
Impact of Mitochondrial A3243G Heteroplasmy on Mitochondrial Bioenergetics and Dynamics of Directly Reprogrammed MELAS Neurons

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Establish of Primary Skin Fibroblasts
2.2. Measurement of Mutant Heteroplasmy
2.3. Direct Reprogramming of Primary Skin Fibroblasts to iNs
2.4. Western Blotting
2.5. Detection of Mitochondrial Membrane Potential (ΔΨm)
2.6. Measurement of Mitochondrial Reactive Oxygen Species
2.7. Cellular Bioenergetics
2.8. Mitochondria Morphology and Network
2.9. Statistical Analysis
3. Results
3.1. Generation of iNs
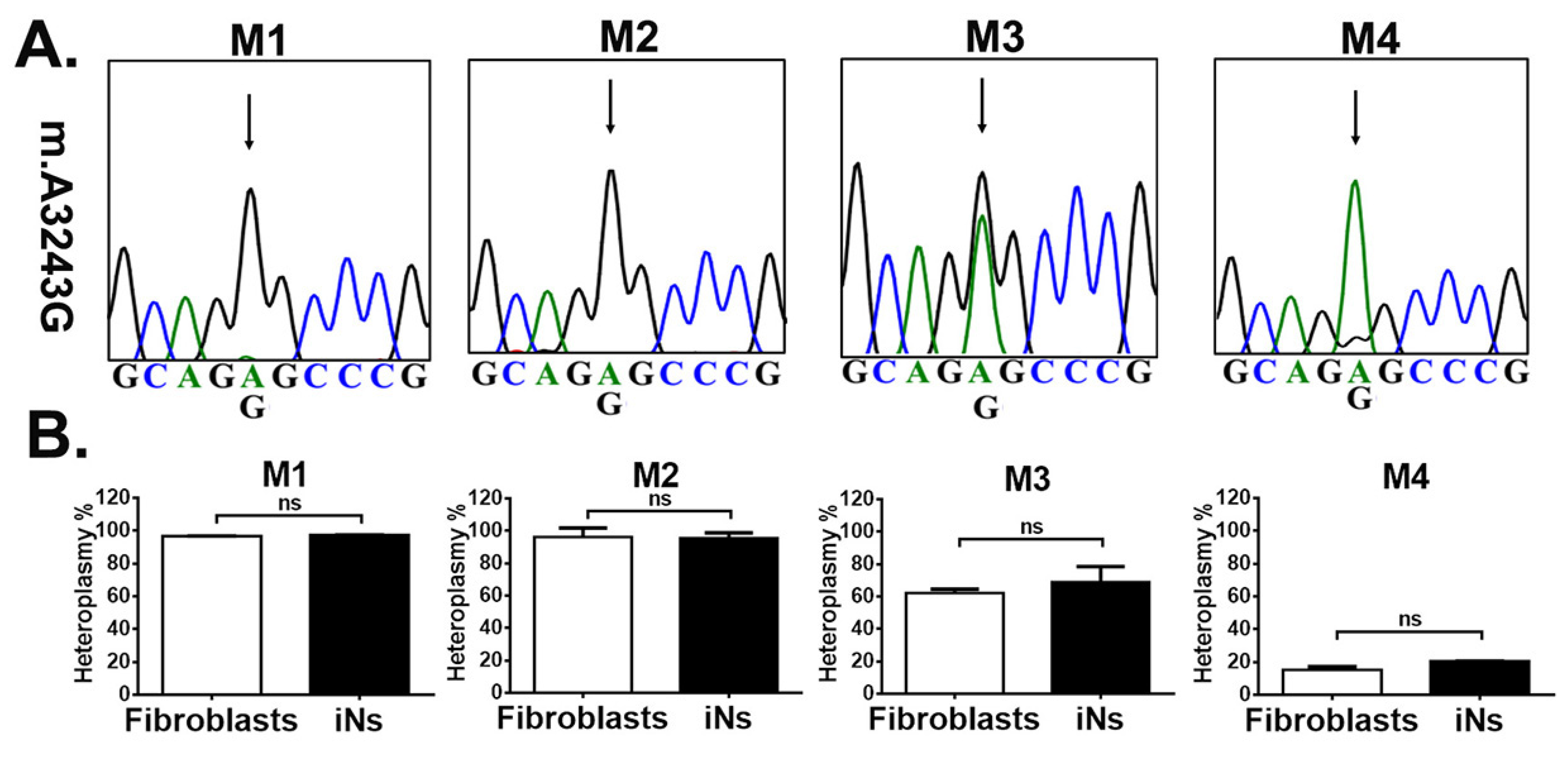
3.2. iNs Retained Stable m.A3243G Heteroplasmy
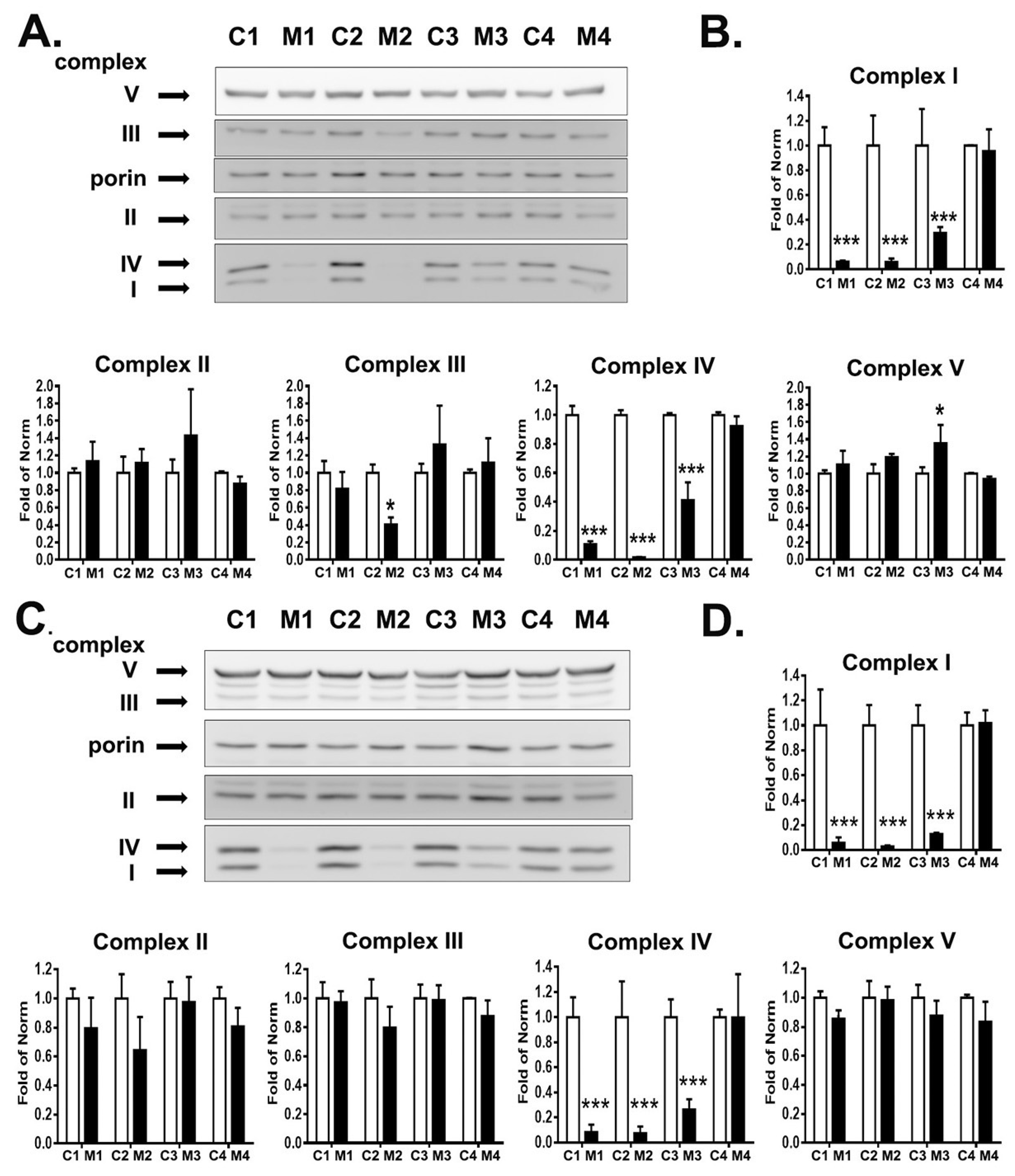
3.3. RC Complexes Deficiency in MELAS iNs
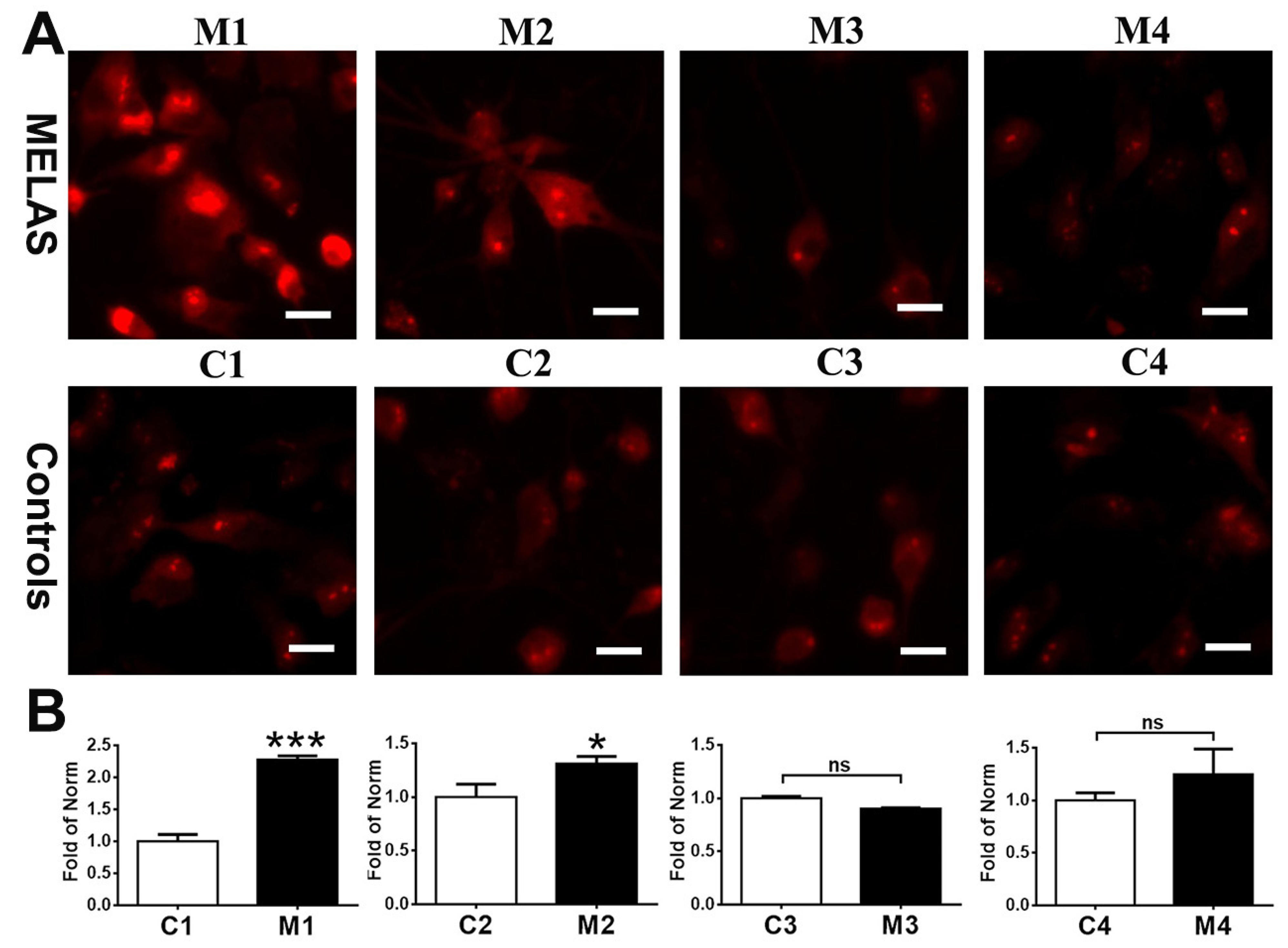
3.4. High Heteroplasmy Increased ROS Production
3.5. Reduction in ΔΨm in iNs with High Heteroplasmy
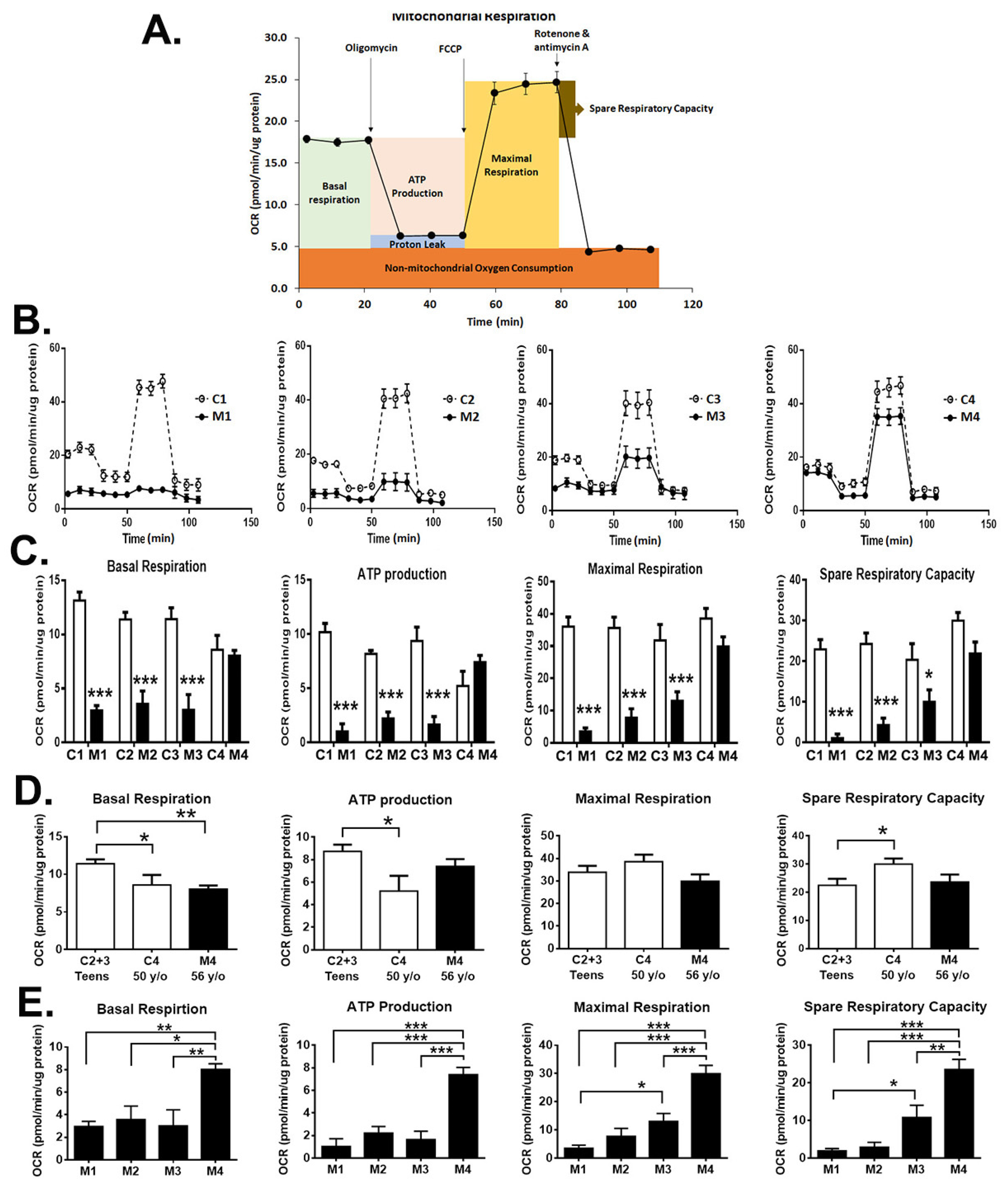
3.6. Impact of Heteroplasmy on the Bioenergetics
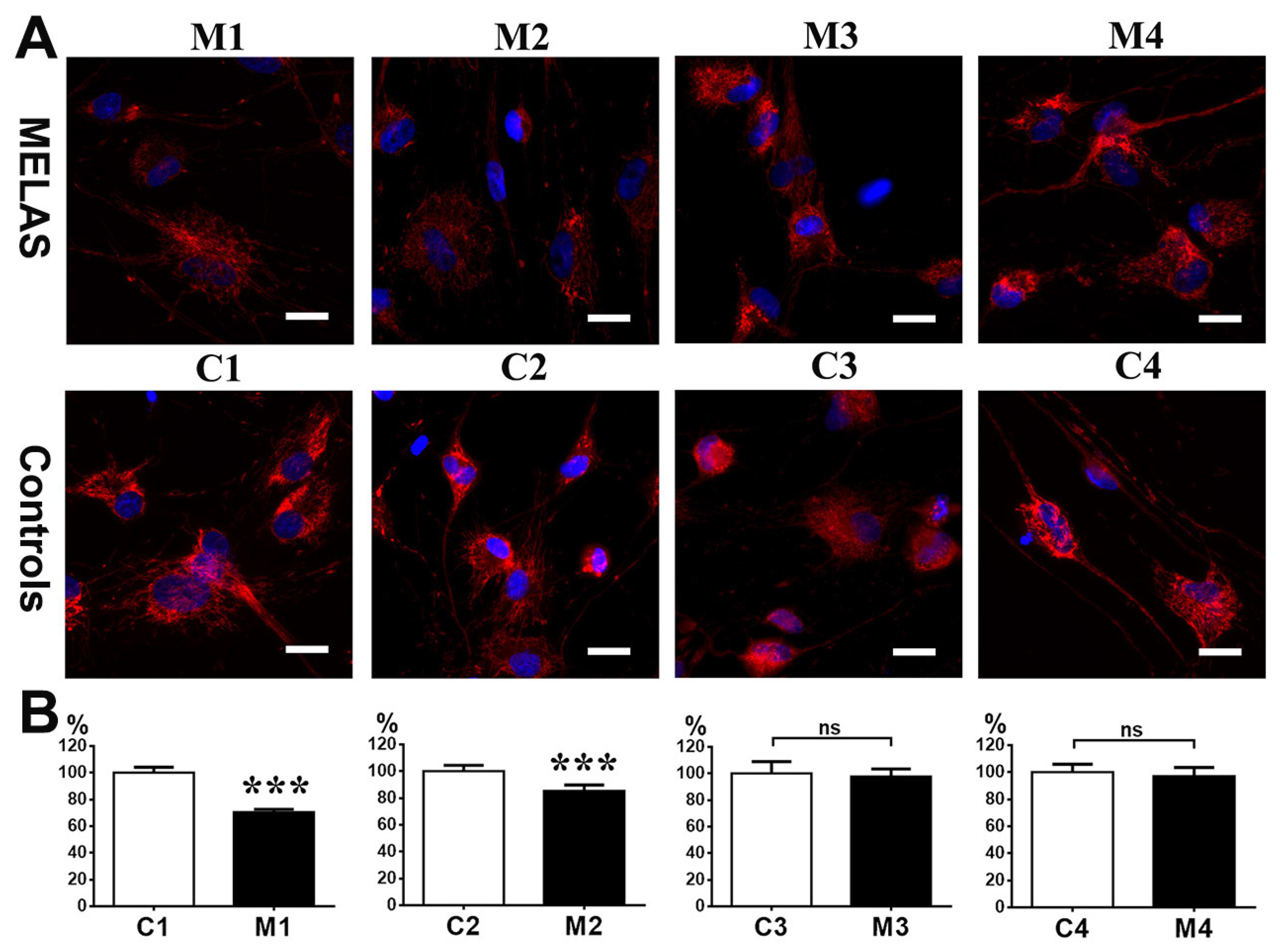
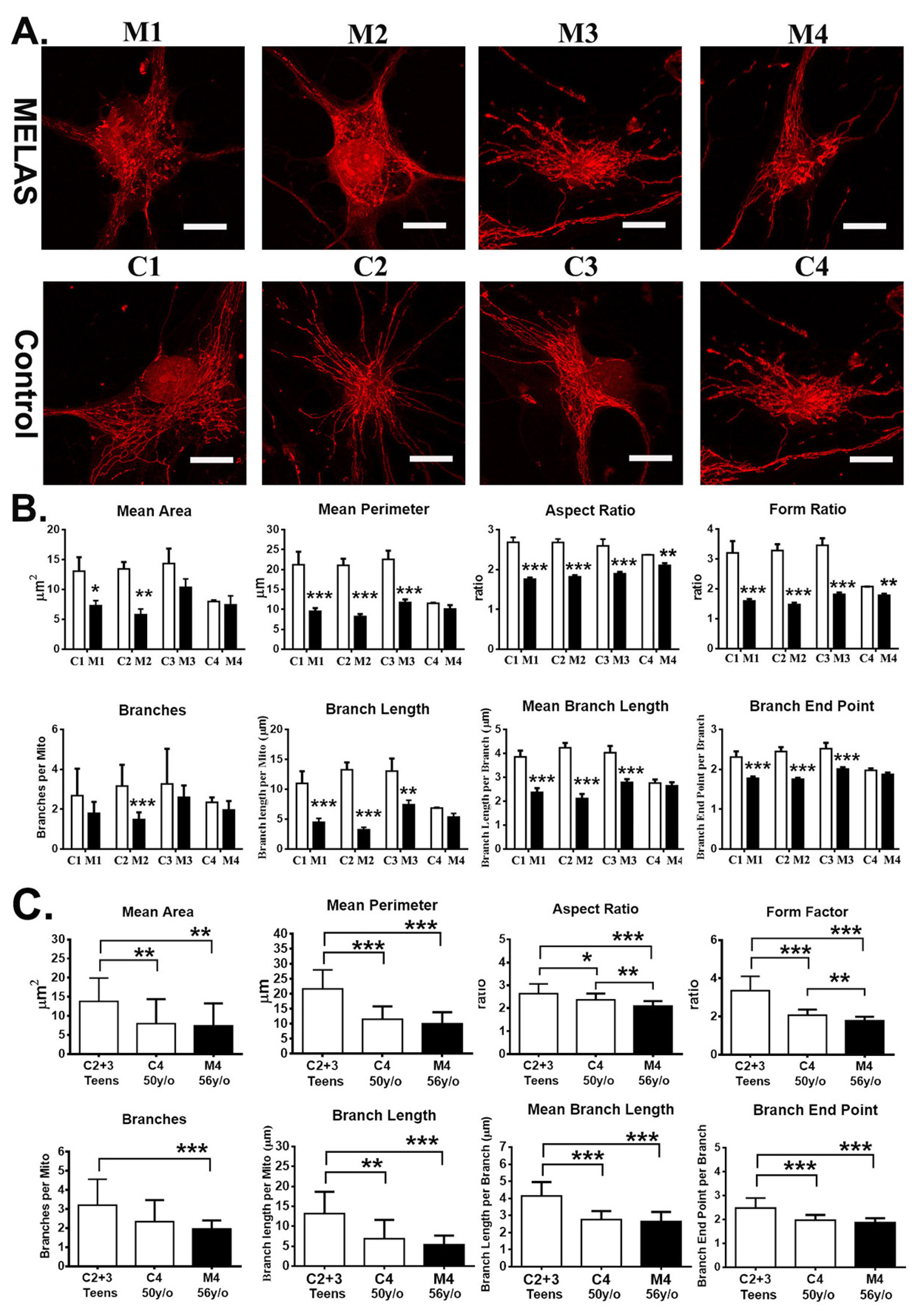
3.7. Impact of Heteroplasmy on Mitochondria Dynamics
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brandon, M.C.; Lott, M.T.; Nguyen, K.C.; Spolim, S.; Navathe, S.B.; Baldi, P.; Wallace, D.C. MITOMAP: A human mitochondrial genome database—2004 update. Nucleic Acids Res. 2005, 33, D611–D613. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Nonaka, I.; Horai, S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar] [CrossRef]
- Ruiz-Pesini, E.; Lott, M.T.; Procaccio, V.; Poole, J.C.; Brandon, M.C.; Mishmar, D.; Yi, C.; Kreuziger, J.; Baldi, P.; Wallace, D.C. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007, 35, D823–D828. [Google Scholar] [CrossRef] [PubMed]
- Sasarman, F.; Antonicka, H.; Shoubridge, E.A. The A3243G tRNALeu(UUR) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially suppressed by overexpression of EFTu and EFG2. Hum. Mol. Genet. 2008, 17, 3697–3707. [Google Scholar] [CrossRef]
- Mariotti, C.; Savarese, N.; Suomalainen, A.; Rimoldi, M.; Comi, G.; Prelle, A.; Antozzi, C.; Servidei, S.; Jarre, L.; DiDonato, S.; et al. Genotype to phenotype correlations in mitochondrial encephalomyopathies associated with the A3243G mutation of mitochondrial DNA. J. Neurol. 1995, 242, 304–312. [Google Scholar] [CrossRef]
- Chomyn, A.; Martinuzzi, A.; Yoneda, M.; Daga, A.; Hurko, O.; Johns, D.; Lai, S.T.; Nonaka, I.; Angelini, C.; Attardi, G. MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc. Natl. Acad. Sci. USA 1992, 89, 4221–4225. [Google Scholar] [CrossRef]
- Janssen, G.M.; Maassen, J.A.; van Den Ouweland, J.M. The diabetes-associated 3243 mutation in the mitochondrial tRNA(Leu(UUR)) gene causes severe mitochondrial dysfunction without a strong decrease in protein synthesis rate. J. Biol. Chem. 1999, 274, 29744–29748. [Google Scholar] [CrossRef]
- Dunbar, D.R.; Moonie, P.A.; Jacobs, H.T.; Holt, I.J. Different cellular backgrounds confer a marked advantage to either mutant or wild-type mitochondrial genomes. Proc. Natl. Acad. Sci. USA 1995, 92, 6562–6566. [Google Scholar] [CrossRef]
- van den Ouweland, J.M.; Maechler, P.; Wollheim, C.B.; Attardi, G.; Maassen, J.A. Functional and morphological abnormalities of mitochondria harbouring the tRNA(Leu)(UUR) mutation in mitochondrial DNA derived from patients with maternally inherited diabetes and deafness (MIDD) and progressive kidney disease. Diabetologia 1999, 42, 485–492. [Google Scholar] [CrossRef]
- Sandhu, J.K.; Sodja, C.; McRae, K.; Li, Y.; Rippstein, P.; Wei, Y.H.; Lach, B.; Lee, F.; Bucurescu, S.; Harper, M.E.; et al. Effects of nitric oxide donors on cybrids harbouring the mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) A3243G mitochondrial DNA mutation. Biochem. J. 2005, 391, 191–202. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Desquiret-Dumas, V.; Gueguen, N.; Barth, M.; Chevrollier, A.; Hancock, S.; Wallace, D.C.; Amati-Bonneau, P.; Henrion, D.; Bonneau, D.; Reynier, P.; et al. Metabolically induced heteroplasmy shifting and l-arginine treatment reduce the energetic defect in a neuronal-like model of MELAS. Biochim. Biophys. Acta 2012, 1822, 1019–1029. [Google Scholar] [CrossRef]
- Hamalainen, R.H.; Manninen, T.; Koivumaki, H.; Kislin, M.; Otonkoski, T.; Suomalainen, A. Tissue- and cell-type-specific manifestations of heteroplasmic mtDNA 3243A > G mutation in human induced pluripotent stem cell-derived disease model. Proc. Natl. Acad. Sci. USA 2013, 110, E3622–E3630. [Google Scholar] [CrossRef]
- Povea-Cabello, S.; Villanueva-Paz, M.; Suarez-Rivero, J.M.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Sanchez-Alcazar, J.A. Advances in mt-tRNA Mutation-Caused Mitochondrial Disease Modeling: Patients’ Brain in a Dish. Front. Genet. 2020, 11, 610764. [Google Scholar] [CrossRef] [PubMed]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Sudhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.P.; Yang, N.; Vierbuchen, T.; Ostermeier, A.; Fuentes, D.R.; Yang, T.Q.; Citri, A.; Sebastiano, V.; Marro, S.; Sudhof, T.C.; et al. Induction of human neuronal cells by defined transcription factors. Nature 2011, 476, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Ladewig, J.; Mertens, J.; Kesavan, J.; Doerr, J.; Poppe, D.; Glaue, F.; Herms, S.; Wernet, P.; Kogler, G.; Muller, F.J.; et al. Small molecules enable highly efficient neuronal conversion of human fibroblasts. Nat. Methods 2012, 9, 575–578. [Google Scholar] [CrossRef]
- Povea-Cabello, S.; Villanueva-Paz, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Alvarez-Cordoba, M.; Suarez-Rivero, J.M.; Montes, M.A.; Rodriguez-Moreno, A.; Andrade-Talavera, Y.; Armengol, J.A.; et al. Modeling Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Episodes Syndrome Using Patient-Derived Induced Neurons Generated by Direct Reprogramming. Cell. Reprogram. 2022, 24, 294–303. [Google Scholar] [CrossRef]
- Bai, R.K.; Wong, L.J. Detection and quantification of heteroplasmic mutant mitochondrial DNA by real-time amplification refractory mutation system quantitative PCR analysis: A single-step approach. Clin. Chem. 2004, 50, 996–1001. [Google Scholar] [CrossRef]
- Lim, S.M.; Choi, W.J.; Oh, K.W.; Xue, Y.; Choi, J.Y.; Kim, S.H.; Nahm, M.; Kim, Y.E.; Lee, J.; Noh, M.Y.; et al. Directly converted patient-specific induced neurons mirror the neuropathology of FUS with disrupted nuclear localization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2016, 11, 8. [Google Scholar] [CrossRef]
- Hu, W.; Qiu, B.; Guan, W.; Wang, Q.; Wang, M.; Li, W.; Gao, L.; Shen, L.; Huang, Y.; Xie, G.; et al. Direct Conversion of Normal and Alzheimer’s Disease Human Fibroblasts into Neuronal Cells by Small Molecules. Cell Stem Cell 2015, 17, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, B.A.; Ge, S.X.; Studlack, P.E.; Polster, B.M. Low micromolar concentrations of the superoxide probe MitoSOX uncouple neural mitochondria and inhibit complex IV. Free Radic. Biol. Med. 2015, 86, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, M.E.; Kauffman, M.K.; Traore, K.; Zhu, H.; Trush, M.A.; Jia, Z.; Li, Y.R. MitoSOX-Based Flow Cytometry for Detecting Mitochondrial ROS. React. Oxyg. Species Apex 2016, 2, 361–370. [Google Scholar] [CrossRef]
- Lin, D.S.; Huang, Y.W.; Ho, C.S.; Hung, P.L.; Hsu, M.H.; Wang, T.J.; Wu, T.Y.; Lee, T.H.; Huang, Z.D.; Chang, P.C.; et al. Oxidative Insults and Mitochondrial DNA Mutation Promote Enhanced Autophagy and Mitophagy Compromising Cell Viability in Pluripotent Cell Model of Mitochondrial Disease. Cells 2019, 8, 65. [Google Scholar] [CrossRef]
- Chaudhry, A.; Shi, R.; Luciani, D.S. A pipeline for multidimensional confocal analysis of mitochondrial morphology, function, and dynamics in pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E87–E101. [Google Scholar] [CrossRef]
- Yeom, K.H.; Mitchell, S.; Linares, A.J.; Zheng, S.; Lin, C.H.; Wang, X.J.; Hoffmann, A.; Black, D.L. Polypyrimidine tract-binding protein blocks miRNA-124 biogenesis to enforce its neuronal-specific expression in the mouse. Proc. Natl. Acad. Sci. USA 2018, 115, E11061–E11070. [Google Scholar] [CrossRef]
- Janssen, G.M.; Hensbergen, P.J.; van Bussel, F.J.; Balog, C.I.; Maassen, J.A.; Deelder, A.M.; Raap, A.K. The A3243G tRNALeu(UUR) mutation induces mitochondrial dysfunction and variable disease expression without dominant negative acting translational defects in complex IV subunits at UUR codons. Hum. Mol. Genet. 2007, 16, 2472–2481. [Google Scholar] [CrossRef]
- Zhu, D.; Li, X.; Tian, Y. Mitochondrial-to-nuclear communication in aging: An epigenetic perspective. Trends Biochem. Sci. 2022, 47, 645–659. [Google Scholar] [CrossRef]
- Kim, Y.; Zheng, X.; Ansari, Z.; Bunnell, M.C.; Herdy, J.R.; Traxler, L.; Lee, H.; Paquola, A.C.M.; Blithikioti, C.; Ku, M.; et al. Mitochondrial Aging Defects Emerge in Directly Reprogrammed Human Neurons due to Their Metabolic Profile. Cell Rep. 2018, 23, 2550–2558. [Google Scholar] [CrossRef]
- Lin, D.S.; Kao, S.H.; Ho, C.S.; Wei, Y.H.; Hung, P.L.; Hsu, M.H.; Wu, T.Y.; Wang, T.J.; Jian, Y.R.; Lee, T.H.; et al. Inflexibility of AMPK-mediated metabolic reprogramming in mitochondrial disease. Oncotarget 2017, 8, 73627–73639. [Google Scholar] [CrossRef] [PubMed]
- Herbers, E.; Kekalainen, N.J.; Hangas, A.; Pohjoismaki, J.L.; Goffart, S. Tissue specific differences in mitochondrial DNA maintenance and expression. Mitochondrion 2019, 44, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.F.; Rorbach, J.; Van Haute, L.; D’Souza, A.R.; Rebelo-Guiomar, P.; Powell, C.A.; Brierley, I.; Firth, A.E.; Minczuk, M. Maturation of selected human mitochondrial tRNAs requires deadenylation. Elife 2017, 6, 27596. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Cotan, D.; Cordero, M.D.; Garrido-Maraver, J.; Oropesa-Avila, M.; Rodriguez-Hernandez, A.; Gomez Izquierdo, L.; De la Mata, M.; De Miguel, M.; Lorite, J.B.; Infante, E.R.; et al. Secondary coenzyme Q10 deficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. FASEB J. 2011, 25, 2669–2687. [Google Scholar] [CrossRef]
- Chung, C.Y.; Singh, K.; Kotiadis, V.N.; Valdebenito, G.E.; Ahn, J.H.; Topley, E.; Tan, J.; Andrews, W.D.; Bilanges, B.; Pitceathly, R.D.S.; et al. Constitutive activation of the PI3K-Akt-mTORC1 pathway sustains the m.3243 A > G mtDNA mutation. Nat. Commun. 2021, 12, 6409. [Google Scholar] [CrossRef]
- Rovcanin, B.; Jancic, J.; Pajic, J.; Rovcanin, M.; Samardzic, J.; Djuric, V.; Nikolic, B.; Ivancevic, N.; Novakovic, I.; Kostic, V. Oxidative Stress Profile in Genetically Confirmed Cases of Leber’s Hereditary Optic Neuropathy. J. Mol. Neurosci. 2021, 71, 1070–1081. [Google Scholar] [CrossRef]
- Chakrabarty, S.; Govindaraj, P.; Sankaran, B.P.; Nagappa, M.; Kabekkodu, S.P.; Jayaram, P.; Mallya, S.; Deepha, S.; Ponmalar, J.N.J.; Arivinda, H.R.; et al. Contribution of nuclear and mitochondrial gene mutations in mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome. J. Neurol. 2021, 268, 2192–2207. [Google Scholar] [CrossRef]
- Distelmaier, F.; Visch, H.J.; Smeitink, J.A.; Mayatepek, E.; Koopman, W.J.; Willems, P.H. The antioxidant Trolox restores mitochondrial membrane potential and Ca2+ -stimulated ATP production in human complex I deficiency. J. Mol. Med. 2009, 87, 515–522. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, H.; Kang, X.; Liang, Y.; Lan, T.; Li, T.; Tan, T.; Peng, J.; Zhang, Q.; An, G.; et al. Targeted elimination of mutant mitochondrial DNA in MELAS-iPSCs by mitoTALENs. Protein Cell 2018, 9, 283–297. [Google Scholar] [CrossRef]
- Klein Gunnewiek, T.M.; Van Hugte, E.J.H.; Frega, M.; Guardia, G.S.; Foreman, K.; Panneman, D.; Mossink, B.; Linda, K.; Keller, J.M.; Schubert, D.; et al. m.3243A > G-Induced Mitochondrial Dysfunction Impairs Human Neuronal Development and Reduces Neuronal Network Activity and Synchronicity. Cell Rep. 2020, 31, 107538. [Google Scholar] [CrossRef] [PubMed]
- Galloway, C.A.; Lee, H.; Yoon, Y. Mitochondrial morphology-emerging role in bioenergetics. Free Radic Biol. Med. 2012, 53, 2218–2228. [Google Scholar] [CrossRef]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Tokuyama, T.; Hirai, A.; Shiiba, I.; Ito, N.; Matsuno, K.; Takeda, K.; Saito, K.; Mii, K.; Matsushita, N.; Fukuda, T.; et al. Mitochondrial Dynamics Regulation in Skin Fibroblasts from Mitochondrial Disease Patients. Biomolecules 2020, 10, 450. [Google Scholar] [CrossRef] [PubMed]
- Duvezin-Caubet, S.; Jagasia, R.; Wagener, J.; Hofmann, S.; Trifunovic, A.; Hansson, A.; Chomyn, A.; Bauer, M.F.; Attardi, G.; Larsson, N.G.; et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J. Biol. Chem. 2006, 281, 37972–37979. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Smith, H.J.; Yao, P.; Mair, W.B. Causal roles of mitochondrial dynamics in longevity and healthy aging. EMBO Rep. 2019, 20, e48395. [Google Scholar] [CrossRef]
- Koopman, W.J.; Verkaart, S.; Visch, H.J.; van Emst-de Vries, S.; Nijtmans, L.G.; Smeitink, J.A.; Willems, P.H. Human NADH:ubiquinone oxidoreductase deficiency: Radical changes in mitochondrial morphology? Am. J. Physiol. Cell. Physiol. 2007, 293, C22–C29. [Google Scholar] [CrossRef] [PubMed]
- Muliyil, S.; Narasimha, M. Mitochondrial ROS regulates cytoskeletal and mitochondrial remodeling to tune cell and tissue dynamics in a model for wound healing. Dev. Cell 2014, 28, 239–252. [Google Scholar] [CrossRef]
- Fan, X.; Hussien, R.; Brooks, G.A. H2O2-induced mitochondrial fragmentation in C2C12 myocytes. Free Radic. Biol. Med. 2010, 49, 1646–1654. [Google Scholar] [CrossRef]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef]
- Chang, J.C.; Hoel, F.; Liu, K.H.; Wei, Y.H.; Cheng, F.C.; Kuo, S.J.; Tronstad, K.J.; Liu, C.S. Peptide-mediated delivery of donor mitochondria improves mitochondrial function and cell viability in human cybrid cells with the MELAS A3243G mutation. Sci. Rep. 2017, 7, 10710. [Google Scholar] [CrossRef] [PubMed]
- Vergani, L.; Malena, A.; Sabatelli, P.; Loro, E.; Cavallini, L.; Magalhaes, P.; Valente, L.; Bragantini, F.; Carrara, F.; Leger, B.; et al. Cultured muscle cells display defects of mitochondrial myopathy ameliorated by anti-oxidants. Brain 2007, 130, 2715–2724. [Google Scholar] [CrossRef] [PubMed][Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, D.-S.; Huang, Y.-W.; Ho, C.-S.; Huang, T.-S.; Lee, T.-H.; Wu, T.-Y.; Huang, Z.-D.; Wang, T.-J. Impact of Mitochondrial A3243G Heteroplasmy on Mitochondrial Bioenergetics and Dynamics of Directly Reprogrammed MELAS Neurons. Cells 2023, 12, 15. https://doi.org/10.3390/cells12010015
Lin D-S, Huang Y-W, Ho C-S, Huang T-S, Lee T-H, Wu T-Y, Huang Z-D, Wang T-J. Impact of Mitochondrial A3243G Heteroplasmy on Mitochondrial Bioenergetics and Dynamics of Directly Reprogrammed MELAS Neurons. Cells. 2023; 12(1):15. https://doi.org/10.3390/cells12010015
Chicago/Turabian StyleLin, Dar-Shong, Yu-Wen Huang, Che-Sheng Ho, Tung-Sun Huang, Tsung-Han Lee, Tsu-Yen Wu, Zon-Darr Huang, and Tuan-Jen Wang. 2023. "Impact of Mitochondrial A3243G Heteroplasmy on Mitochondrial Bioenergetics and Dynamics of Directly Reprogrammed MELAS Neurons" Cells 12, no. 1: 15. https://doi.org/10.3390/cells12010015
APA StyleLin, D.-S., Huang, Y.-W., Ho, C.-S., Huang, T.-S., Lee, T.-H., Wu, T.-Y., Huang, Z.-D., & Wang, T.-J. (2023). Impact of Mitochondrial A3243G Heteroplasmy on Mitochondrial Bioenergetics and Dynamics of Directly Reprogrammed MELAS Neurons. Cells, 12(1), 15. https://doi.org/10.3390/cells12010015