 2008, 14, 2249–2252. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef]
- Jutzi, J.S.; Bogeska, R.; Nikoloski, G.; Schmid, C.; Seeger, T.S.; Stegelmann, F.; Schwemmers, S.; Gründer, A.; Peeken, J.C.; Gothwal, M.; et al. MPN patients harbor recurrent truncating mutations in transcription factor NF-E2. J. Exp. Med. 2013, 210, 1003–1019. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Pacilli, A.; Coltro, G.; Mannelli, F.; Mannelli, L.; Contini, E.; Rotunno, G.; Bartalucci, N.; Fiaccabrino, S.; Sordi, B.; et al. Characteristics and clinical correlates of NFE2 mutations in chronic Myeloproliferative neoplasms. Am. J. Hematol. 2019, 95, E23–E26. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.D.; Miller, P.G.; Silver, A.J.; Sellar, R.S.; Bhatt, S.; Gibson, C.; McConkey, M.; Adams, D.; Mar, B.; Mertins, P.; et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 2018, 132, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Boddu, P.; Chihara, D.; Masarova, L.; Pemmaraju, N.; Patel, K.P.; Verstovsek, S. The co-occurrence of driver mutations in chronic myeloproliferative neoplasms. Ann. Hematol. 2018, 97, 2071–2080. [Google Scholar] [CrossRef]
- Zamora, L.; Xicoy, B.; Cabezón, M.; Fernandez, C.; Marcé, S.; Velez, P.; Xandri, M.; Gallardo, D.; Millá, F.; Feliu, E.; et al. Co-existence of JAK2 V617F and CALR mutations in primary myelofibrosis. Leuk. Lymphoma 2015, 56, 2973–2974. [Google Scholar] [CrossRef]
- Beucher, A.; Dib, M.; Orvain, C.; Bouvier, A.; Jouanneau-Courville, R.; Dobo, I.; Cottin, L.; Guardiola, P.; Rousselet, M.; Paz, D.L.; et al. Next generation sequencing redefines a triple negative essential thombocythaemia as double-positive with rare mutations on JAK2 V617 and MPL W515 hotspots. Br. J. Haematol. 2019, 186, 785–788. [Google Scholar] [CrossRef]
- Acha, P.; Xandri, M.; Fuster-Tormo, F.; Palomo, L.; Xicoy, B.; Cabezón, M.; Marcé, S.; Granada, I.; Vela, D.; Sagüés, M.; et al. Diagnostic and prognostic contribution of targeted NGS in patients with triple-negative myeloproliferative neoplasms. Am. J. Hematol. 2019, 94, E264–E267. [Google Scholar] [CrossRef]
- Tefferi, A. Primary myelofibrosis: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2018, 93, 1551–1560. [Google Scholar] [CrossRef]
- Dupont, S.; Massé, A.; James, C.; Teyssandier, I.; Lécluse, Y.; Larbret, F.; Ugo, V.; Saulnier, P.; Koscielny, S.; Le Couédic, J.P.; et al. The JAK2 617V>F mutation triggers erythropoietin hypersensitivity and terminal erythroid amplification in primary cells from patients with polycythemia vera. Blood 2007, 110, 1013–1021. [Google Scholar] [CrossRef]
- Ancochea, À.; Álvarez-Larrán, A.; Morales-Indiano, C.; García-Pallarols, F.; Martínez-Avilés, L.; Angona, A.; Senín, A.; Bellosillo, B.; Besses, C. The role of serum erythropoietin level and JAK2 V617F allele burden in the diagnosis of polycythaemia vera. Br. J. Haematol. 2014, 167, 411–417. [Google Scholar] [CrossRef]
- Vytrva, N.; Stacher, E.; Regitnig, P.; Zinke-Cerwenka, W.; Hojas, S.; Hubmann, E.; Porwit, A.; Bjorkholm, M.; Hoefler, G.; Beham-Schmid, C. Megakaryocytic Morphology and Clinical Parameters in Essential Thrombocythemia, Polycythemia Vera, and Primary Myelofibrosis With and Without JAK2V617F. Arch. Pathol. Lab. Med. 2014, 138, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Warshawsky, I.; Mularo, F.; Hren, C.; Jakubowski, M. Failure of the Ipsogen MutaScreen kit to detect the JAK2 617V>F mutation in samples with additional rare exon 14 mutations: Implications for clinical testing and report of a novel 618C>F mutation in addition to 617V>F. Blood 2010, 115, 3175–3176. [Google Scholar] [CrossRef] [PubMed]
- Tiong, I.S.; Casolari, D.A.; Moore, S.; Nguyen, T.; Van Velzen, M.J.; Zantomio, D.; Scott, H.S.; D’Andrea, R.J.; Hahn, C.N.; Ross, D.M. Apparent ‘JAK2-negative’ polycythaemia vera due to compound mutations in exon 14. Br. J. Haematol. 2017, 178, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Camps, C.; Petousi, N.; Bento, C.; Cario, H.; Copley, R.R.; McMullin, M.F.; Van Wijk, R.; Ratcliffe, P.; Robbins, P.A.; Taylor, J.C. Gene panel sequencing improves the diagnostic work-up of patients with idiopathic erythrocytosis and identifies new mutations. Haematologica 2016, 101, 1306–1318. [Google Scholar] [CrossRef]
- Steensma, D.P. Clinical consequences of clonal hematopoiesis of indeterminate potential. Blood Adv. 2018, 2, 3404–3410. [Google Scholar] [CrossRef]
- Hussein, K.; Bock, O.; Theophile, K.; von Neuhoff, N.; Buhr, T.; Schlué, J.; Büsche, G.; Kreipe, H. JAK2V617F allele burden discriminates essential thrombocythemia from a subset of prefibrotic-stage primary myelofibrosis. Exp. Hematol. 2009, 37, 1186–1193.e7. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Ferretti, V.V.; Klampfl, T.; Harutyunyan, A.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef]
- Tefferi, A.; Wassie, E.A.; Guglielmelli, P.; Gangat, N.; Belachew, A.A.; Lasho, T.L.; Finke, C.; Ketterling, R.; Hanson, C.A.; Pardanani, A.; et al. Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: A collaborative study of 1027 patients. Am. J. Hematol. 2014, 89, E121–E124. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef]
- Buhr, T.; Hebeda, K.; Kaloutsi, V.; Porwit, A.; Van Der Walt, J.; Kreipe, H. European Bone Marrow Working Group trial on reproducibility of World Health Organization criteria to discriminate essential thrombocythemia from prefibrotic primary myelofibrosis. Haematologica 2011, 97, 360–365. [Google Scholar] [CrossRef]
- Wilkins, B.S.; Erber, W.; Bareford, D.; Buck, G.; Wheatley, K.; East, C.L.; Paul, B.; Harrison, C.N.; Green, A.R.; Campbell, P.J. Bone marrow pathology in essential thrombocythemia: Interobserver reliability and utility for identifying disease subtypes. Blood 2008, 111, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Newberry, K.J.; Patel, K.; Masarova, L.; Luthra, R.; Manshouri, T.; Jabbour, E.; Bose, P.; Daver, N.; Cortes, J.; Kantarjian, H.; et al. Clonal evolution and outcomes in myelofibrosis after ruxolitinib discontinuation. Blood 2017, 130, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, A.J.; Rampal, R.K.; Levine, R.L. Leukemia secondary to myeloproliferative neoplasms. Blood 2020, 136, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef] [PubMed]
- Baade, P.D.; Ross, D.M.; Anderson, L.A.; Forsyth, C.; Fritschi, L. Changing incidence of myeloproliferative neoplasms in Australia, 2003–2014. Am. J. Hematol. 2019, 94, E107–E109. [Google Scholar] [CrossRef]
- Gowin, K.; Ballen, K.; Ahn, K.W.; Hu, Z.-H.; Ali, H.; Arcasoy, M.O.; Devlin, R.; Coakley, M.; Gerds, A.T.; Green, M.; et al. Survival following allogeneic transplant in patients with myelofibrosis. Blood Adv. 2020, 4, 1965–1973. [Google Scholar] [CrossRef]
- Brecqueville, M.; Rey, J.; Bertucci, F.; Coppin, E.; Finetti, P.; Carbuccia, N.; Gelsi-Boyer, V.; Arnoulet, C.; Gisserot, O.; Murati, A.; et al. Mutation analysis of ASXL1, CBL, DNMT3A, IDH1, IDH2, JAK2, MPL, NF1, SF3B1, SUZ12, and TET2 in myeloproliferative neoplasms. Genes. Chromosomes Cancer 2012, 51, 743–755. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Valle, V.D.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Bernard, O.A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Lasho, T.L.; Finke, C.M.; Hanson, C.A.; Jimma, T.; Knudson, R.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. SF3B1 mutations in primary myelofibrosis: Clinical, histopathology and genetic correlates among 155 patients. Leukemia 2012, 26, 1135–1137. [Google Scholar] [CrossRef]
- Lasho, T.L.; Jimma, T.; Finke, C.M.; Patnaik, M.; Hanson, C.A.; Ketterling, R.; Pardanani, A.; Tefferi, A. SRSF2 mutations in primary myelofibrosis: Significant clustering with IDH mutations and independent association with inferior overall and leukemia-free survival. Blood 2012, 120, 4168–4171. [Google Scholar] [CrossRef] [PubMed]
- Grand, F.H.; Hidalgo-Curtis, C.E.; Ernst, T.; Zoi, K.; Zoi, C.; McGuire, C.; Kreil, S.; Jones, A.; Score, J.; Metzgeroth, G.; et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood 2009, 113, 6182–6192. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Pardanani, A.; Patel, J.; Wadleigh, M.; Lasho, T.; Heguy, A.; Beran, M.; Gilliland, D.G.; Levine, R.L.; Tefferi, A. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia 2011, 25, 1200–1202. [Google Scholar] [CrossRef]
- Raza, S.; Viswanatha, D.; Frederick, L.; Lasho, T.; Finke, C.; Knudson, R.; Ketterling, R.; Pardanani, A.; Tefferi, A. TP53 mutations and polymorphisms in primary myelofibrosis. Am. J. Hematol. 2011, 87, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Venton, G.; Courtier, F.; Charbonnier, A.; D’Incan, E.; Saillard, C.; Mohty, B.; Mozziconacci, M.-J.; Birnbaum, D.; Murati, A.; Vey, N.; et al. Impact of gene mutations on treatment response and prognosis of acute myeloid leukemia secondary to myeloproliferative neoplasms. Am. J. Hematol. 2017, 93, 330–338. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).