
Study of the Regulatory Mechanism of miR-26a-5p in Allergic Asthma
Abstract
:1. Introduction
2. Materials and Methods
2.1. Main Materials and Reagents
2.2. Construction of an Allergic Asthma Model in Mice
2.3. qRT-PCR
2.4. Screening for Target Genes
2.5. Western Blot
2.6. Dual Luciferase Reporter Gene Test
2.7. Infection and Identification of miR-26a-5p Lentivirus
2.8. HE Staining
2.9. Masson Staining
2.10. TUNEL Staining
2.11. ELISA
2.12. Immunohistochemistry
2.13. Construction and Characterization of a Cellular Allergic Asthma Model
2.14. CCK8
2.15. EdU Staining
2.16. Flow Cytometry
2.17. Flow Antigen Testing
2.18. Statistical Data Analysis
3. Results
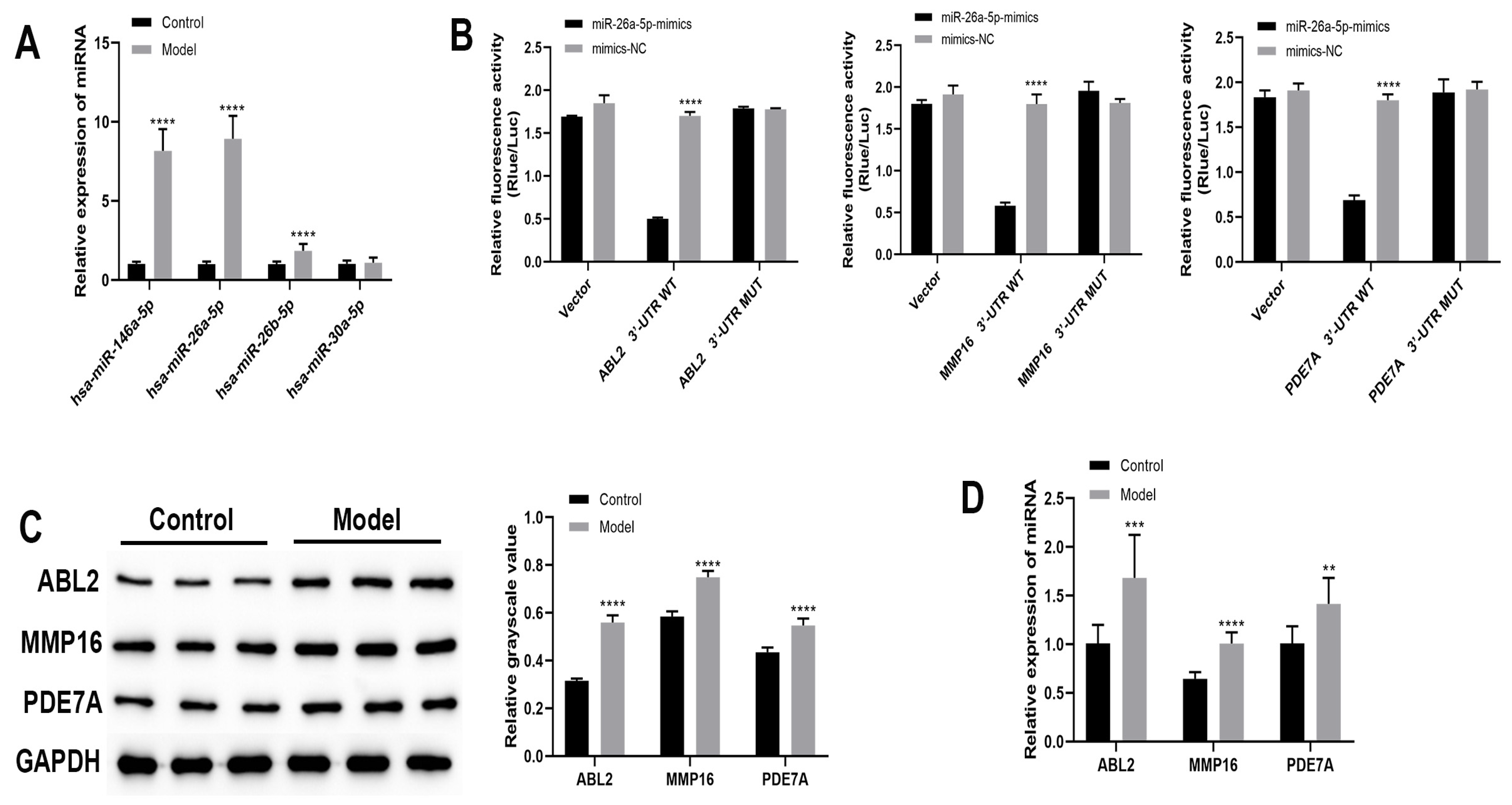
3.1. Bioinformatic Analysis of mRNAs Interacting with miRNAs
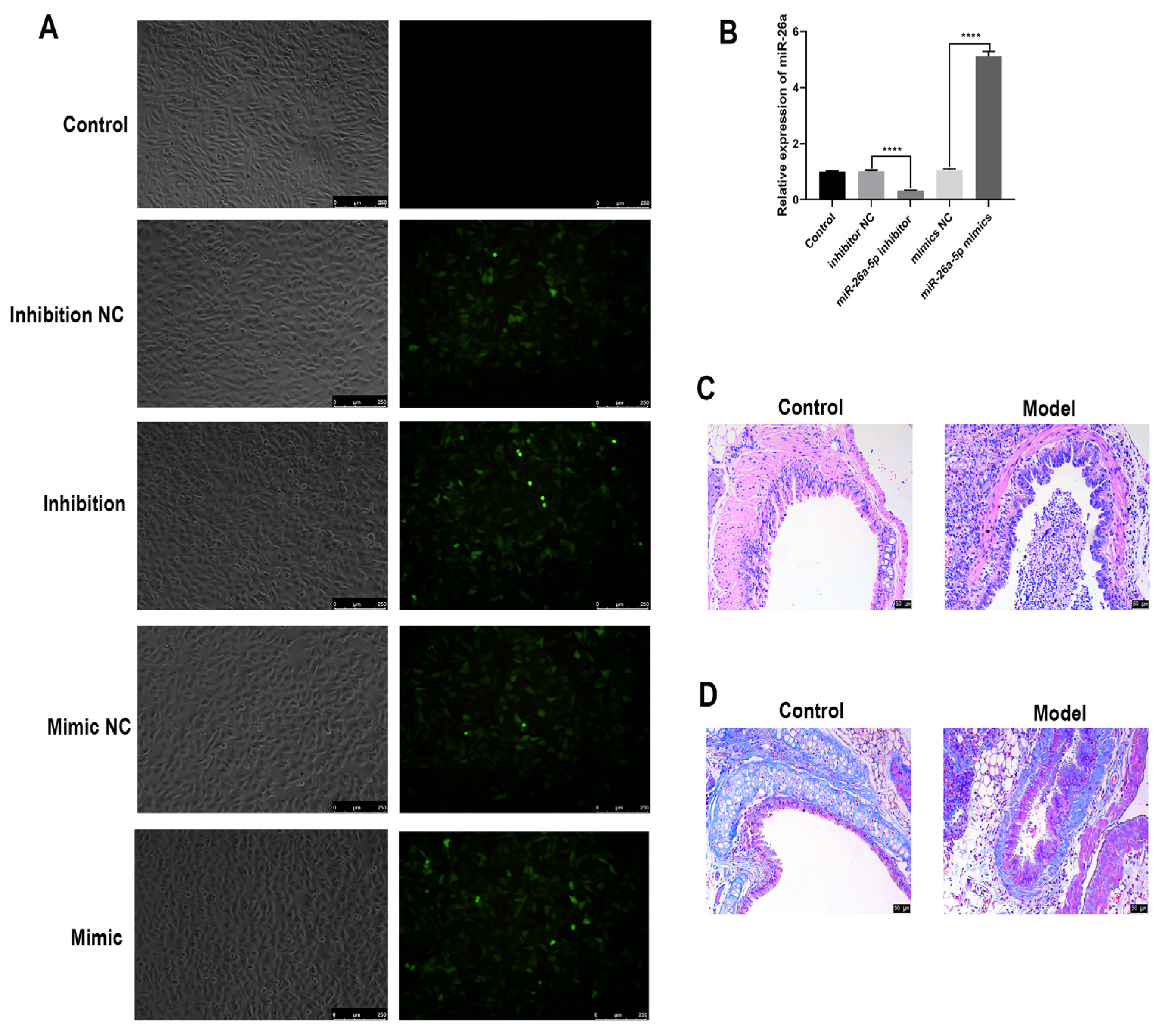
3.2. Identification of Allergic Asthma Models in Mice
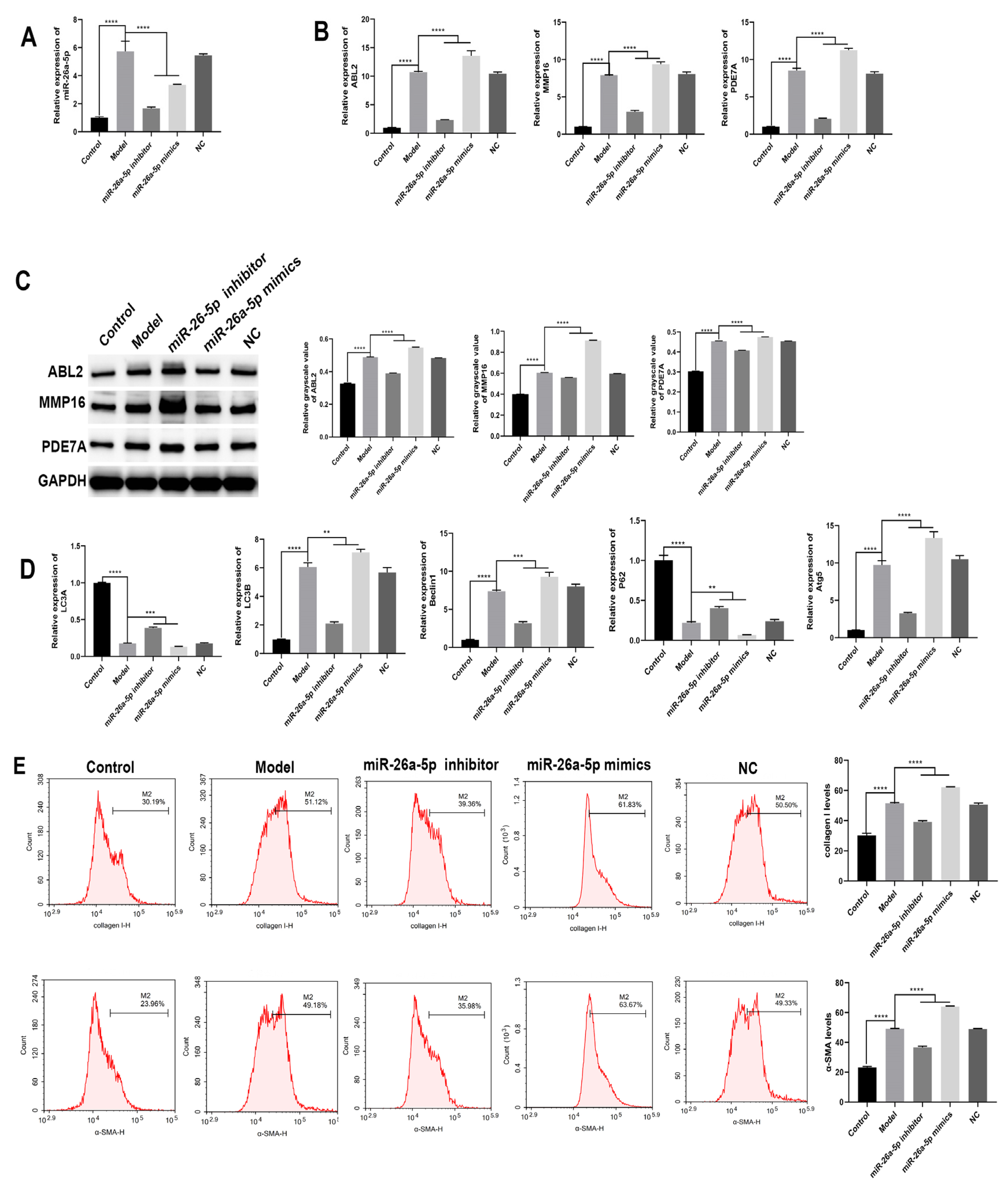
3.3. The Regulatory Role of miR-26a-5p in a Mouse Model of Allergic Asthma
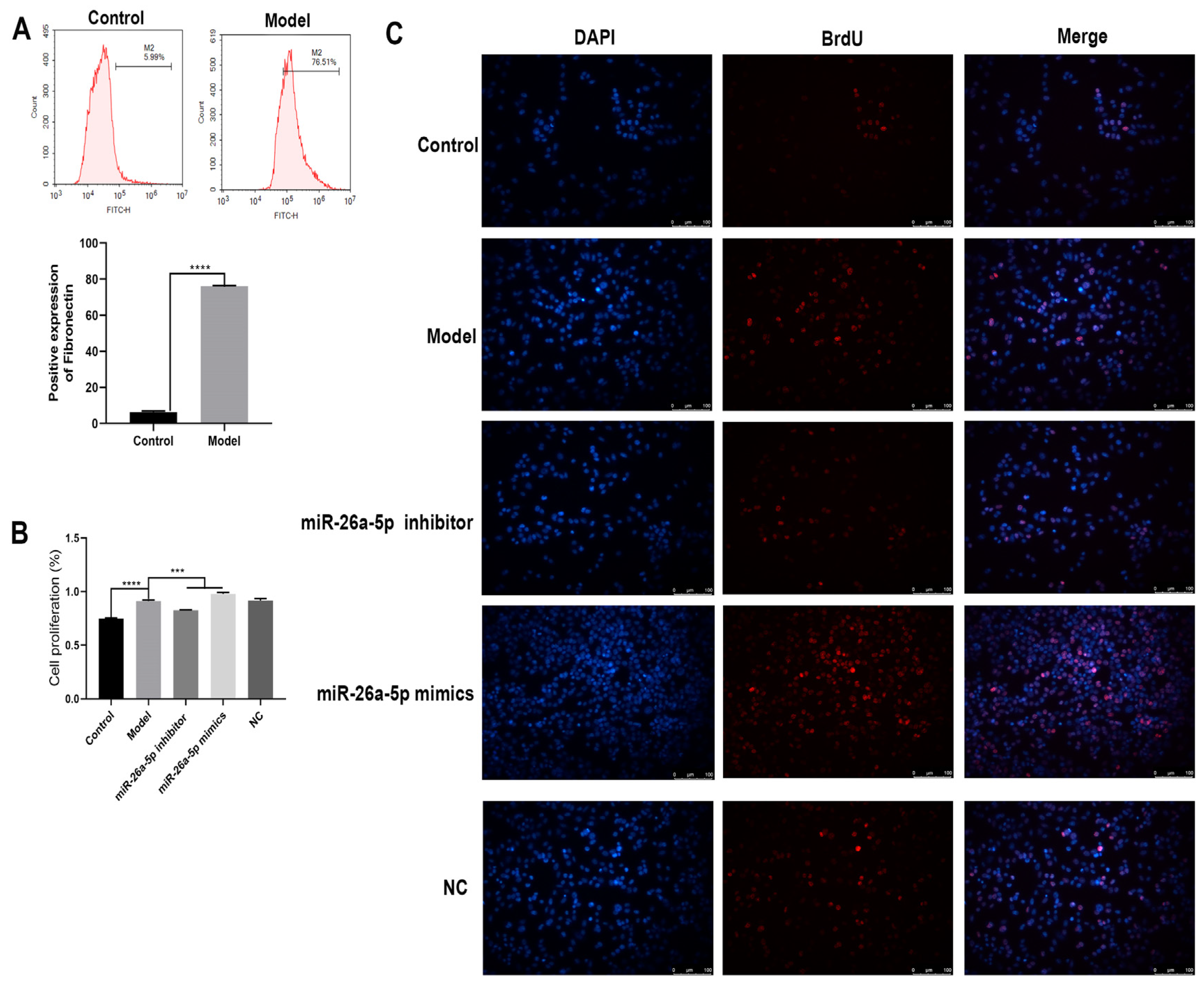
3.4. Construction and Characterization of a Cellular Allergic Asthma Model
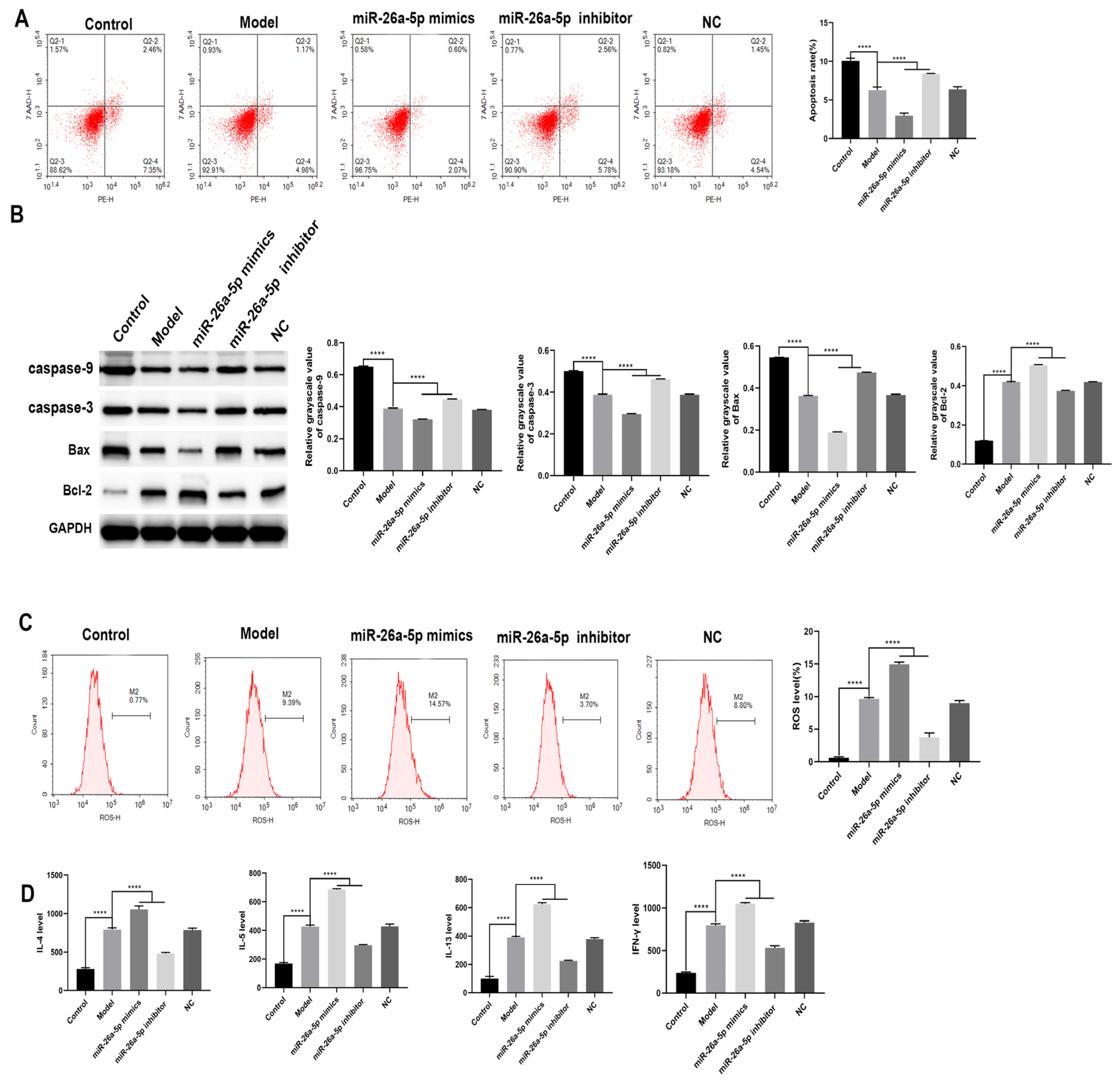
3.5. Effect of miR-26a-5p on Apoptosis and Inflammation in a Model of Allergic Asthma
3.6. Effect of miR-26a-5p on Cellular Autophagy in a Model of Allergic Asthma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schoettler, N.; Strek, M.E. Recent Advances in Severe Asthma: From Phenotypes to Personalized Medicine. Chest 2020, 157, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbach, H.; Wagner, C.; Wegmann, M. Airway remodeling in asthma: What really matters. Cell Tissue Res. 2017, 367, 551–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camoretti-Mercado, B.; Lockey, R.F. Airway smooth muscle pathophysiology in asthma. J. Allergy Clin. Immunol. 2021, 147, 1983–1995. [Google Scholar] [CrossRef] [PubMed]
- Kota, A.; Deshpande, D.A.; Haghi, M.; Oliver, B.; Sharma, P. Autophagy and airway fibrosis: Is there a link? F1000Research 2017, 6, 409. [Google Scholar] [CrossRef] [PubMed]
- Ramelli, S.C.; Gerthoffer, W.T. MicroRNA Targets for Asthma Therapy. Adv. Exp. Med. Biol. 2021, 1303, 89–105. [Google Scholar] [CrossRef] [PubMed]
- James, B.N.; Oyeniran, C.; Sturgill, J.L.; Newton, J.; Martin, R.K.; Bieberich, E.; Weigel, C.; Maczis, M.A.; Palladino, E.N.D.; Lownik, J.C.; et al. Ceramide in apoptosis and oxidative stress in allergic inflammation and asthma. J. Allergy Clin. Immunol. 2021, 147, 1936–1948. [Google Scholar] [CrossRef]
- White, S.R. Apoptosis and the airway epithelium. J. Allergy 2011, 2011, 948406. [Google Scholar] [CrossRef] [Green Version]
- Juncadella, I.J.; Kadl, A.; Sharma, A.K.; Shim, Y.M.; Hochreiter-Hufford, A.; Borish, L.; Ravichandran, K.S. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature 2013, 493, 547–551. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Li, Y.; Zhong, W.; Gao, P.; Hu, C. Recent developments in the role of reactive oxygen species in allergic asthma. J. Thorac. Dis. 2017, 9, E32–E43. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.; Li, K.; Hu, Z. Asthma and Autophagy. Adv. Exp. Med. Biol. 2020, 1207, 581–584. [Google Scholar] [CrossRef]
- Maneechotesuwan, K. Role of microRNA in severe asthma. Respir. Investig. 2019, 57, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Shirai, T.; Shimoshikiryo, T.; Ueda, M.; Gon, Y.; Maruoka, S.; Itoh, K. Circulating microRNA-15b-5p as a biomarker for asthma-COPD overlap. Allergy 2021, 76, 766–774. [Google Scholar] [CrossRef] [PubMed]
- ElKashef, S.; Ahmad, S.E.; Soliman, Y.M.A.; Mostafa, M.S. Role of microRNA-21 and microRNA-155 as biomarkers for bronchial asthma. Innate Immun. 2021, 27, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Panganiban, R.; Kho, A.T.; McGeachie, M.J.; Farnam, L.; Chase, R.P.; Weiss, S.T.; Lu, Q.; Tantisira, K.G. Circulating MicroRNAs and Treatment Response in Childhood Asthma. Am. J. Respir. Crit. Care Med. 2020, 202, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Weidner, J.; Bartel, S.; Kılıç, A.; Zissler, U.M.; Renz, H.; Schwarze, J.; Schmidt-Weber, C.B.; Maes, T.; Rebane, A.; Krauss-Etschmann, S.; et al. Spotlight on microRNAs in allergy and asthma. Allergy 2021, 76, 1661–1678. [Google Scholar] [CrossRef] [PubMed]
- Lou, L.; Tian, M.; Chang, J.; Li, F.; Zhang, G. MiRNA-192-5p attenuates airway remodeling and autophagy in asthma by targeting MMP-16 and ATG7. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 122, 109692. [Google Scholar] [CrossRef]
- Chen, H.; Xu, X.; Cheng, S.; Xu, Y.; Xuefei, Q.; Cao, Y.; Xie, J.; Wang, C.Y.; Xu, Y.; Xiong, W. Small interfering RNA directed against microRNA-155 delivered by a lentiviral vector attenuates asthmatic features in a mouse model of allergic asthma. Exp. Ther. Med. 2017, 14, 4391–4396. [Google Scholar] [CrossRef]
- Yin, H.; Zhang, S.; Sun, Y.; Li, S.; Ning, Y.; Dong, Y.; Shang, Y.; Bai, C. MicroRNA-34/449 targets IGFBP-3 and attenuates airway remodeling by suppressing Nur77-mediated autophagy. Cell Death Dis. 2017, 8, e2998. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Chong, L.; Lin, P.; Li, H.; Zhu, L.; Wu, Q.; Li, C. MicroRNA-133a alleviates airway remodeling in asthtama through PI3K/AKT/mTOR signaling pathway by targeting IGF1R. J. Cell. Physiol. 2019, 234, 4068–4080. [Google Scholar] [CrossRef]
- Lötvall, J.; Akdis, C.A.; Bacharier, L.B.; Bjermer, L.; Casale, T.B.; Custovic, A.; Lemanske, R.F., Jr.; Wardlaw, A.J.; Wenzel, S.E.; Greenberger, P.A. Asthma endotypes: A new approach to classification of disease entities within the asthma syndrome. J. Allergy Clin. Immunol. 2011, 127, 355–360. [Google Scholar] [CrossRef]
- Arbes, S.J., Jr.; Gergen, P.J.; Vaughn, B.; Zeldin, D.C. Asthma cases attributable to atopy: Results from the Third National Health and Nutrition Examination Survey. J. Allergy Clin. Immunol. 2007, 120, 1139–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akar-Ghibril, N.; Casale, T.; Custovic, A.; Phipatanakul, W. Allergic Endotypes and Phenotypes of Asthma. J. Allergy Clin. Immunol. Pract. 2020, 8, 429–440. [Google Scholar] [CrossRef]
- Roffel, M.P.; Boudewijn, I.M.; van Nijnatten, J.L.L.; Faiz, A.; Vermeulen, C.J.; van Oosterhout, A.J.; Affleck, K.; Timens, W.; Bracke, K.R.; Maes, T.; et al. Identification of asthma-associated microRNAs in bronchial biopsies. Eur. Respir. J. 2022, 59, 2101294. [Google Scholar] [CrossRef]
- Karam, R.A.; Abd Elrahman, D.M. Differential expression of miR-155 and Let-7a in the plasma of childhood asthma: Potential biomarkers for diagnosis and severity. Clin. Biochem. 2019, 68, 30–36. [Google Scholar] [CrossRef]
- Ilmarinen, P.; Kankaanranta, H. Eosinophil apoptosis as a therapeutic target in allergic asthma. Basic Clin. Pharmacol. Toxicol. 2014, 114, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.; Taillé, C.; Laveneziana, P.; Bourdin, A.; Chanez, P.; Humbert, M. Anti-interleukin-5 therapy in severe asthma. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2013, 22, 251–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, H.F.; Phipps, S.; Foster, P.S. Eosinophil trafficking in allergy and asthma. J. Allergy Clin. Immunol. 2007, 119, 1303–1310; quiz 1311–1302. [Google Scholar] [CrossRef]
- Barnes, P.J. Reactive oxygen species and airway inflammation. Free. Radic. Biol. Med. 1990, 9, 235–243. [Google Scholar] [CrossRef]
- Zhang, Y.; Do, D.C.; Hu, X.; Wang, J.; Zhao, Y.; Mishra, S.; Zhang, X.; Wan, M.; Gao, P. CaMKII oxidation regulates cockroach allergen-induced mitophagy in asthma. J. Allergy Clin. Immunol. 2021, 147, 1464–1477.e11. [Google Scholar] [CrossRef]
- Sachdeva, K.; Do, D.C.; Zhang, Y.; Hu, X.; Chen, J.; Gao, P. Environmental Exposures and Asthma Development: Autophagy, Mitophagy, and Cellular Senescence. Front. Immunol. 2019, 10, 2787. [Google Scholar] [CrossRef] [Green Version]
- Poon, A.H.; Chouiali, F.; Tse, S.M.; Litonjua, A.A.; Hussain, S.N.; Baglole, C.J.; Eidelman, D.H.; Olivenstein, R.; Martin, J.G.; Weiss, S.T.; et al. Genetic and histologic evidence for autophagy in asthma pathogenesis. J. Allergy Clin. Immunol. 2012, 129, 569–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.N.; Suh, D.H.; Trinh, H.K.; Chwae, Y.J.; Park, H.S.; Shin, Y.S. The role of autophagy in allergic inflammation: A new target for severe asthma. Exp. Mol. Med. 2016, 48, e243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Wang, X.; Dai, L.; Jia, L.; Jing, X.; Liu, Y.; Wang, H.; Li, P.; An, L.; Liu, M. Suppression of microRNA-384 enhances autophagy of airway smooth muscle cells in asthmatic mouse. Oncotarget 2017, 8, 67933–67941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghavami, S.; Mutawe, M.M.; Schaafsma, D.; Yeganeh, B.; Unruh, H.; Klonisch, T.; Halayko, A.J. Geranylgeranyl transferase 1 modulates autophagy and apoptosis in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L420–L428. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.; Sharma, P.; Shah, S.D.; Deshpande, D.A. Bitter taste receptor agonists alter mitochondrial function and induce autophagy in airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L154–L165. [Google Scholar] [CrossRef] [Green Version]
- Neill, T.; Schaefer, L.; Iozzo, R.V. Instructive roles of extracellular matrix on autophagy. Am. J. Pathol. 2014, 184, 2146–2153. [Google Scholar] [CrossRef] [Green Version]
- Poon, A.H.; Choy, D.F.; Chouiali, F.; Ramakrishnan, R.K.; Mahboub, B.; Audusseau, S.; Mogas, A.; Harris, J.M.; Arron, J.R.; Laprise, C.; et al. Increased Autophagy-Related 5 Gene Expression Is Associated with Collagen Expression in the Airways of Refractory Asthmatics. Front. Immunol. 2017, 8, 355. [Google Scholar] [CrossRef] [Green Version]
- Jessop, F.; Hamilton, R.F.; Rhoderick, J.F.; Shaw, P.K.; Holian, A. Autophagy deficiency in macrophages enhances NLRP3 inflammasome activity and chronic lung disease following silica exposure. Toxicol. Appl. Pharmacol. 2016, 309, 101–110. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence | Amplified Fragment Size (bp) |
|---|---|---|
| ABL2-F | CCGCCGTCGTGTTACTTT | 119 |
| ABL2-R | ATGGTTCTCGCCCACTAGG | |
| MMP16-F | CGTGCCTTTGATGTGTGG | 184 |
| MMP16-R | CGGGTCCAGGGAAATAAG | |
| PDE7A-F | AGTCTCCGCCAGGAACAT | 125 |
| PDE7A-R | CTTGTCCATTGTAATCCTCATC | |
| miR-146a-5p-F | CTGGTGTCGTGGAGTCGG | 57 |
| miR-146a-5p-R | GGGGTGAGAACTGAATTCCA | |
| miR-26a-5p-F | CTGGTGTCGTGGAGTCGG | 57 |
| miR-26a-5p-R | GGGGTTCAAGTAATCCAGGA | |
| miR-26b-5p-F | CTGGTGTCGTGGAGTCGG | 57 |
| miR-26b-5p-R | GGGGGTTCAAGTAATTCAGG | |
| miR-30a-5p-F | CTGGTGTCGTGGAGTCGG | 57 |
| miR-30a-5p-R | GGGGTGTAAACATCCTCGAC | |
| U6-F | CTCGCTTCGGCAGCACATATACT | 93 |
| U6-R | ACGCTTCACGAATTTGCGTGTC | |
| LC3A-F | CTGTCCTGGATAAGACCAAGTTT | 171 |
| LC3A-R | CCTGTTCATAGATGTCAGCGAT | |
| LC3B-F | ACCAAGCCTTCTTCCTCC | 94 |
| LC3B-R | CCGTCTTCATCTCTCTCACTC | |
| Beclin1-F | GAGAGACCCAGGAGGAAGA | 116 |
| Beclin1-R | GGGACTGAGGAATAGTAAGCA | |
| P62-F | TTGCCTTTTCCAGTGATGA | 196 |
| P62-R | TAGCGAGTTCCCACCACA | |
| Atg5-F | GGCTCACTTTATGTCGTGTATG | 93 |
| Atg5-R | AGCTGCTTGTGGTCTTTTTT | |
| GAPDH-F | CCTTCCGTGTTCCTAC | 152 |
| GAPDH-R | GACAACCTGGTCCTCA |
| Gene Symbol | Gene Description |
|---|---|
| ABL2 | c-abl oncogene 2, non-receptor tyrosine kinase |
| MMP16 | matrix metallopeptidase 16 (membrane-inserted) |
| PDE7A | phosphodiesterase 7A |
| Gene Symbol | GO Description | ||
|---|---|---|---|
| Bp | Cc | Mf | |
| ABL2 | signal transduction | actin cytoskeleton | phosphotyrosine residue binding |
| MMP16 | extracellular matrix disassembly | integral component of plasma membrane | metalloaminopeptidase activity |
| PDE7A | cAMP-mediated signaling | cytosol | 3′,5′-cyclic-nucleotide phosphodiesterase activity |
| Gene Symbol | Pathway Name |
|---|---|
| ABL2 | Ras signaling pathway |
| MMP16 | Activation of matrix metalloproteinases |
| PDE7A | GPCR downstream signaling |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, J.; Liu, M.; Chen, S.; Liu, S.; Li, F.; Li, C. Study of the Regulatory Mechanism of miR-26a-5p in Allergic Asthma. Cells 2023, 12, 38. https://doi.org/10.3390/cells12010038
Zhong J, Liu M, Chen S, Liu S, Li F, Li C. Study of the Regulatory Mechanism of miR-26a-5p in Allergic Asthma. Cells. 2023; 12(1):38. https://doi.org/10.3390/cells12010038
Chicago/Turabian StyleZhong, Jinnan, Min Liu, Shi Chen, Shuang Liu, Fajiu Li, and Chenghong Li. 2023. "Study of the Regulatory Mechanism of miR-26a-5p in Allergic Asthma" Cells 12, no. 1: 38. https://doi.org/10.3390/cells12010038