
Identification of Transcription Factor Networks during Mouse Hindlimb Development
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Weighted Gene Co-Expression Network Analysis
2.3. Identification of Significant Modules and Hub Genes
2.4. Construction of Transcription Factor Network
2.5. Validation in Mice Limb Tissue and ATDC5 Cells
3. Results
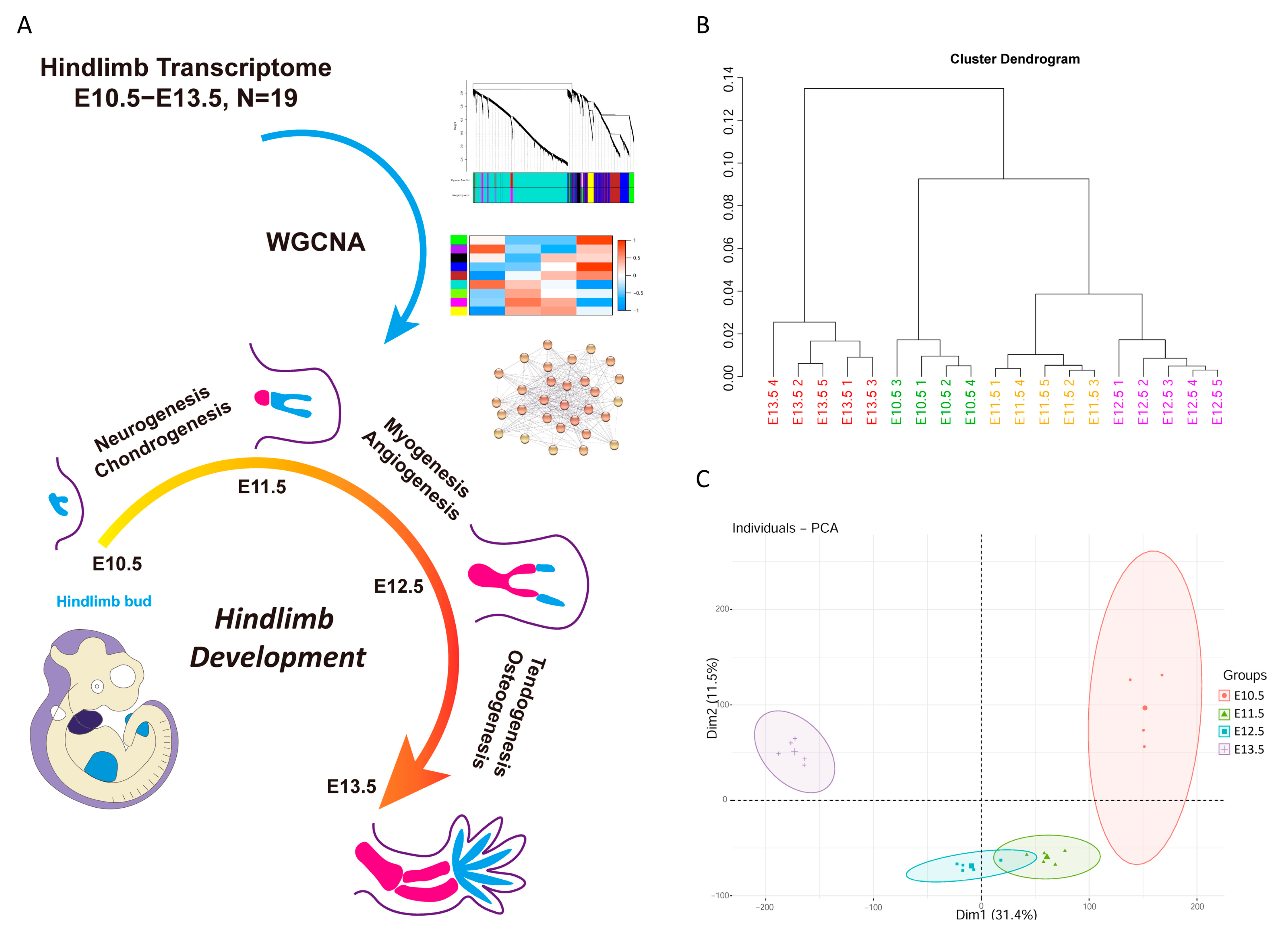
3.1. Quality Control of Embryonic Mice Hindlimb Transcriptome
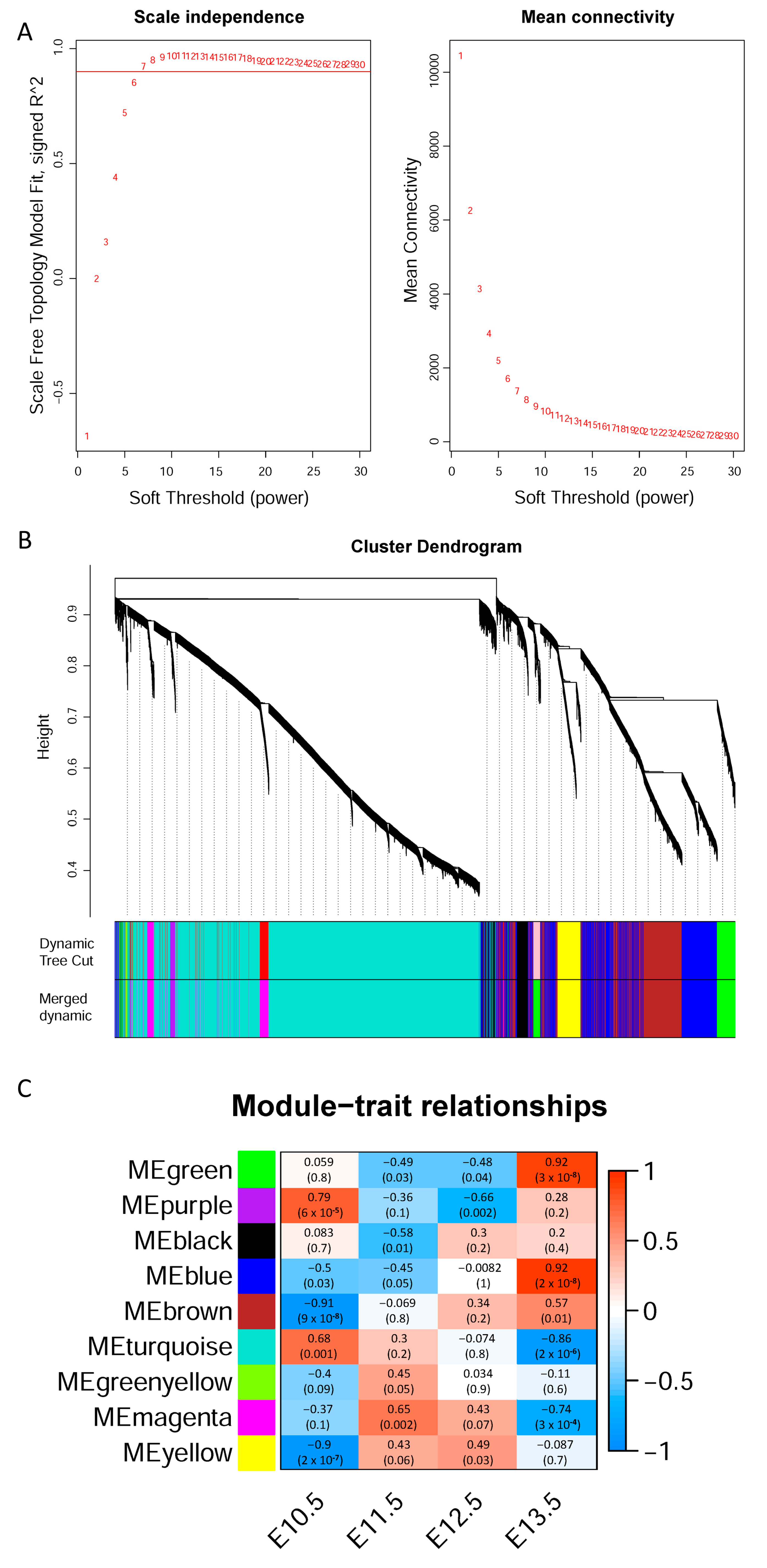
3.2. Weighted Gene Co-Expression Network Analysis
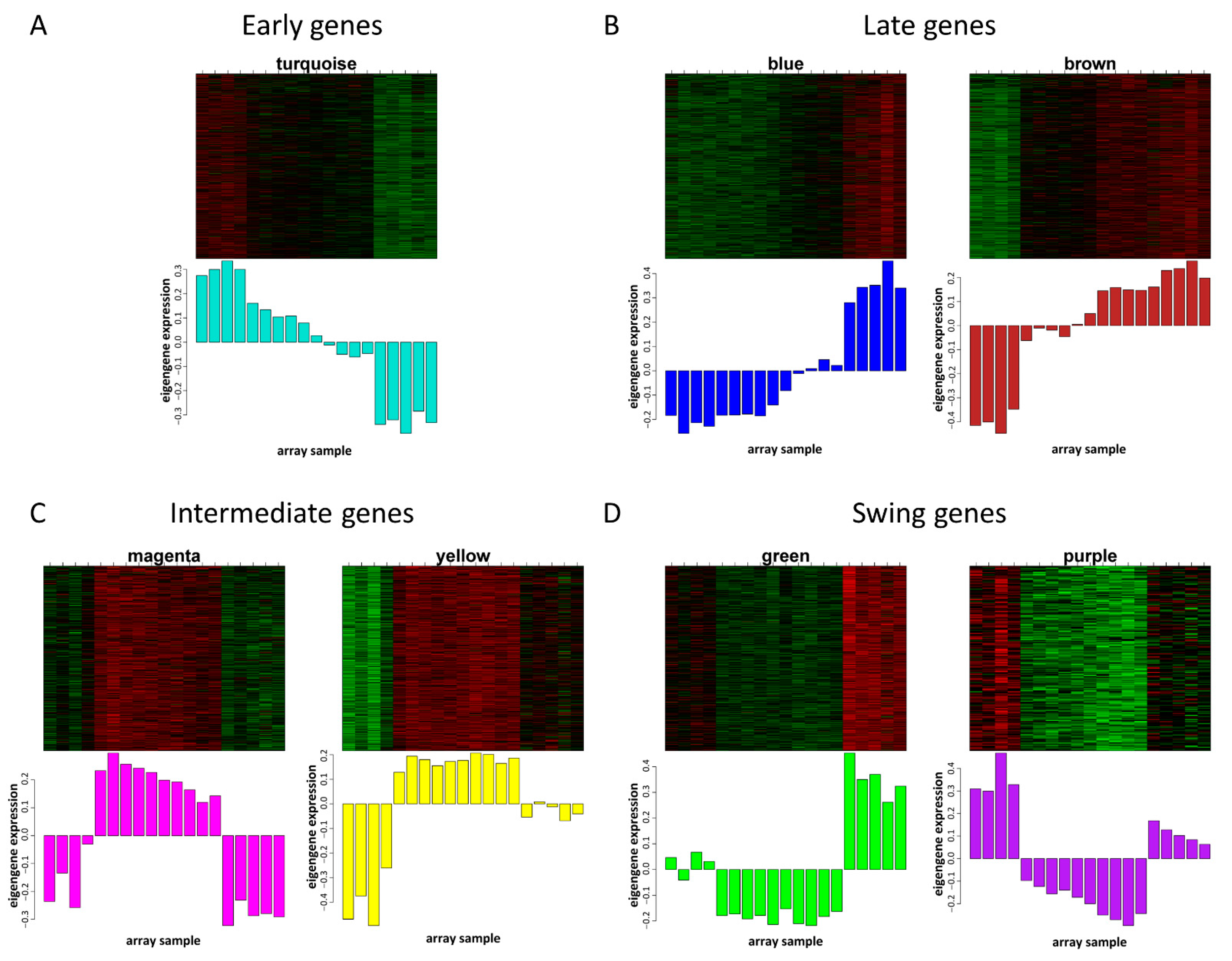
3.3. Gene Expression Patterns in Different Modules
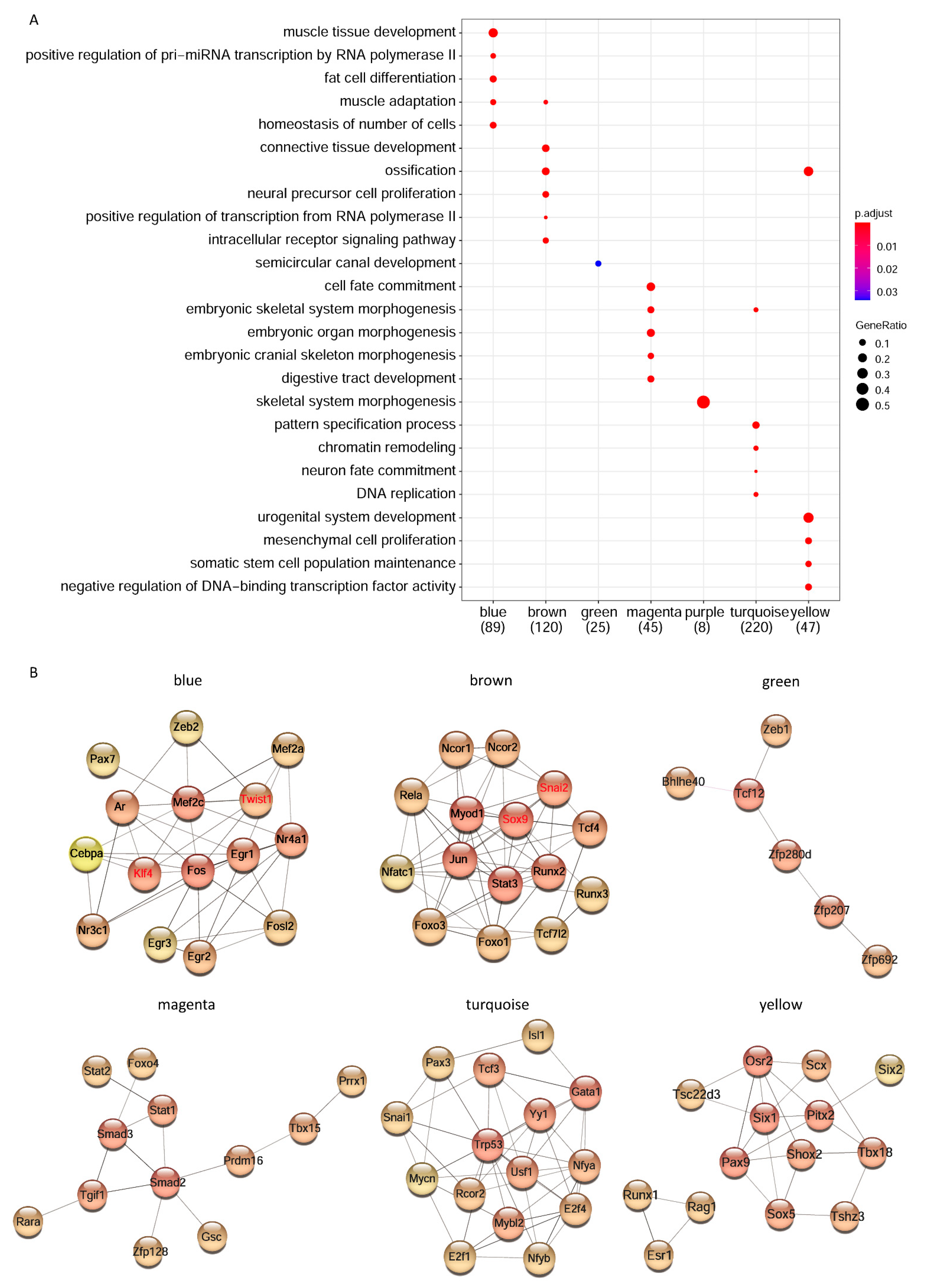
3.4. Enrichment of Transcription Factor and Network Construction
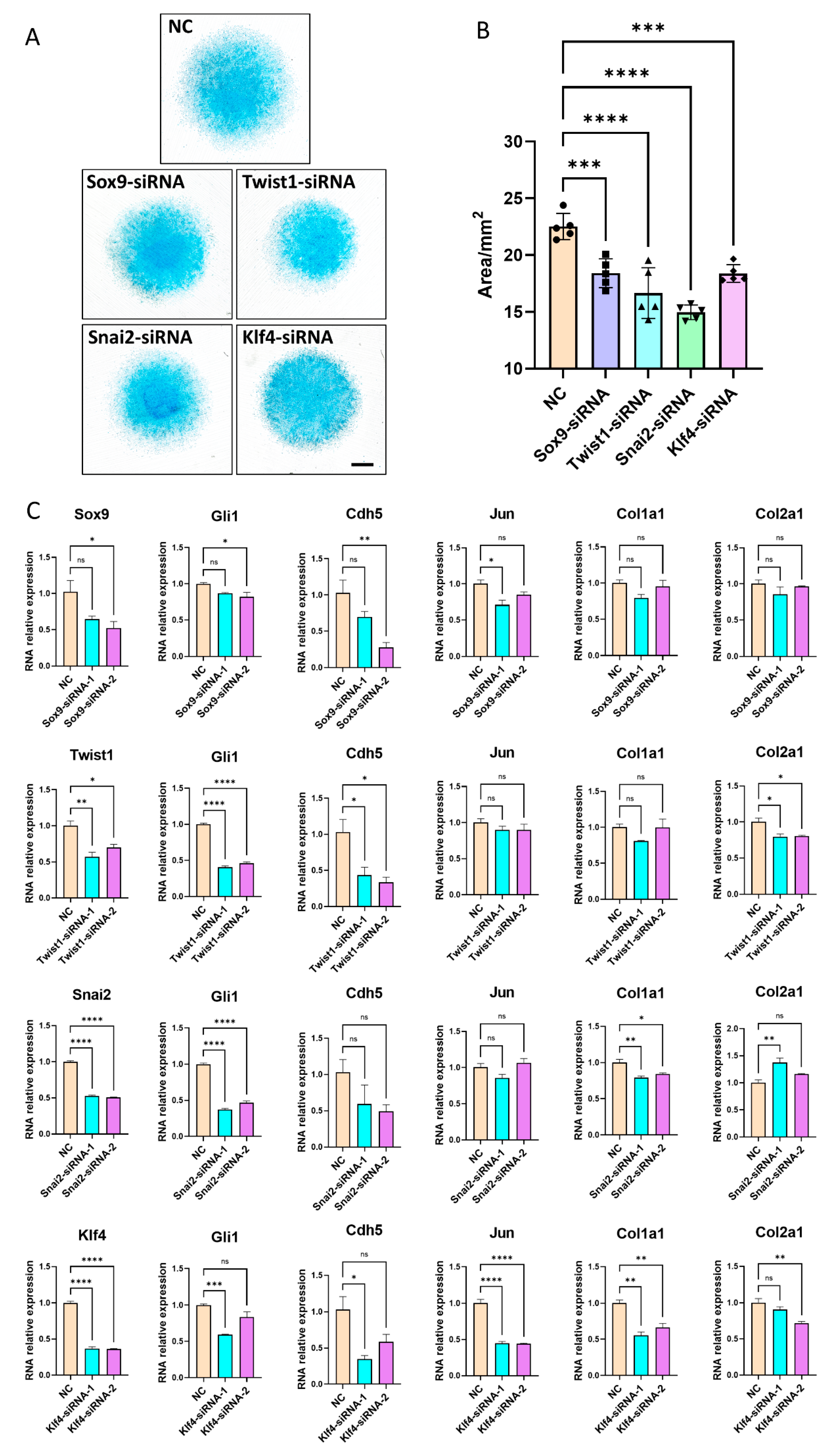
3.5. Inhibition of Key Transcription Factors Lead to Disruption of Limb Development Signals
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petit, F.; Sears, K.E.; Ahituv, N. Limb development: A paradigm of gene regulation. Nat. Rev. Genet. 2017, 18, 245–258. [Google Scholar] [CrossRef]
- Duboc, V.; Logan, M.P. Regulation of limb bud initiation and limb-type morphology. Dev. Dyn. 2011, 240, 1017–1027. [Google Scholar] [CrossRef]
- Sermeus, Y.; Vangheel, J.; Geris, L.; Smeets, B.; Tylzanowski, P. Mechanical Regulation of Limb Bud Formation. Cells 2022, 11, 420. [Google Scholar] [CrossRef]
- Delgado, I.; Torres, M. Coordination of limb development by crosstalk among axial patterning pathways. Dev. Biol. 2017, 429, 382–386. [Google Scholar] [CrossRef]
- Sheth, R.; Grégoire, D.; Dumouchel, A.; Scotti, M.; Pham, J.M.T.; Nemec, S.; Bastida, M.F.; Ros, M.A.; Kmita, M. Decoupling the function of Hox and Shh in developing limb reveals multiple inputs of Hox genes on limb growth. Development 2013, 140, 2130–2138. [Google Scholar] [CrossRef] [Green Version]
- Beederman, M.; Lamplot, J.D.; Nan, G.; Wang, J.; Liu, X.; Yin, L.; Li, R.; Shui, W.; Zhang, H.; Kim, S.H. BMP signaling in mesenchymal stem cell differentiation and bone formation. J. Biomed. Sci. Eng. 2013, 6, 32. [Google Scholar] [CrossRef] [Green Version]
- Zakany, J.; Duboule, D. The role of Hox genes during vertebrate limb development. Curr. Opin. Genet. Dev. 2007, 17, 359–366. [Google Scholar] [CrossRef]
- Chiang, C.; Litingtung, Y.; Harris, M.P.; Simandl, B.K.; Li, Y.; Beachy, P.A.; Fallon, J.F. Manifestation of the limb prepattern: Limb development in the absence of sonic hedgehog function. Dev. Biol. 2001, 236, 421–435. [Google Scholar] [CrossRef] [Green Version]
- Geetha-Loganathan, P.; Nimmagadda, S.; Scaal, M. Wnt signaling in limb organogenesis. Organogenesis 2008, 4, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Parker, S.E.; Mai, C.T.; Canfield, M.A.; Rickard, R.; Wang, Y.; Meyer, R.E.; Anderson, P.; Mason, C.A.; Collins, J.S.; Kirby, R.S. Updated national birth prevalence estimates for selected birth defects in the United States, 2004–2006. Birth Defects Res. Part A Clin. Mol. Teratol. 2010, 88, 1008–1016. [Google Scholar] [CrossRef]
- Gurrieri, F.; Kjaer, K.W.; Sangiorgi, E.; Neri, G. Limb anomalies: Developmental and evolutionary aspects. Am. J. Med. Genet. 2002, 115, 231–244. [Google Scholar] [CrossRef]
- Taher, L.; Collette, N.M.; Murugesh, D.; Maxwell, E.; Ovcharenko, I.; Loots, G.G. Global gene expression analysis of murine limb development. PLoS ONE 2011, 6, e28358. [Google Scholar] [CrossRef] [Green Version]
- Havis, E.; Bonnin, M.-A.; Olivera-Martinez, I.; Nazaret, N.; Ruggiu, M.; Weibel, J.; Durand, C.; Guerquin, M.-J.; Bonod-Bidaud, C.; Ruggiero, F.; et al. Transcriptomic analysis of mouse limb tendon cells during development. Development 2014, 141, 3683–3696. [Google Scholar] [CrossRef] [Green Version]
- Desanlis, I.; Paul, R.; Kmita, M. Transcriptional Trajectories in Mouse Limb Buds Reveal the Transition from Anterior-Posterior to Proximal-Distal Patterning at Early Limb Bud Stage. J. Dev. Biol. 2020, 8, 31. [Google Scholar] [CrossRef]
- Liu, H.; Xu, J.; Liu, C.-F.; Lan, Y.; Wylie, C.; Jiang, R. Whole transcriptome expression profiling of mouse limb tendon development by using RNA-seq. J. Orthop. Res. 2015, 33, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Henry, V.J.; Bandrowski, A.E.; Pepin, A.-S.; Gonzalez, B.J.; Desfeux, A. OMICtools: An informative directory for multi-omic data analysis. Database 2014, 2014, bau069. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Duboc, V.; Logan, M.P. Pitx1 is necessary for normal initiation of hindlimb outgrowth through regulation of Tbx4 expression and shapes hindlimb morphologies via targeted growth control. Development 2011, 138, 5301–5309. [Google Scholar] [CrossRef] [Green Version]
- DeLaurier, A.; Schweitzer, R.; Logan, M. Pitx1 determines the morphology of muscle, tendon, and bones of the hindlimb. Dev. Biol. 2006, 299, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Miao, Y.-R.; Jia, L.-H.; Yu, Q.-Y.; Zhang, Q.; Guo, A.-Y. AnimalTFDB 3.0: A comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2019, 47, D33–D38. [Google Scholar] [CrossRef]
- Watanabe, K.; Stringer, S.; Frei, O.; Umićević Mirkov, M.; de Leeuw, C.; Polderman, T.J.; van der Sluis, S.; Andreassen, O.A.; Neale, B.M.; Posthuma, D. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 2019, 51, 1339–1348. [Google Scholar] [CrossRef]
- DeLise, A.M.; Stringa, E.; Woodward, W.A.; Mello, M.A.; Tuan, R.S. Embryonic limb mesenchyme micromass culture as an in vitro model for chondrogenesis and cartilage maturation. Methods Mol. Biol. 2000, 137, 359–375. [Google Scholar] [CrossRef]
- Askary, A.; Xu, P.; Barske, L.; Bay, M.; Bump, P.; Balczerski, B.; Bonaguidi, M.A.; Crump, J.G. Genome-wide analysis of facial skeletal regionalization in zebrafish. Development 2017, 144, 2994–3005. [Google Scholar] [CrossRef] [Green Version]
- Feregrino, C.; Tschopp, P. Assessing evolutionary and developmental transcriptome dynamics in homologous cell types. Dev. Dyn. 2021, 251, 1472–1489. [Google Scholar] [CrossRef]
- Osipovich, A.B.; Dudek, K.D.; Greenfest-Allen, E.; Cartailler, J.-P.; Manduchi, E.; Potter Case, L.; Choi, E.; Chapman, A.G.; Clayton, H.W.; Gu, G. A developmental lineage-based gene co-expression network for mouse pancreatic β-cells reveals a role for Zfp800 in pancreas development. Development 2021, 148, dev196964. [Google Scholar] [CrossRef]
- Onimaru, K.; Tatsumi, K.; Tanegashima, C.; Kadota, M.; Nishimura, O.; Kuraku, S. Developmental hourglass and heterochronic shifts in fin and limb development. eLife 2021, 10, e62865. [Google Scholar] [CrossRef]
- Rabinowitz, A.H.; Vokes, S.A. Integration of the transcriptional networks regulating limb morphogenesis. Dev. Biol. 2012, 368, 165–180. [Google Scholar] [CrossRef] [Green Version]
- Sheeba, C.; Logan, M. The roles of T-box genes in vertebrate limb development. Curr. Top. Dev. Biol. 2017, 122, 355–381. [Google Scholar]
- Almalki, S.G.; Agrawal, D.K. Key transcription factors in the differentiation of mesenchymal stem cells. Differentiation 2016, 92, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Buckingham, M.; Relaix, F. The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 645–673. [Google Scholar] [CrossRef]
- Hirsch, N.; Eshel, R.; Bar Yaacov, R.; Shahar, T.; Shmulevich, F.; Dahan, I.; Levaot, N.; Kaplan, T.; Lupiáñez, D.G.; Birnbaum, R.Y. Unraveling the transcriptional regulation of TWIST1 in limb development. PLoS Genet. 2018, 14, e1007738. [Google Scholar] [CrossRef]
- Komori, T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res. 2010, 339, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Yap, S.P.; Xing, X.; Kraus, P.; Sivakamasundari, V.; Chan, H.Y.; Lufkin, T. Generation of mice with a novel conditional null allele of the Sox9 gene. Biotechnol. Lett. 2011, 33, 1551–1558. [Google Scholar] [CrossRef]
- Takagi, T.; Moribe, H.; Kondoh, H.; Higashi, Y. DeltaEF1, a zinc finger and homeodomain transcription factor, is required for skeleton patterning in multiple lineages. Development 1998, 125, 21–31. [Google Scholar] [CrossRef]
- Wang, W.; Song, B.; Anbarchian, T.; Shirazyan, A.; Sadik, J.E.; Lyons, K.M. Smad2 and Smad3 regulate chondrocyte proliferation and differentiation in the growth plate. PLoS Genet. 2016, 12, e1006352. [Google Scholar] [CrossRef] [Green Version]
- Ota, S.; Zhou, Z.-Q.; Keene, D.R.; Knoepfler, P.; Hurlin, P.J. Activities of N-Myc in the developing limb link control of skeletal size with digit separation. Development 2007, 134, 1583–1592. [Google Scholar] [CrossRef] [Green Version]
- Bobick, B.E.; Cobb, J. Shox2 regulates progression through chondrogenesis in the mouse proximal limb. J. Cell Sci. 2012, 125, 6071–6083. [Google Scholar] [CrossRef] [Green Version]
- Ye, W.; Song, Y.; Huang, Z.; Osterwalder, M.; Ljubojevic, A.; Xu, J.; Bobick, B.; Abassah-Oppong, S.; Ruan, N.; Shamby, R. A unique stylopod patterning mechanism by Shox2-controlled osteogenesis. Development 2016, 143, 2548–2560. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, Y.; Rodriguez-León, J.; Belmonte, J.C.I. The role of TGFβs and Sox9 during limb chondrogenesis. Curr. Opin. Cell Biol. 2006, 18, 723–729. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Jiang, R.; Tan, G.; Liu, Z.; Tang, C.; Wu, R.; Shi, D. Identification of Transcription Factor Networks during Mouse Hindlimb Development. Cells 2023, 12, 28. https://doi.org/10.3390/cells12010028
Wang M, Jiang R, Tan G, Liu Z, Tang C, Wu R, Shi D. Identification of Transcription Factor Networks during Mouse Hindlimb Development. Cells. 2023; 12(1):28. https://doi.org/10.3390/cells12010028
Chicago/Turabian StyleWang, Maochun, Ruiyang Jiang, Guihua Tan, Zizheng Liu, Chen Tang, Rui Wu, and Dongquan Shi. 2023. "Identification of Transcription Factor Networks during Mouse Hindlimb Development" Cells 12, no. 1: 28. https://doi.org/10.3390/cells12010028