
Pharmacotherapy Evolution in Alzheimer’s Disease: Current Framework and Relevant Directions

 ,
,  ,
,  and
and 
Abstract
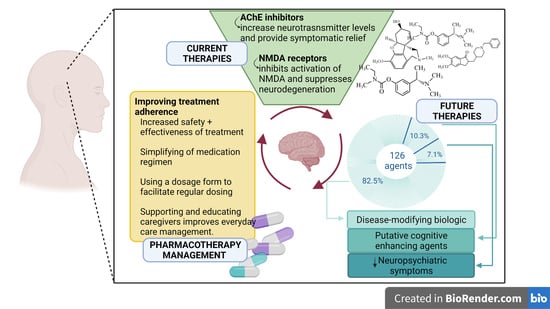
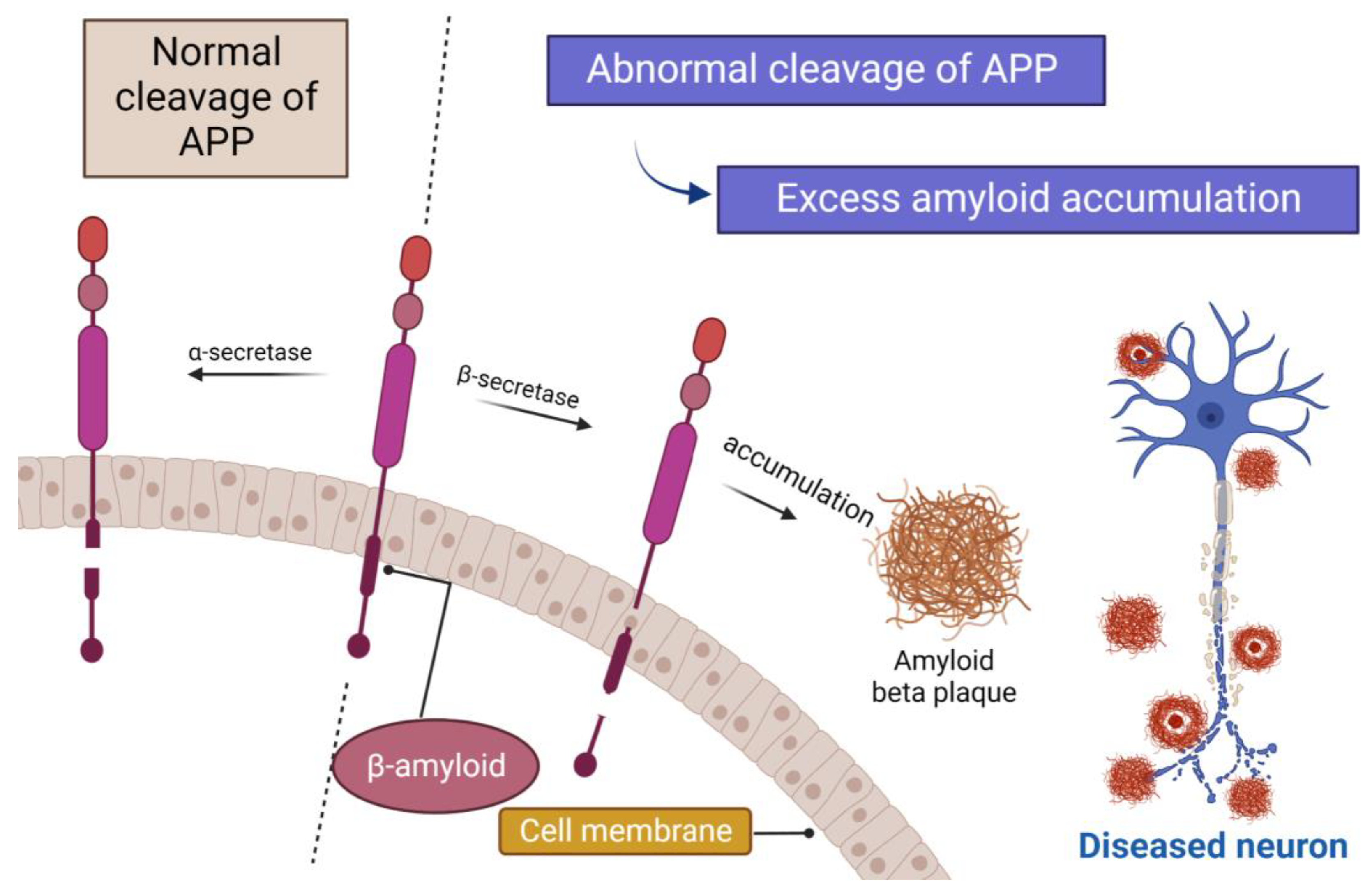
1. Introduction
2. Treatment of Alzheimer’s Disease
2.1. Acetylcholine and Acetylcholinesterase
2.2. AChE Enzyme Sites
2.3. Response to Treatment with AChE Inhibitors
2.4. Side Effects of AChE Inhibitors
3. Cholinesterase Inhibitors
3.1. Donepezil
3.2. Rivastigmine
3.3. Galantamine
4. Glutamate Antagonists
Memantine
5. Recent Progress in Medicinal Development
5.1. Exploring the Molecular and Cellular Pathways in Alzheimer’s Treatment
5.2. Phase I Studies
5.3. Phase II Studies
5.4. Phase III Studies
6. Exploring Alternative Treatments for AD
7. Challenges
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Disease. Available online: https://www.alzheimers.org.uk/about-dementia/types-dementia/alzheimers-disease (accessed on 26 May 2022).
- Anand, K.S.; Dhikav, V. Hippocampus in health and disease: An overview. Ann. Indian Acad. Neurol. 2012, 15, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Basics of Alzheimer’s Disease and Dementia What Is Alzheimer’s Disease? Available online: https://www.nia.nih.gov/health/what-alzheimers-disease (accessed on 27 May 2022).
- Ritchie, C.; Smailagic, N.; Noel-Storr, A.H.; Ukoumunne, O.; Ladds, E.C.; Martin, S. CSF tau and the CSF tau/ABeta ratio for the diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst. Rev. 2017, 3, CD010803. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. The Molecular and Cellular Basis of Neurodegenerative Diseases: Underlying Mechanisms; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Phillips, M.A.; Stewart, M.A.; Woodling, D.L.; Xie, Z.-R. Has molecular docking ever brought us a medicine? In Molecular Docking; Vlachakis, D.P., Ed.; IntechOpen: London, UK, 2018; Available online: https://www.intechopen.com/chapters/59054 (accessed on 29 May 2022). [CrossRef]
- Trott, O.; Olson, A.J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- BIOVIA, D.S. BIOVIA Discovery Studio Visualizer. Softw. Version 2017, 20, 779. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Samanta, S.; Ramesh, M.; Govindaraju, T. Alzheimer’s is a Multifactorial Disease. In Alzheimer’s Disease: Recent Findings in Pathophysiology, Diagnostic and Therapeutic Modalities; Govindaraju T., Ed.; Royal Society of Chemistry; 2022; pp. 1–34. Available online: https://pubs.rsc.org/en/content/chapterhtml/2022/bk9781839162305-00001?isbn=978-1-83916-230-5&sercode=bk (accessed on 27 May 2022).
- Larson, E.B.; Kukull, W.A.; Katzman, R.L. Cognitive impairment: Dementia and Alzheimer’s disease. Annu. Rev. Public Health 1992, 13, 431–449. [Google Scholar] [CrossRef]
- Roy, K. Computational Modeling of Drugs Against Alzheimer’s Disease; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- McCorry, L.K. Physiology of the autonomic nervous system. Am. J. Pharm. Educ. 2007, 71, 78. [Google Scholar] [CrossRef]
- Mushtaq, G.; Greig, N.H.; Khan, J.A.; Kamal, M.A. Status of acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease and type 2 diabetes mellitus. CNS Neurol. Disord. Drug Targets 2014, 13, 1432–1439. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, S.; Zhang, Y. Catalytic reaction mechanism of acetylcholinesterase determined by Born-Oppenheimer ab initio QM/MM molecular dynamics simulations. J. Phys. Chem. B 2010, 114, 8817–8825. [Google Scholar] [CrossRef]
- Xu, Y.; Cheng, S.; Sussman, J.L.; Silman, I.; Jiang, H. Computational Studies on Acetylcholinesterases. Molecules 2017, 22, 1324. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.A.; Kiametis, A.S.; Treptow, W. Donepezil Inhibits Acetylcholinesterase via Multiple Binding Modes at Room Temperature. J. Chem. Inf. Model. 2020, 60, 3463–3471. [Google Scholar] [CrossRef] [PubMed]
- Svobodova, B.; Mezeiova, E.; Hepnarova, V.; Hrabinova, M.; Muckova, L.; Kobrlova, T.; Jun, D.; Soukup, O.; Jimeno, M.L.; Marco-Contelles, J.; et al. Exploring Structure-Activity Relationship in Tacrine-Squaramide Derivatives as Potent Cholinesterase Inhibitors. Biomolecules 2019, 9, 379. [Google Scholar] [CrossRef] [PubMed]
- Galantamine Derivatives as Acetylcholinesterase Inhibitors: Docking, Design, Synthesis, and Inhibitory Activity. Available online: https://experiments.springernature.com/articles/10.1007/978-1-4939-7404-7_6 (accessed on 19 May 2022).
- Ambure, P.; Kar, S.; Roy, K. Pharmacophore mapping-based virtual screening followed by molecular docking studies in search of potential acetylcholinesterase inhibitors as anti-Alzheimer’s agents. Biosystems 2014, 116, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Seniya, C.; Khan, G.J.; Uchadia, K. Identification of potential herbal inhibitor of acetylcholinesterase associated Alzheimer’s disorders using molecular docking and molecular dynamics simulation. Biochem. Res. Int. 2014, 2014, 705451. [Google Scholar] [CrossRef][Green Version]
- Pinto, T.C.C.; Machado, L.; Bulgacov, T.M.; Rodrigues-Júnior, A.L.; Costa, M.L.G.; Ximenes, R.C.C.; Sougey, E.B. Is the Montreal Cognitive Assessment (MoCA) screening superior to the Mini-Mental State Examination (MMSE) in the detection of mild cognitive impairment (MCI) and Alzheimer’s Disease (AD) in the elderly? Int. Psychogeriatr. 2019, 31, 491–504. [Google Scholar] [CrossRef]
- Scharre, D.W.; Chang, S.i.; Nagaraja, H.N.; Wheeler, N.C.; Kataki, M. Self-Administered Gerocognitive Examination: Longitudinal cohort testing for the early detection of dementia conversion. Alzheimer’s Res. Ther. 2021, 13, 192. [Google Scholar] [CrossRef]
- Gill, S.S.; Anderson, G.M.; Fischer, H.D.; Bell, C.M.; Li, P.; Normand, S.-L.T.; Rochon, P.A. Syncope and Its Consequences in Patients With Dementia Receiving Cholinesterase Inhibitors: A Population-Based Cohort Study. Arch. Intern. Med. 2009, 169, 867–873. [Google Scholar] [CrossRef]
- McGleenon, B.M.; Dynan, K.B.; Passmore, A.P. Acetylcholinesterase inhibitors in Alzheimer’s disease. Br. J. Clin. Pharmacol. 1999, 48, 471–480. [Google Scholar] [CrossRef]
- Waldemar, G.; Burns, A. Alzheimer’s Disease; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- Singer, M.; Romero, B.; Koenig, E.; Förstl, H.; Brunner, H. Nightmares in patients with Alzheimer’s disease caused by donepezil. Therapeutic effect depends on the time of intake. Nervenarzt 2005, 76, 1127–1129. [Google Scholar] [CrossRef]
- Budson, A.E.; Solomon, P.R. Memory Loss, Alzheimer’s Disease, and Dementia-E-Book: A Practical Guide for Clinicians; Elsevier Health Sciences: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Galli, A.; Mori, F.; Benini, L.; Cacciarelli, N. Acetylcholinesterase protection and the anti-diisopropylfluorophosphate efficacy of E2020. Eur. J. Pharm. 1994, 270, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Donepezil. Available online: https://go.drugbank.com/drugs/DB00843 (accessed on 22 May 2022).
- Kumar, A.; Gupta, V.; Sharma, S. Donepezil. In StatPearls; StatPearls Publishing Copyright © 2022; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Nochi, S.; Asakawa, N.; Sato, T. Kinetic study on the inhibition of acetylcholinesterase by 1-benzyl-4-[(5,6-dimethoxy-1-indanon)-2-yl] methylpiperidine hydrochloride (E2020). Biol. Pharm. Bull. 1995, 18, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E.; Zhu, X.D.; Williams, E.; Sherman, K.A. The effect of the selective reversible acetylcholinesterase inhibitor E2020 on extracellular acetylcholine and biogenic amine levels in rat cortex. Neuropharmacology 1996, 35, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Ogura, H.; Arai, Y.; Limura, Y.; Yamanishi, Y. Research and development of donepezil hydrochloride, a new type of acetylcholinesterase inhibitor. Jpn. J. Pharm. 2002, 89, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, A.; Mihara, M.; Kamakura, H.; Tomono, Y.; Hasegawa, J.; Yamazaki, K.; Morishita, N.; Tanaka, T. Comparison of the pharmacokinetics of E2020, a new compound for Alzheimer’s disease, in healthy young and elderly subjects. J. Clin. Pharm. 1993, 33, 1086–1091. [Google Scholar] [CrossRef]
- Heydorn, W.E. Donepezil (E2020): A new acetylcholinesterase inhibitor. Review of its pharmacology, pharmacokinetics, and utility in the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 1997, 6, 1527–1535. [Google Scholar] [CrossRef]
- Geerts, H.; Guillaumat, P.O.; Grantham, C.; Bode, W.; Anciaux, K.; Sachak, S. Brain levels and acetylcholinesterase inhibition with galantamine and donepezil in rats, mice, and rabbits. Brain Res. 2005, 1033, 186–193. [Google Scholar] [CrossRef]
- Cacabelos, R. Donepezil in Alzheimer’s disease: From conventional trials to pharmacogenetics. Neuropsychiatr. Dis. Treat. 2007, 3, 303–333. [Google Scholar]
- Rosenbloom, M.H.; Finley, R.; Scheinman, M.M.; Feldman, M.D.; Miller, B.L.; Rabinovici, G.D. Donepezil-associated bradyarrhythmia in a patient with dementia with Lewy bodies (DLB). Alzheimer Dis. Assoc. Disord. 2010, 24, 209–211. [Google Scholar] [CrossRef]
- Agboton, C.; Mahdavian, S.; Singh, A.; Ghazvini, P.; Hill, A.; Sweet, R. Impact of nighttime donepezil administration on sleep in the older adult population: A retrospective study. Ment. Health Clin. 2014, 4, 257–259. [Google Scholar] [CrossRef]
- Jackson, S.; Ham, R.J.; Wilkinson, D. The safety and tolerability of donepezil in patients with Alzheimer’s disease. Br. J. Clin. Pharmacol. 2004, 58 (Suppl. S1), 1–8. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Corey-Bloom, J.; Zhang, R.; Li, H.; Ieni, J.; Schindler, R. Safety and tolerability of donepezil at doses up to 20 mg/day: Results from a pilot study in patients with Alzheimer’s disease. Drugs Aging 2008, 25, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Grossberg, G. Review of rivastigmine and its clinical applications in Alzheimer’s disease and related disorders. Expert Opin. Pharm. 2001, 2, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Grantham, D.L.; Hopkins, D.A. Distribution of butyrylcholinesterase in the human amygdala and hippocampal formation. J. Comp. Neurol. 1998, 393, 374–390. [Google Scholar] [CrossRef]
- Onor, M.L.; Trevisiol, M.; Aguglia, E. Rivastigmine in the treatment of Alzheimer’s disease: An update. Clin. Interv. Aging 2007, 2, 17–32. [Google Scholar] [CrossRef]
- Cummings, J.; Lefèvre, G.; Small, G.; Appel-Dingemanse, S. Pharmacokinetic rationale for the rivastigmine patch. Neurology 2007, 69, S10–S13. [Google Scholar] [CrossRef]
- Lefèvre, G.; Pommier, F.; Sedek, G.; Allison, M.; Huang, H.L.; Kiese, B.; Ho, Y.Y.; Appel-Dingemanse, S. Pharmacokinetics and bioavailability of the novel rivastigmine transdermal patch versus rivastigmine oral solution in healthy elderly subjects. J. Clin. Pharm. 2008, 48, 246–252. [Google Scholar] [CrossRef]
- Farlow, M.R.; Grossberg, G.T.; Sadowsky, C.H.; Meng, X.; Somogyi, M. A 24-week, randomized, controlled trial of rivastigmine patch 13.3 mg/24 h versus 4.6 mg/24 h in severe Alzheimer’s dementia. CNS Neurosci. Ther. 2013, 19, 745–752. [Google Scholar] [CrossRef]
- Sadowsky, C.H.; Micca, J.L.; Grossberg, G.T.; Velting, D.M. Rivastigmine from capsules to patch: Therapeutic advances in the management of Alzheimer’s disease and Parkinson’s disease dementia. Prim. Care Companion CNS Disord. 2014, 16, 10.4088. [Google Scholar] [CrossRef]
- Rivastigmine. Available online: https://go.drugbank.com/drugs/DB00989 (accessed on 25 May 2021).
- Birks, J.S.; Chong, L.Y.; Grimley Evans, J. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2015, 9, Cd001191. [Google Scholar] [CrossRef]
- Jhee, S.S.; Shiovitz, T.; Hartman, R.D.; Messina, J.; Anand, R.; Sramek, J.; Cutler, N.R. Centrally acting antiemetics mitigate nausea and vomiting in patients with Alzheimer’s disease who receive rivastigmine. Clin. Neuropharmacol. 2002, 25, 122–123. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Davis, K.L.; Gonzalez, R.B.; Wilkinson, D.G. Practical Pharmacology for Alzheimer’s Disease; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Exelon. Available online: https://www.rxlist.com/exelon-drug.htm#description (accessed on 21 May 2022).
- Ale, I.; Lachapelle, J.M.; Maibach, H.I. Skin tolerability associated with transdermal drug delivery systems: An overview. Adv. Ther. 2009, 26, 920–935. [Google Scholar] [CrossRef] [PubMed]
- Wohlrab, J.; Kreft, B.; Tamke, B. Skin tolerability of transdermal patches. Expert Opin. Drug Deliv. 2011, 8, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Bores, G.M.; Huger, F.P.; Petko, W.; Mutlib, A.E.; Camacho, F.; Rush, D.K.; Selk, D.E.; Wolf, V.; Kosley, R.W., Jr.; Davis, L.; et al. Pharmacological evaluation of novel Alzheimer’s disease therapeutics: Acetylcholinesterase inhibitors related to galanthamine. J Pharm. Exp 1996, 277, 728–738. [Google Scholar]
- Thomsen, T.; Kewitz, H. Selective inhibition of human acetylcholinesterase by galanthamine in vitro and in vivo. Life Sci. 1990, 46, 1553–1558. [Google Scholar] [CrossRef]
- Nordberg, A.; Darreh-Shori, T.; Peskind, E.; Soininen, H.; Mousavi, M.; Eagle, G.; Lane, R. Different cholinesterase inhibitor effects on CSF cholinesterases in Alzheimer patients. Curr. Alzheimer Res. 2009, 6, 4–14. [Google Scholar] [CrossRef]
- Piotrovsky, V.; Van Peer, A.; Van Osselaer, N.; Armstrong, M.; Aerssens, J. Galantamine population pharmacokinetics in patients with Alzheimer’s disease: Modeling and simulations. J. Clin. Pharm. 2003, 43, 514–523. [Google Scholar] [CrossRef]
- Farlow, M.R. Clinical pharmacokinetics of galantamine. Clin. Pharm. 2003, 42, 1383–1392. [Google Scholar] [CrossRef]
- Galantamine. Available online: https://go.drugbank.com/drugs/DB00674 (accessed on 20 May 2022).
- Prvulovic, D.; Hampel, H.; Pantel, J. Galantamine for Alzheimer’s disease. Expert Opin. Drug Metab. Toxicol. 2010, 6, 345–354. [Google Scholar] [CrossRef]
- Aronson, S.; Van Baelen, B.; Kavanagh, S.; Schwalen, S. Optimal dosing of galantamine in patients with mild or moderate Alzheimer’s disease: Post Hoc analysis of a randomized, double-blind, placebo-controlled trial. Drugs Aging 2009, 26, 231–239. [Google Scholar] [CrossRef]
- Folch, J.; Busquets, O.; Ettcheto, M.; Sánchez-López, E.; Castro-Torres, R.D.; Verdaguer, E.; Garcia, M.L.; Olloquequi, J.; Casadesús, G.; Beas-Zarate, C.; et al. Memantine for the Treatment of Dementia: A Review on its Current and Future Applications. J. Alzheimers Dis. 2018, 62, 1223–1240. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [PubMed]
- Riedel, G.; Platt, B.; Micheau, J. Glutamate receptor function in learning and memory. Behav. Brain Res. 2003, 140, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Moreno, A.; Banerjee, A.; Paulsen, O. Presynaptic NMDA receptors and spike timing-dependent long-term depression at cortical synapses. Front. Synaptic Neurosci. 2010, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P.; Caruso, C.; Lasaga, M. Memantine agonist action at dopamine D2High receptors. Synapse 2008, 62, 149–153. [Google Scholar] [CrossRef]
- Wesnes, K.A.; Aarsland, D.; Ballard, C.; Londos, E. Memantine improves attention and episodic memory in Parkinson’s disease dementia and dementia with Lewy bodies. Int. J. Geriatr. Psychiatry 2015, 30, 46–54. [Google Scholar] [CrossRef]
- Parsons, C.G.; Danysz, W.; Dekundy, A.; Pulte, I. Memantine and cholinesterase inhibitors: Complementary mechanisms in the treatment of Alzheimer’s disease. Neurotox. Res. 2013, 24, 358–369. [Google Scholar] [CrossRef]
- Atri, A.; Molinuevo, J.L.; Lemming, O.; Wirth, Y.; Pulte, I.; Wilkinson, D. Memantine in patients with Alzheimer’s disease receiving donepezil: New analyses of efficacy and safety for combination therapy. Alzheimer’s Res. Ther. 2013, 5, 6. [Google Scholar] [CrossRef]
- Matsunaga, S.; Kishi, T.; Iwata, N. Memantine monotherapy for Alzheimer’s disease: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0123289. [Google Scholar] [CrossRef]
- Memantine. Available online: https://go.drugbank.com/drugs/DB01043 (accessed on 25 May 2022).
- van Marum, R.J. Update on the use of memantine in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2009, 5, 237–247. [Google Scholar] [CrossRef]
- Thomas, S.J.; Grossberg, G.T. Memantine: A review of studies into its safety and efficacy in treating Alzheimer’s disease and other dementias. Clin. Interv. Aging 2009, 4, 367–377. [Google Scholar] [CrossRef] [PubMed]
- 2870 Studies Found for: Alzheimer. Available online: https://www.clinicaltrials.gov/ct2/results?cond=alzheimer+&term=&cntry=&state=&city=&dist=%20ongoing%20clinical%20trials (accessed on 31 May 2022).
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef] [PubMed]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; Strooper, B.D. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Uddin, M.S.; Kabir, M.T.; Rahman, M.S.; Behl, T.; Jeandet, P.; Ashraf, G.M.; Najda, A.; Bin-Jumah, M.N.; El-Seedi, H.R.; Abdel-Daim, M.M. Revisiting the Amyloid Cascade Hypothesis: From Anti-Aβ Therapeutics to Auspicious New Ways for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 5858. [Google Scholar] [CrossRef]
- Trushina, N.I.; Bakota, L.; Mulkidjanian, A.Y.; Brandt, R. The Evolution of Tau Phosphorylation and Interactions. Front. Aging Neurosci. 2019, 11, 256. [Google Scholar] [CrossRef]
- Noble, W.; Hanger, D.P.; Miller, C.C.; Lovestone, S. The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol. 2013, 4, 83. [Google Scholar] [CrossRef]
- Liu, S.L.; Wang, C.; Jiang, T.; Tan, L.; Xing, A.; Yu, J.T. The Role of Cdk5 in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 4328–4342. [Google Scholar] [CrossRef]
- Bittar, A.; Bhatt, N.; Kayed, R. Advances and considerations in AD tau-targeted immunotherapy. Neurobiol. Dis. 2020, 134, 104707. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S.; Alam, J.; Fillit, H.; Iwatsubo, T.; Liu-Seifert, H.; Sabbagh, M.; Salloway, S.; Sampaio, C.; Sims, J.; Sperling, B. Combination therapy for Alzheimer’s disease: Perspectives of the EU/US CTAD Task Force. J. Prev. Alzheimer’s Dis. 2019, 6, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, G.; Sehgal, A.; Bhardwaj, S.; Singh, S.; Buhas, C.; Judea-Pusta, C.; Uivarosan, D.; Munteanu, M.A.; Bungau, S. Multifaceted role of matrix metalloproteinases in neurodegenerative diseases: Pathophysiological and therapeutic perspectives. Int. J. Mol. Sci. 2021, 22, 1413. [Google Scholar] [CrossRef]
- Dheen, S.T.; Kaur, C.; Ling, E.A. Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Fratila, O.; Brata, R.; Bungau, S. Exploring the potential of therapeutic agents targeted towards mitigating the events associated with amyloid-β cascade in Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 7443. [Google Scholar] [CrossRef] [PubMed]
- Kaur, D.; Behl, T.; Sehgal, A.; Singh, S.; Sharma, N.; Bungau, S. Multifaceted Alzheimer’s Disease: Building a Roadmap for Advancement of Novel Therapies. Neurochem. Res. 2021, 46, 2832–2851. [Google Scholar] [CrossRef]
- Bajda, M.; Guzior, N.; Ignasik, M.; Malawska, B. Multi-target-directed ligands in Alzheimer’s disease treatment. Curr. Med. Chem. 2011, 18, 4949–4975. [Google Scholar] [CrossRef]
- Agis-Torres, A.; Sollhuber, M.; Fernandez, M.; Sanchez-Montero, J. Multi-target-directed ligands and other therapeutic strategies in the search of a real solution for Alzheimer’s disease. Curr. Neuropharmacol. 2014, 12, 2–36. [Google Scholar] [CrossRef]
- Bhute, S.; Sarmah, D.; Datta, A.; Rane, P.; Shard, A.; Goswami, A.; Borah, A.; Kalia, K.; Dave, K.R.; Bhattacharya, P. Molecular pathogenesis and interventional strategies for Alzheimer’s disease: Promises and pitfalls. ACS Pharmacol. Transl. Sci. 2020, 3, 472–488. [Google Scholar] [CrossRef] [PubMed]
- Oxford, A.E.; Stewart, E.S.; Rohn, T.T. Clinical Trials in Alzheimer’s Disease: A Hurdle in the Path of Remedy. Int. J. Alzheimers Dis. 2020, 2020, 5380346. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline. Alzheimers Dement. 2020, 6, e12050. [Google Scholar]
- Sub-Lingual Dexmedetomidine in Agitation Associated with Dementia (TRANQUILITY). Available online: https://clinicaltrials.gov/ct2/show/NCT04251910 (accessed on 30 May 2022).
- Rosenzweig, A.B.; Sittambalam, C.D. A new approach to the prevention and treatment of delirium in elderly patients in the intensive care unit. J. Community Hosp. Intern. Med. Perspect. 2015, 5, 27950. [Google Scholar] [CrossRef]
- MK-1942/Donepezil Interactions in Participants with Alzheimer’s Disease (MK-1942-005) (DDI). Available online: https://clinicaltrials.gov/ct2/show/NCT04308304 (accessed on 29 May 2022).
- Mycose AdminiStration for HealIng Alzheimer NEuropathy (MASHIANE). Available online: https://clinicaltrials.gov/ct2/show/NCT04663854 (accessed on 28 May 2022).
- Khalifeh, M.; Read, M.I.; Barreto, G.E.; Sahebkar, A. Trehalose against Alzheimer’s Disease: Insights into a Potential Therapy. BioEssays 2020, 42, 1900195. [Google Scholar] [CrossRef]
- Benito-Cuesta, I.; Ordoñez-Gutierrez, L.; Wandosell, F. Trehalose Reduces the Secreted Beta-Amyloid Levels in Primary Neurons Independently of Autophagy Induction. Metabolites 2021, 11, 421. [Google Scholar] [CrossRef]
- Safety and Immunogenicity of Repeated Doses of ABvac40 in Patients with a-MCI or Vm-AD. Available online: https://clinicaltrials.gov/ct2/show/NCT03461276 (accessed on 26 May 2022).
- Jia, Q.; Deng, Y.; Qing, H. Potential Therapeutic Strategies for Alzheimer’s Disease Targeting or Beyond β-Amyloid: Insights from Clinical Trials. BioMed Res. Int. 2014, 2014, 837157. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, Q.; Zhang, Y.-W.; Xu, H. Proteolytic processing of Alzheimer’s β-amyloid precursor protein. J. Neurochem. 2012, 120 (Suppl. S1), 9–21. [Google Scholar] [CrossRef]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef]
- Alector Announces First Participant Dosed in Phase 2 Study Evaluating AL002 in Individuals with Early Alzheimer’s Disease. Available online: https://investors.alector.com/node/7616/pdf (accessed on 25 May 2022).
- ACI-35. Available online: https://alzheimersnewstoday.com/aci-35/ (accessed on 28 May 2022).
- Winblad, B.; Graf, A.; Riviere, M.-E.; Andreasen, N.; Ryan, J.M. Active immunotherapy options for Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 7. [Google Scholar] [CrossRef]
- THERAPEUTICS, ACI-35. Available online: https://www.alzforum.org/therapeutics/aci-35 (accessed on 30 May 2022).
- Cassava Sciences Initiates a Phase 3 Efficacy Trial of Simufilam for the Treatment of Patients with Alzheimer’s Disease. Available online: https://www.cassavasciences.com/news-releases/news-release-details/cassava-sciences-initiates-phase-3-efficacy-trial-simufilam (accessed on 31 May 2022).
- Status of Cassava Sciences New Alzheimer’s Drug: Simufilam. Available online: https://www.dementiacarecentral.com/aboutdementia/treating/simufilam/ (accessed on 22 May 2022).
- Silverman, M.; Wallner, B.; Key, C.; Duggan, S.M.; Reynolds, L. A single-and multiple-ascending dose study to evaluate the safety and pharmacokinetics of oral PU-AD, an epichaperome inhibitor to treat Alzheimer’s disease: Human/Trial design. Alzheimer’s Dement. 2020, 16, e041144. [Google Scholar] [CrossRef]
- Therapeutics, PU-AD. Available online: https://www.alzforum.org/therapeutics/pu-ad (accessed on 11 November 2022).
- Silvestro, S.; Valeri, A.; Mazzon, E. Aducanumab and Its Effects on Tau Pathology: Is This the Turning Point of Amyloid Hypothesis? Int. J. Mol. Sci. 2022, 23, 2011. [Google Scholar] [CrossRef] [PubMed]
- Athar, T.; Al Balushi, K.; Khan, S.A. Recent advances on drug development and emerging therapeutic agents for Alzheimer’s disease. Mol. Biol. Rep. 2021, 48, 5629–5645. [Google Scholar] [CrossRef] [PubMed]
- FDA’s Decision to Approve New Treatment for Alzheimer’s Disease. Available online: https://www.fda.gov/drugs/news-events-human-drugs/fdas-decision-approve-new-treatment-alzheimers-disease (accessed on 19 May 2022).
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol. 2010, 9, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Effect of CAFfeine on Cognition in Alzheimer’s Disease (CAFCA). Available online: https://www.clinicaltrials.gov/ct2/show/NCT04570085?term=caffeine&cond=alzheimer&draw=2&rank=1 (accessed on 29 May 2022).
- #AAIC21—Phase 3 Trial of Anti-Inflammatory NE3107 Begins Enrolling. Available online: https://alzheimersnewstoday.com/2021/08/12/aaic21-phase-3-trial-ne3107-therapy-inflammation-insulin-sensitivity-enrolls-1st-patient/ (accessed on 30 May 2022).
- A Phase 3 Study of NE3107 in Probable Alzheimer’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT04669028 (accessed on 20 May 2022).
- Hey, J.A.; Kocis, P.; Hort, J.; Abushakra, S.; Power, A.; Vyhnálek, M.; Yu, J.Y.; Tolar, M. Discovery and Identification of an Endogenous Metabolite of Tramiprosate and Its Prodrug ALZ-801 that Inhibits Beta Amyloid Oligomer Formation in the Human Brain. CNS Drugs 2018, 32, 849–861. [Google Scholar] [CrossRef]
- Hey, J.A.; Yu, J.Y.; Versavel, M.; Abushakra, S.; Kocis, P.; Power, A.; Kaplan, P.L.; Amedio, J.; Tolar, M. Clinical Pharmacokinetics and Safety of ALZ-801, a Novel Prodrug of Tramiprosate in Development for the Treatment of Alzheimer’s Disease. Clin Pharm. 2018, 57, 315–333. [Google Scholar] [CrossRef]
- Alternative Treatments. Available online: https://www.alz.org/alzheimers-dementia/treatments/alternative-treatments (accessed on 14 December 2022).
- Dietary Supplements. Available online: https://www.fda.gov/consumers/consumer-updates/dietary-supplements (accessed on 14 December 2022).
- Is That Supplement Safe to Take with Your Medications? Available online: https://www.alzdiscovery.org/cognitive-vitality/blog/is-that-supplement-safe-to-take-with-your-medications (accessed on 18 December 2022).
- Dietary Supplements and Cognitive Function, Dementia, and Alzheimer’s Disease. Available online: https://www.nccih.nih.gov/health/providers/digest/dietary-supplements-and-cognitive-function-dementia-and-alzheimers-disease (accessed on 29 November 2022).
- Behl, T.; Makkar, R.; Sehgal, A.; Singh, S.; Sharma, N.; Zengin, G.; Bungau, S.; Andronie-Cioara, F.L.; Munteanu, M.A.; Brisc, M.C. Current trends in neurodegeneration: Cross talks between oxidative stress, cell death, and inflammation. Int. J. Mol. Sci. 2021, 22, 7432. [Google Scholar] [CrossRef]
- Abdel-Daim, M.M.; Abo-El-Sooud, K.; Aleya, L.; Bungǎu, S.G.; Najda, A.; Saluja, R. Alleviation of Drugs and Chemicals Toxicity: Biomedical Value of Antioxidants. Oxidative Med. Cell. Longev. 2018, 2018, 6276438. [Google Scholar] [CrossRef]
- Ben Amar, M. Cannabinoids in medicine: A review of their therapeutic potential. J. Ethnopharmacol. 2006, 105, 1–25. [Google Scholar] [CrossRef]
- Abate, G.; Uberti, D.; Tambaro, S. Potential and Limits of Cannabinoids in Alzheimer’s Disease Therapy. Biology 2021, 10, 542. [Google Scholar] [CrossRef]
- Ferrer, I. Cannabinoids for treatment of Alzheimer’s disease: Moving toward the clinic. Front. Pharmacol. 2014, 5, 37. [Google Scholar] [CrossRef]
- Schubert, D.; Kepchia, D.; Liang, Z.; Dargusch, R.; Goldberg, J.; Maher, P. Efficacy of Cannabinoids in a Pre-Clinical Drug-Screening Platform for Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 7719–7730. [Google Scholar] [CrossRef] [PubMed]
- Cannabis and Cannabis-Derived Products. Available online: https://www.alz.org/media/documents/cannabis-and-cannabis-derived-products-statement-updated-feb-2020.pdf (accessed on 16 December 2022).
- Music Therapy. Available online: https://my.clevelandclinic.org/health/treatments/8817-music-therapy (accessed on 17 December 2022).
- Art and Music. Available online: https://www.alz.org/help-support/caregiving/daily-care/art-music (accessed on 11 December 2022).
- Leggieri, M.; Thaut, M.H.; Fornazzari, L.; Schweizer, T.A.; Barfett, J.; Munoz, D.G.; Fischer, C.E. Music Intervention Approaches for Alzheimer’s Disease: A Review of the Literature. Front. Neurosci. 2019, 13, 132. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Morales, C.; Calero, R.; Moreno-Morales, P.; Pintado, C. Music Therapy in the Treatment of Dementia: A Systematic Review and Meta-Analysis. Front. Med. 2020, 7, 160. [Google Scholar] [CrossRef] [PubMed]
- Gómez Gallego, M.; Gómez García, J. Music therapy and Alzheimer’s disease: Cognitive, psychological, and behavioural effects. Neurologia 2017, 32, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Alternative Treatments for Dementia. Available online: https://alzheimer.ca/en/about-dementia/how-can-i-treat-dementia/alternative-treatments-dementia (accessed on 10 December 2022).
- Viggo Hansen, N.; Jørgensen, T.; Ørtenblad, L. Massage and touch for dementia. Cochrane Database Syst. Rev. 2006, 2006, Cd004989. [Google Scholar] [CrossRef] [PubMed]
- Lai, N.M.; Chang, S.M.W.; Ng, S.S.; Tan, S.L.; Chaiyakunapruk, N.; Stanaway, F. Animal-assisted therapy for dementia. Cochrane Database Syst. Rev. 2019, 2019, CD013243. [Google Scholar]
- Dubois, B.; Padovani, A.; Scheltens, P.; Rossi, A.; Dell’Agnello, G. Timely diagnosis for Alzheimer’s disease: A literature review on benefits and challenges. J. Alzheimer’s Dis. 2016, 49, 617–631. [Google Scholar] [CrossRef]
- Dietary Supplements. Available online: https://www.fda.gov/food/dietary-supplements (accessed on 27 November 2022).
- Akram, M.; Nawaz, A. Effects of medicinal plants on Alzheimer’s disease and memory deficits. Neural Regen. Res. 2017, 12, 660–670. [Google Scholar] [CrossRef]
- Akter, R.; Rahman, M.H.; Behl, T.; Chowdhury, M.A.R.; Manirujjaman, M.; Bulbul, I.J.; Elshenaw, S.E.; Tit, D.M.; Bungau, S. Prospective Role of Polyphenolic Compounds in the Treatment of Neurodegenerative Diseases. CNS Neurol. Disord. Drug Targets 2021, 20, 430–450. [Google Scholar] [CrossRef]
- O’Kelly, J.W. Music therapy and neuroscience: Opportunities and challenges. Voices A World Forum Music. Ther. 2016, 16. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
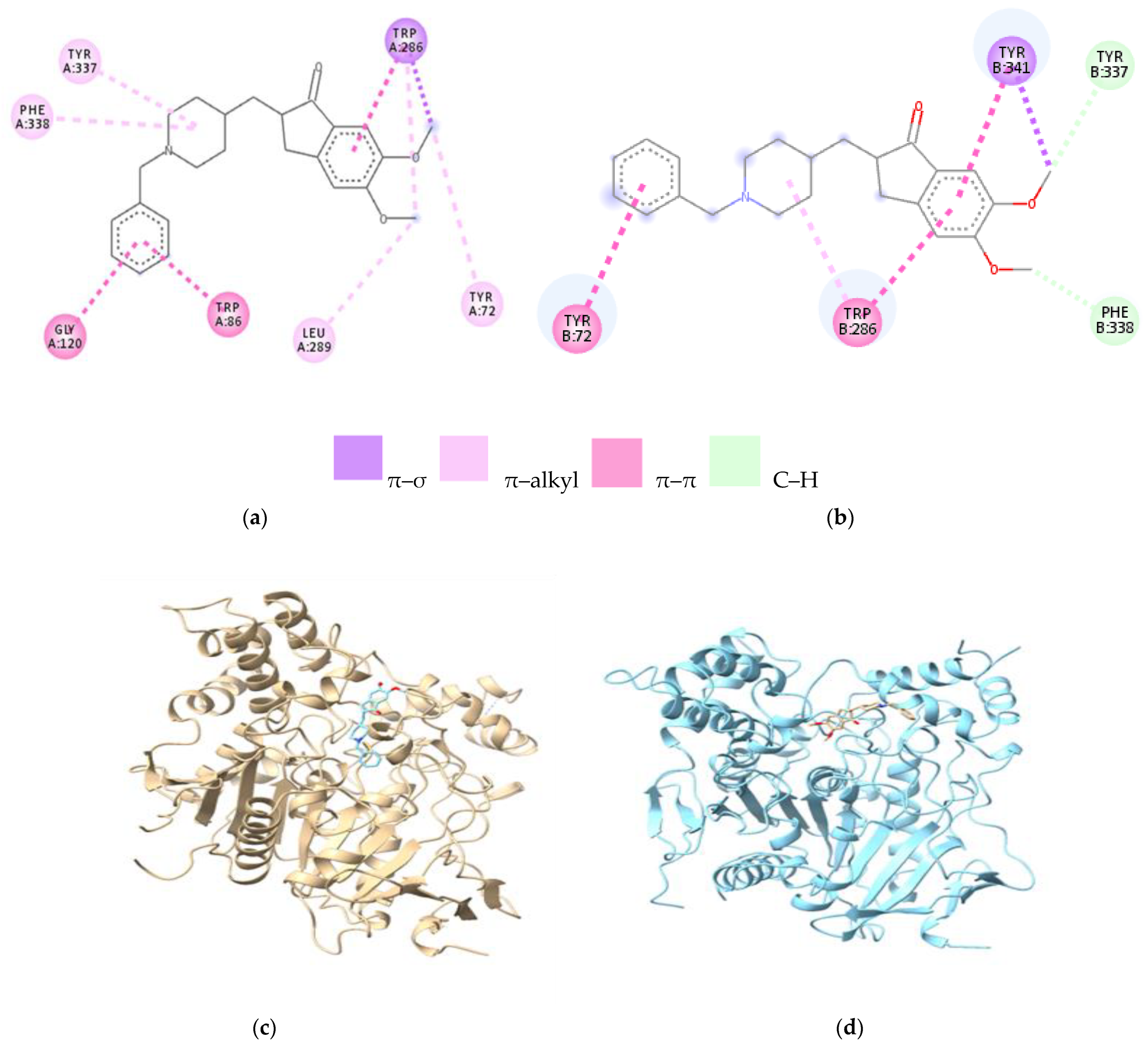
| Amino Acids of Chain A | Distance Ligand–Protein (Å) | Types of Bonds | Amino Acids of Chain B | Distance Ligand–Protein (Å) | Types of Bonds |
|---|---|---|---|---|---|
| Gly120 | 3.95 | π–amide | Trp286 | 4.97 | π–alkyl |
| Trp86 | 4.92 | π–π | 4.46 | π–π | |
| 4.38 | π–π | Tyr337 | 3.58 | C–H | |
| Phe338 | 5.02 | π–alkyl | Tyr341 | 4.45 | π–π |
| Tyr337 | 3.58 | π–alkyl | 3.64 | π–σ | |
| Trp286 | 4.34 | π–π | Tyr72 | 4.89 | π–π |
| 4.60 | π–Alkyl | Phe338 | 3.40 | C–H | |
| 3.93 | π–σ | - | - | - | |
| Tyr72 | 5.17 | π–alkyl | - | - | - |
| Leu289 | 5.15 | Alkyl | - | - | - |
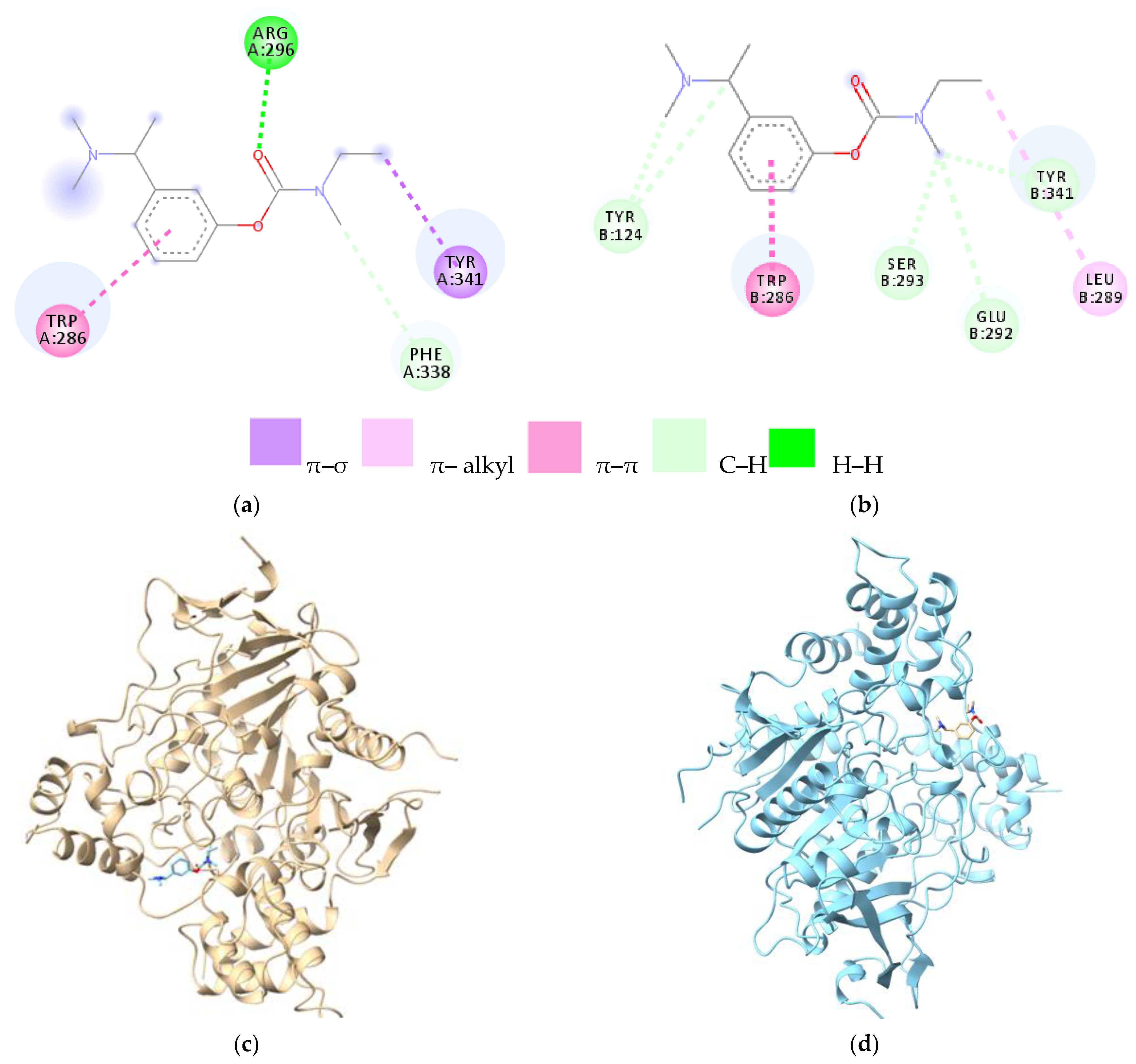
| Amino Acids of Chain A | Distance Ligand–Protein (Å) | Types of Bonds | Amino Acids of Chain B | Distance Ligand–Protein (Å) | Types of Bonds |
|---|---|---|---|---|---|
| Trp286 | 4.87 | π–π | Trp286 | 4.01 | π–π |
| Arg296 | 2.32 | leg. de H | Tyr124 | 3.55 | C–H |
| Tyr341 | 3.62 | π–σ | 3.66 | C–H | |
| Phe338 | 3.61 | C–H | Ser293 | 3.50 | C–H |
| - | - | - | Glu292 | 3.59 | C–H |
| - | - | - | Leu289 | 4.80 | Alkyl |
| - | - | - | Tyr341 | 3.58 | C–H |
| Amino Acids of Chain A | Distance Ligand–Protein (Å) | Types of Bonds | Amino Acids of Chain B | Distance Ligand–Protein (Å) | Types of Bonds |
|---|---|---|---|---|---|
| Trp286 | 4.70 | π–alkyl | Tyr341 | 3.63 | π–σ |
| 4.78 | π–alkyl | 5.14 | π–alkyl | ||
| Phe295 | 2.87 | leg. de H | Leu76 | 4.45 | Alkyl |
| Phe297 | 4.99 | π–alkyl | Trp286 | 4.92 | π–π |
| - | - | - | 4.08 | π–π | |
| - | - | - | 4.79 | Alkyl |
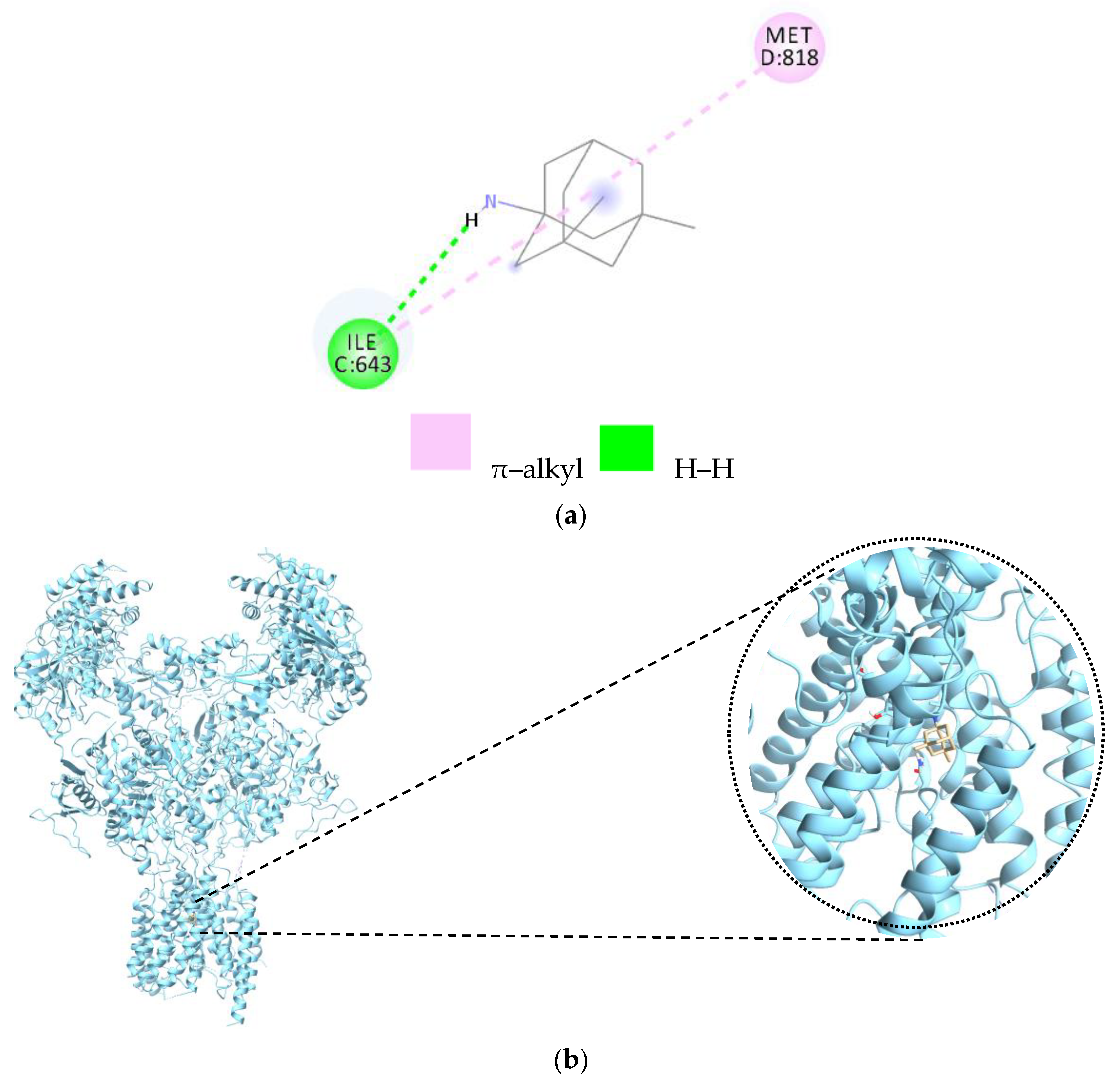
| Amino Acids | Distance Ligand–Protein (Å) | Types of Bonds |
|---|---|---|
| Ile643 | 2.86 | H |
| Met818 | 4.71 | Alkyl |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miculas, D.C.; Negru, P.A.; Bungau, S.G.; Behl, T.; Hassan, S.S.u.; Tit, D.M. Pharmacotherapy Evolution in Alzheimer’s Disease: Current Framework and Relevant Directions. Cells 2023, 12, 131. https://doi.org/10.3390/cells12010131
Miculas DC, Negru PA, Bungau SG, Behl T, Hassan SSu, Tit DM. Pharmacotherapy Evolution in Alzheimer’s Disease: Current Framework and Relevant Directions. Cells. 2023; 12(1):131. https://doi.org/10.3390/cells12010131
Chicago/Turabian StyleMiculas, Denisa Claudia, Paul Andrei Negru, Simona Gabriela Bungau, Tapan Behl, Syed Shams ul Hassan, and Delia Mirela Tit. 2023. "Pharmacotherapy Evolution in Alzheimer’s Disease: Current Framework and Relevant Directions" Cells 12, no. 1: 131. https://doi.org/10.3390/cells12010131
APA StyleMiculas, D. C., Negru, P. A., Bungau, S. G., Behl, T., Hassan, S. S. u., & Tit, D. M. (2023). Pharmacotherapy Evolution in Alzheimer’s Disease: Current Framework and Relevant Directions. Cells, 12(1), 131. https://doi.org/10.3390/cells12010131