1. Introduction
Chronic obstructive pulmonary disease (COPD), a heterogeneous condition that encompasses chronic bronchitis and emphysema, is the third leading cause of death worldwide [
1,
2]. The disease is progressive, and currently incurable, with the treatment options limited to those controlling symptoms. The main cause of COPD is tobacco smoking, or chronic exposure to some other noxious gases, in complex interaction with genetic and developmental factors. In addition, single-gene defect (α1-anti-trypsin deficiency), severe asthma, infections, abnormal lung development and/or failed lung regeneration can cause or contribute to COPD development [
3,
4,
5,
6]. The underlying molecular mechanisms of COPD pathology include increased oxidative and nitrosative stress, an imbalance between proteolytic activity and anti-proteolytic defense, persistent inflammation in the lung, uncontrolled autophagy, enhanced apoptosis and/or accelerated lung aging [
4,
7]. In addition to respiratory symptoms, most COPD patients suffer from mild to moderate pulmonary hypertension (PH) [
8,
9]. We have previously described the essential role of the inducible nitric oxide synthase (iNOS) in the development and reversal of tobacco smoke-induced emphysema and PH in mice [
10]. More recently, we demonstrated that iNOS inhibition ameliorates parenchymal destruction and promotes reverse remodeling of the pulmonary vasculature, even in a severe model of elastase-induced emphysema, characterized by prominent parenchymal damage similar to the lesions found in lungs of end-stage COPD patients [
11]. Regarding the molecular mechanism, evidence was provided that protein nitration, prompted by the formation of peroxynitrite from iNOS-derived nitric oxide (NO) and superoxide produced by the NADPH oxidase comprising the NADPH oxidase organizer (NoxO)-1 subunit, may drive lung emphysema development [
10,
12]. Interestingly, while it was clear that both vascular and alveolar changes occur in an iNOS-dependent manner in mice, we observed that they are associated with iNOS deregulation in different cell types. In particular, we showed that the presence of iNOS in myeloid cells leads to pulmonary vascular remodeling but does not contribute to emphysema development [
10,
13]. However, it remained unclear which pulmonary vascular, stromal, or epithelial cell type was the main source of the iNOS activity linked to pathological changes in the lung parenchyma. In this regard, Boyer and colleagues published that alveolar epithelial type II cells (AECII) are characterized by marked iNOS upregulation in the elastase model of emphysema and by a more prominent accumulation of nitrated proteins than any other lung cell type [
14]. While the authors did not observe any beneficial effect of preventive iNOS inhibition on the emphysema development monitored up to 20 days after elastase instillation, the abovementioned recent work from our group showed that a longer 12-week treatment with an iNOS inhibitor can ameliorate fully established emphysema and PH in this animal model [
11]. This finding suggests that iNOS inhibition may be a viable option for the treatment of severe emphysema in mice if present during a longer period of time. However, it was unresolved which cell type was driving lung regeneration upon iNOS inhibition in this model. As the delineation of the exact signaling pathway through which iNOS exerts its pathological effects is of vast importance for potential future transfer to the human disease, and might impact the route of administration, we decided to further investigate which cell type is behind pathological iNOS activity and the reversal of this disease. Previously, strong evidence was provided that AECll have stem-cell features in the adult lung, and proliferate to repopulate and repair the injured epithelium [
15,
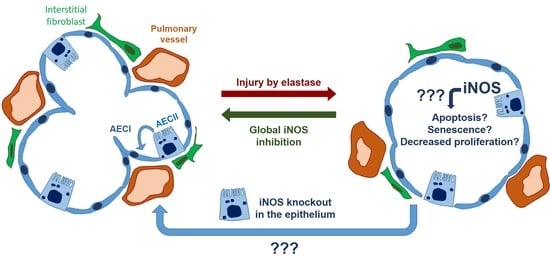
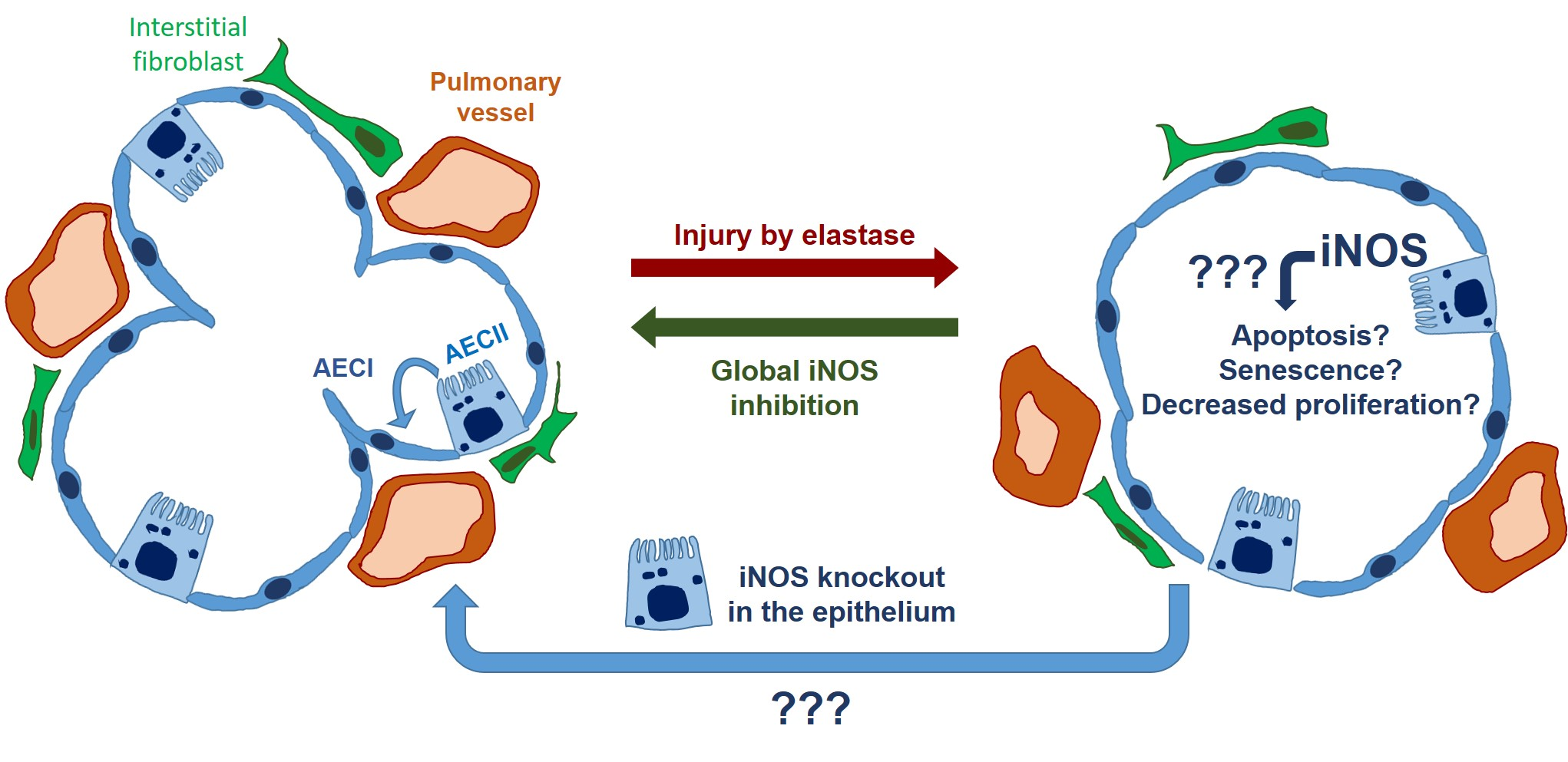
16]. We thus hypothesized that increased iNOS activity, the subsequent formation of peroxynitrite, and the accumulation of nitrated proteins in AECII cells could impair their potential for self-renewal, and disable the regeneration of respiratory epithelium. Therefore, this study aimed to investigate whether the deletion of iNOS in the AECII can promote the reversal of established severe emphysema if the knockout is present during the 12 weeks post-injury in the elastase mouse model.
2. Materials and Methods
2.1. Experimental Design and Elastase Application
The mice were housed in individually ventilated cages, with a 12-h dark/light cycle and food and water supplied ad libitum. Male and female iNos CCSP rtTA2s-M2 LC1 (B6. Nos2tm2904.1Arte C(Cg)-Tg(Scgb1a1-rtTA)38-Tg(tetO-cre)LC1Bjd) (Figure 1B), 3–4 months old, were randomly allocated to four experimental groups: saline-instilled control mice, saline-instilled doxycycline-treated mice, porcine pancreatic elastase (PPE)-instilled control or PPE-instilled doxycycline-treated mice. First, 100 µL saline or elastase (E7885; Merck KGaA, Darmstadt, Germany) dissolved in saline (24 U/kg body weight) was delivered intratracheally to the anesthetized animals (3% isoflurane in 100% O2) using a MicroSprayer® Aerosolizer (Model IA-1C; Penn-Century, Inc., Wyndmoor, PA, USA) with a high-pressure syringe (Model FMJ-250; Penn-Century, Inc., Wyndmoor, PA, USA). Then, from the 14th to the 28th day post instillation, mice from the appropriate experimental groups were fed with chow containing doxycycline (600 mg/kg supplemented with 2% saccharose; Altromin Spezialfutter GmbH & Co., Lage, Germany). After this point, the mice were given an additional 12 weeks for possible recovery, prior to the in vivo measurements: µCT, echocardiography, hemodynamic, and lung function measurements (Figure 1F). The final numbers in these measurements may vary because of technical problems, e.g., with the placement of the hemodynamic catheter or the echocardiographic assessment. All analyses of the in vivo experiments were performed at the end of the observation period, and the histological and molecular biology analyses were performed using material thereof. For confirmation of iNOS knockout induction in AECII by doxycycline, lungs were harvested from the mice immediately or 12 weeks after termination of feeding with doxycycline-containing chow. Isolated AECII from these lungs or whole-lung sections were investigated for luciferase and iNOS expression, respectively. All animal experiments were approved by the regional authorities for animal welfare (Regierungspräsidium Giessen, Germany), in accordance with the German animal welfare law and the European legislation for the protection of animals used for scientific purposes (2010/63/EU).
2.2. µCT and Echocardiography
Micro-computed tomography (µCT) and echocardiography were carried out under isoflurane (Baxter Deutschland GmbH, Unterschleiβheim, Germany) anesthesia by a blinded single observer, as described previously [
17]. The following parameters were measured: functional residual capacity (FRC) and lung density using µCT, and pulmonary acceleration time, pulmonary ejection time, tricuspid annular plane systolic excursion, and right ventricular (RV) wall thickness using echocardiography.
2.3. Bronchoalveolar Lavage and Lung Tissue Processing
After the hemodynamic measurement, the thoracic cavity was opened, and bronchoalveolar lavage (BAL) was carried out three times, using 700 µL ice-cold PBS buffer. The collected BAL was then centrifuged for 10 min at 4 °C at a speed of 2490× g (Micro 200R, Hettich, Germany), and separated into BAL fluid (BALF), stored at −80 °C) and BAL cells (resuspended in FBS with 10% DMSO and kept at −80 °C). The blood was flushed out of the lungs through the pulmonary artery with the saline solution, at a pressure of 22 cmH2O during continuous ventilation (Minivent, Hugo Sachs Electronik, March, Germany). The right lung was harvested for molecular biology analyses, while the left lung was fixed under simultaneous vascular perfusion (pressure: 22 cmH2O) and inflation (pressure: 12 cmH2O) with formalin.
2.4. Lung Function and Hemodynamic Measurements
Lung function measurement and hemodynamic measurements were carried out under isoflurane anesthesia, as previously reported. Deep inflation was used as a recruitment maneuver, followed by single-frequency forced oscillation and broadband forced oscillation, and the measurement of a respiratory pressure–volume (P–V) loop [
13,
17]. After lung function assessment, a micro-tip catheter (Millar Instruments, Houston, TX, USA) was placed in the right ventricle through the surgically prepared jugular vein for measurement of the right ventricular systolic pressure (RVSP). The animals were sacrificed under anesthesia by exsanguination through the carotid artery.
2.5. Right-Heart-Hypertrophy Assessment
After lung fixation with formalin, the heart was removed, dissected into the RV and the left ventricle plus septum (LV + S), and weighed. The Fulton index (RV/(LV + S)) was calculated as a measure of RV hypertrophy.
2.6. Alveolar Morphometry
For alveolar morphometry, formalin-fixed and paraffin-embedded 3 µm thick lung sections were stained with hematoxylin and eosin (H&E). The sections were analyzed using the Qwin alveolar morphometry software, as previously described [
10,
11,
13], to assess alveolar wall thickness, airspace, and mean linear intercept. The bronchi and vessels were excluded from the analysis.
2.7. Isolation of Murine Primary AECII and Metabolic Activity Assay
Primary AECII were isolated from the lungs of mice fed with either standard or doxycycline-containing chow, in accordance with the previously described protocol [
12]. The cells were either immediately lysed and proteins extracted, or they were seeded in 96-well plates at density 35,000 cells/well in DMEM (high glucose, Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA), supplemented with 10% (
v/v) fetal bovine serum (FBS, Sigma Aldrich, Munich, Germany), 2% (
v/v) L-Glutamine, 100 IU/mL penicillin, and 100 µg/mL streptomycin (all from Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA), and left overnight in a humidified atmosphere of 5% CO
2, at 37 °C. The cells were treated with different concentrations of cigarette smoke extract (CSE) with or without 10 µM (L-NIL) for 6 h, and then the medium was replaced with fresh medium containing 10% (
v/v) alamarBlue Cell Viability Reagent (Thermo Fisher Scientific Inc., Waltham, MA, USA). Metabolic activity was measured 24 h after the start of CSE treatment.
2.8. Metabolic Activity Assay
The MLE 12 (ATCC CRL-2110) cells were seeded in 96 well plates at 10,000 cells/well in DMEM/F-12 (Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA), supplemented with 10% (v/v) FBS (Sigma Aldrich, Munich, Germany), 100 IU/mL penicillin, and 100 µg/mL streptomycin (both from Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA), and cultured overnight in a humidified atmosphere of 5% CO2, at 37 °C. The cells were then serum-starved for 24 h. For the experiments with CSE, the next day, the cells were treated with different CSE concentrations with or without 10 µM L-NIL for 6 h, and then kept for the additional 16 h after the treatment removal and addition of a fresh culture medium. For experiments with BALF, the cells were treated with BALF diluted 1:10 in a serum-free medium for 24 h. PBS served as a control. The experiment was repeated 18 times in total, by treating the cells in three different experiments with BALF from six different animals per group. Afterwards, the BALF treatment was removed and a new culture medium with 10% (v/v) alamarBlue Cell Viability Reagent (Thermo Fisher Scientific Inc., Waltham, MA, USA) was added. The metabolic activity of the cells was determined after 4 h, in accordance with the manufacturer’s instructions.
2.9. Cell Proliferation Assay
The cells were seeded and serum-starved, as described for the metabolic activity assay. Then, the cells were treated with different concentrations of CSE with or without 10 µM L-NIL, for 6 h. The medium was replaced with a fresh medium containing BrdU labeling solution (Roche, Basel, Switzerland), and the cells were cultured for an additional 16 h. For the experiments with BALF, a serum-free medium containing BrdU labeling solution (Roche, Basel, Switzerland) and either BALF diluted 1:10 or PBS (as a control) was added, and the cells were cultured for 16 h. Next, the cells were fixed and proliferation was determined, in accordance with the manufacturer’s instructions. The absorbance was measured in a microplate reader at 370 nm (reference wavelength: 492 nm). The O.D. value for each experimental sample was standardized to the value measured in the control group.
2.10. Cell Apoptosis Assay
The cells were seeded in 96-well plates at 10,000 cells/well and left overnight in a humidified atmosphere of 5% CO2, at 37 °C. The next day, the cells were treated with different concentrations of CSE with or without 10 µM L-NIL for 6 h, and then cultured in a new, fresh medium with or without L-NIL, in the presence of propidium iodide and Annexin XII-based polarity-sensitive probe pSIVATM from the apoptosis kinetic assay (Abcam, Cambridge, UK), for an additional 16 h. For the BALF experiments, fresh serum-free medium containing either BALF diluted 1:10 or PBS was added, together with propidium iodide and Annexin XII-based polarity-sensitive probe pSIVATM (Abcam, Cambridge, UK). The assay was carried out in accordance with the manufacturer’s instructions, using an IncuCyte ZOOM device (Essen BioScience, Ann Arbor, MI, USA) for visualization.
2.11. Western-Blot
The isolation of proteins from mouse lung homogenates, protein concentration measurement, and Western-blot analysis were carried out as previously reported [
11,
13]. The antibodies used were as follows: anti-MMP8 (Cat#78423), anti-cyclin D1 (Cat#134175) anti-cytochrome c (Cat#90529), anti-Bcl2 (Cat#182858) from Abcam, Cambridge, UK, and Bax (Cat#2772S9, Cell Signaling Technology, Danvers, MA, USA), all at a dilution of 1:1000.
2.12. Immunofluorescent Staining of Human and Mouse Lung Sections
The immunofluorescent staining of mouse lung sections was carried out as described previously, with slight modifications [
13]. Briefly, 3 µm thick sections of formalin-fixed, paraffin-embedded left lung lobes were deparaffinized and rehydrated, and heat-mediated antigen retrieval was carried out using Rodent Decloaker (Biocare Medical, Pacheco, USA). Blocking of non-specific antibody binding was carried out using 10% BSA for 1 h at RT and Rodent M Blocker (Biocare Medical, Pacheco, USA). The sections were incubated overnight at 4 °C with the following primary antibodies: anti-iNOS (Cat#3523, Abcam, Cambridge, UK, 1:200), and anti-vWF (Cat#a0082, Dako, Hamburg, Germany, 1:500). The next day, the slides were washed with 1XPBS and incubated for 2 h at RT with the following fluorescently labeled primary and/or secondary antibodies: anti-Cy3-labeled α-smooth muscle actin antibody (Cat#C6198, Sigma-Aldrich, Munich, Germany 1:400), Alexa fluor488-labeled goat anti-rabbit secondary antibody (Cat#A-11034, Thermo Fisher Scientific Inc., Waltham, MA, USA, 1:400), Zenon™ Rabbit IgG Labeling Kit Alexa fluor 647 (Cat#Z25308, Thermo Fisher Scientific Inc., Waltham, MA, USA)-labeled anti-pro-surfactant protein C (SPC), (Cat#ab211326, Abcam, Cambridge, UK, 1:200). Finally, the slides were counterstained with Hoechst, mounted using ProLong™ Glass Antifade Mountant (Cat#P36980, Thermo Fisher Scientific Inc., Waltham, MA, USA), with precision cover-glasses thickness No. 1.5H, and analyzed using confocal microscopy. At least three images from randomly chosen fields were taken for each analysis. For the human lungs, heat-mediated antigen retrieval was carried out using citrate buffer (Zytomed Systems, Berlin, Germany) and blocking of unspecific binding was carried out using 10% BSA and Background Punisher (Biocare Medical, Pacheco, CA, USA). The following primary antibodies were used: anti-iNOS (Cat#NB300-605, Novus Biologicals, Littleton, CO, USA, 1:250) and anti-HTII280 (Cat# TB-27AHT2-280, Terrace Biotech, San Francisco, CA, USA, 1:200).
2.13. Automated Western-Blot Analysis
Automated Western-blot analysis was carried out using the chemiluminescent detection system of the Jess Simple Western™ platform (ProteinSimple, San Jose, CA, USA), in accordance with the manufacturer’s instructions, using an anti-luciferase antibody (Cat#ab21176, Abcam, Cambridge, UK, 1:40).
2.14. Statistical Analysis
Statistical analyses were performed using the GraphPad Prism 9 (LaJolla, CA, USA). All data are expressed as means ± standard error of the mean (SEM). A comparison between multiple groups was performed using analysis of variance (a one- or two-way ANOVA for comparing the results of experiments with one or two independent variables, respectively, and Tukey’s multiple comparison post-hoc test). An independent t-test was used for comparing the equality of means between the two groups, and p-values of < 0.05 were considered statistically significant.
4. Discussion
We investigated whether the induction of iNOS knockout in AECII can promote lung repair or ameliorate pulmonary vascular pathology in mice with fully established elastase-induced emphysema and PH. Our in vivo measurements, performed at the end of a 12-week observation period, revealed statistically significant impairment of heart and lung function and the existence of PH in all elastase-treated mice, irrespective of the iNOS deletion in AECII. These results were corroborated by alveolar morphometry, which showed a prominent, statistically significant development of emphysema in elastase-treated mice that could not be counteracted by iNOS knockout in AECII. Similarly, histological analysis of the small pulmonary vessels indicated that their increased muscularization upon elastase treatment of the lung cannot be reversed by deletion of AECII-derived iNOS. Together, these data demonstrated that iNOS knockout in AECII upon elastase injury in mice does not promote regeneration of alveolar epithelium and reverse remodeling of the pulmonary vasculature. This conclusion was further supported by our in vitro experiments, showing that iNOS inhibition in lung epithelium does not significantly alter the response of these cells to injury, in terms of metabolic activity, proliferation and apoptosis.
We previously demonstrated that iNOS inhibition can prevent and reverse PH and emphysema in mice, both after chronic smoke exposure or intratracheal elastase instillation, and that emphysema development was not dependent on iNOS activity in bone-marrow-derived cells [
10,
11,
13]. Thus, in this study we aimed to identify the specific lung-cell type responsible for iNOS-dependent hindrance of alveolar regeneration in emphysematous lungs. Currently, available treatment options for COPD cannot cure the disease, nor can they stop disease progression. Only an alleviation of symptoms and reduction of future risk of exacerbations is hitherto possible. Interestingly, global inhibition of iNOS promoted lung repair and reverse remodeling of the pulmonary vasculature in two preclinical models of COPD, and might represent a novel treatment option, if transferrable to the human situation. However, for the transfer of such preclinical data to human COPD, delineation of the exact signaling mechanism of lung regeneration upon iNOS inhibition and the identification of responsible cell type(s) is necessary, as they could have a large impact on the therapeutic strategy (e.g., preferred route of administration, duration of treatment). In this regard, it has been suggested that failure of iNOS inhibition strategies in states such as sepsis and pain, might have been in part caused by insufficient understanding of iNOS’ complex functions and the dual modalities of iNOS and NO in a disease state (i.e., concentration-dependent protective versus harmful effects) [
19]. Of note, several clinical trials showed good tolerability of iNOS inhibitors [
20,
21,
22,
23], and recent literature suggests that adult lung regeneration (after pneumonectomy) is possible in humans [
24]. Hence, understanding the exact mechanism by which iNOS inhibition in emphysematous lungs elicits their regeneration and repair and identification of the responsible cell type is an important step toward the transfer of an iNOS inhibition strategy to a clinical trial and potential treatment of emphysema in humans.
Here, we hypothesized that deregulation of iNOS in AECII interferes with their role in lung regeneration, and that consequently, iNOS knockout in this cell type can promote the repair of alveolar epithelium in emphysematous lungs. Among numerous non-bone-marrow lung cell types, we chose AECII as the most likely contributor to the iNOS-dependent impediment of lung repair, based on their role in adult lung regeneration on the one hand and in the pathogenesis of COPD on the other [
25,
26]. Specifically, the proliferation of AECII and their transdifferentiation into AECI play a critical role in the restoration of normal alveolar architecture after injury [
27], while elevated apoptosis of these cells has been linked to emphysema pathology [
28]. Importantly, the previously mentioned study from our group demonstrated that the strong nitrating agent peroxynitrite, known to arise from the reaction of superoxide with iNOS-derived NO, could induce apoptosis of AECII [
10], and Boyer et al. confirmed that emphysematous murine lungs are characterized with the marked upregulation of iNOS and the accumulation of nitrated proteins, predominantly in AECII [
14]. However, to the best of our knowledge, this is the first study to attempt long-term knockout of iNOS, specifically in the AECII, as a therapeutic option for severe emphysema and pulmonary hypertension.
To test whether iNOS deletion in AECII can lead to regeneration of the alveolar epithelium in emphysematous lungs, we employed the
CCSP rtTA2s-M2 LC1 driver line, which enables tight temporal and spatial control of expression of luciferase and Cre recombinase by doxycycline, and was previously reported to efficiently target AECII in the adult lung [
18]. By crossing this line with
iNosflox/flox mice, we achieved inducible, doxycycline-controlled deletion of iNOS, predominantly in AECII. Importantly, a previous study from our group demonstrated that, although doxycycline inhibits MMPs and has anti-inflammatory properties, treatment with this antibiotic alone does not influence the reversal of emphysema and PH in mice [
11]. After the establishment of emphysema in the elastase mouse model, we induced iNOS knockout in AECII and gave an additional 12 weeks for the potential regeneration to occur. We chose this time point because previously Boyer et al. demonstrated that global iNOS knockout did not affect the emphysema severity at shorter time points, while the study from our group showed that long-term (12 weeks) treatment with iNOS inhibitor L-NIL could ameliorate emphysema in the same animal model [
11,
14].
Surprisingly, AECII-specific knockout of iNOS did not promote regeneration of the alveolar epithelium in the elastase mouse model. These data were corroborated by in vitro findings, where inhibition of this enzyme could not significantly alter the response of AECII and MLE-12 cells upon injury. In addition to AECII-specific knockout, the fact that in injured lungs AECII proliferate and give rise to AECI [
25] implies that in our elastase-treated knockouts at least a portion of AECI was also iNOS-deficient. Although our study did not determine exact portions of respective cell types with successful iNOS knockout, the fact that we failed to detect even mild beneficial effects of iNOS knockout implies that a lung compartment other than the alveolar epithelium might be a primary location of detrimental iNOS activity in the context of lung repair. Taken together with previous findings from our laboratory, our current results suggest that another lung cell type, such as pulmonary vascular cells or fibroblasts, may play a critical role in emphysema reversal upon iNOS inhibition. Strong induction of iNOS expression in the pulmonary vasculature of cigarette smoke-exposed mice and COPD patients detected in our previous studies, supports this notion [
10,
13]. Alternatively, the idea should be explored that lung stem cells other than AECII are more important players in the regeneration of emphysematous lungs. Recently, evidence was provided that even AECI can convert to AECII to support lung regrowth after a pneumonectomy [
29]. Moreover, a unique cell population at the bronchioalveolar-duct junctions with stem-cell properties has been identified, and shown to expand upon alveolar injury and differentiate into AECI and AECII [
30]. Nevertheless, even if the knockout of AECII-derived iNOS cannot ameliorate already established severe pathology, the question remains whether iNOS upregulation in alveolar epithelium contributes to the development of emphysema. To test this option, however, another animal model, such as exposure to cigarette smoke, would be more appropriate, as the mechanism of emphysema development in the elastase model relies on the fast enzymatic destruction of the alveolar architecture, and thus does not necessarily mimic the gradual and complex molecular changes driving emphysema in humans [
31].
Similarly, conclusions regarding the potential role of AECII-derived iNOS in reverse remodeling of the pulmonary vasculature have to be drawn with caution, as the scarce data available in the literature suggest that the pulmonary vascular pathology in the elastase model arises from the loss of capillary bed and hypoxemia [
9,
32,
33,
34]. Nevertheless, elastolysis and extracellular matrix remodeling have indeed been suggested as important and early events in PH development [
35,
36,
37]. Similarly, inflammation, another important contributor to pulmonary vascular remodeling, is present in this model as well [
31]. In addition, the previously reported beneficial effect of iNOS inhibition on the pulmonary vasculature in this model argues in favor of its similarity to the situation observed in cigarette smoke-exposed mice and human smokers. Finally, the ability of the iNOS inhibitor L-NIL to ameliorate elastase-induced PH postulates that iNOS knockout would be effective as well if carried out in the respective underlying cell type. We have previously shown that iNOS deregulation in myeloid cells, specifically macrophages, plays an important role in pulmonary vascular pathology in COPD. However, it remained unclear whether the knockout of myeloid cell-derived iNOS is also sufficient to promote the reverse remodeling of pulmonary vasculature. In this regard, it is important to consider the fact that the synergistic/simultaneous effects of iNOS in different cell types could drive lung regeneration. Nevertheless, iNOS knockout in AECII did not produce even a slight, partial improvement in any physiological or histological parameter used for the evaluation of pulmonary vasculature and the right ventricle. This failure to activate reverse remodeling of the pulmonary vasculature with AECII-specific iNOS knockout prompts us to conclude that (an)other lung cell-type(s), either bone-marrow-derived or of another origin, mediate(s) the beneficial effects of iNOS inhibition on remodeled pulmonary vessels in preclinical models of COPD.
As our study focused on the long-term effects of cell-specific iNOS knockout, it does not provide a detailed description of deregulated signaling pathways and physiological processes in early time-points after the injury with elastase, but rather gives insight into the chronic disease state. This is well exemplified by the fact that signaling pathways, such as those governing apoptosis and proliferation, are not deregulated in animals challenged with elastase, contrary to what one would expect after severe lung injury. However, elevated levels of MMP-8 could still be observed in elastase-treated mice, suggesting its possible involvement in the maintenance (or even propagation) of the emphysema pathology. Curiously, among the rare molecular changes we were able to detect in the lungs of elastase-treated mice 12 weeks after the establishment of the pathological phenotype, was a decrease in cytochrome c content that was more prominent in the control than in the AECII-specific iNOS knockout animals. As this change was not accompanied by alterations in caspase 3 activation, we conclude that it is limited to the mitochondrial compartment. The possibility remains that a decrease in the cytochrome c level reflects a reduction in the number of mitochondria, or influences the function of the mitochondrial respiratory chain, but the functional effects and overall significance of this finding remain to be investigated.
Another interesting discovery, warranting further investigation, relates to the in vitro effects of BALF on the apoptosis of the MLE-12 cell line. Namely, the decrease of apoptosis observed in cells treated with BALF from elastase-challenged lungs implies the existence of damage-control mechanisms in these lungs, mediated through as yet unknown soluble factors. Identification of such factors and careful delineation of the signaling pathways they activate might help design novel therapeutic concepts for COPD.



 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





