

CLPP Depletion Causes Diplotene Arrest; Underlying Testis Mitochondrial Dysfunction Occurs with Accumulation of Perrault Proteins ERAL1, PEO1, and HARS2
 , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice and Mouse Embryonic Fibroblasts
2.2. Antibodies
2.3. Light Microscopy and Immunohistology
2.4. Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Analyses
2.5. Fluorescent Immunohistology of Testis Sections
2.6. Fluorescent Immunocytology of Meiotic Spreads
2.7. Transcriptome Re-Analysis
2.8. Global Proteome Profiles by Mass Spec and Label-Free Quantification
2.8.1. Sample Preparation for Liquid Chromatography Mass Spectrometry (LC–MS)
2.8.2. High pH Reverse Phase Fractionation
2.8.3. LC–MS
2.8.4. Data Analysis
2.9. Quantitative PCR of mtDNA in Testis Tissues
2.10. Reverse Transcriptase Quantitative PCR
2.11. Subcellular Fractionation of Testis Tissues
2.12. Quantitative Immunoblots
2.13. cGAMP ELISA
2.14. Statistical Analyses
3. Results
3.1. Analyses of Male Meiotic Propase Progression in CLPP-Null Mice
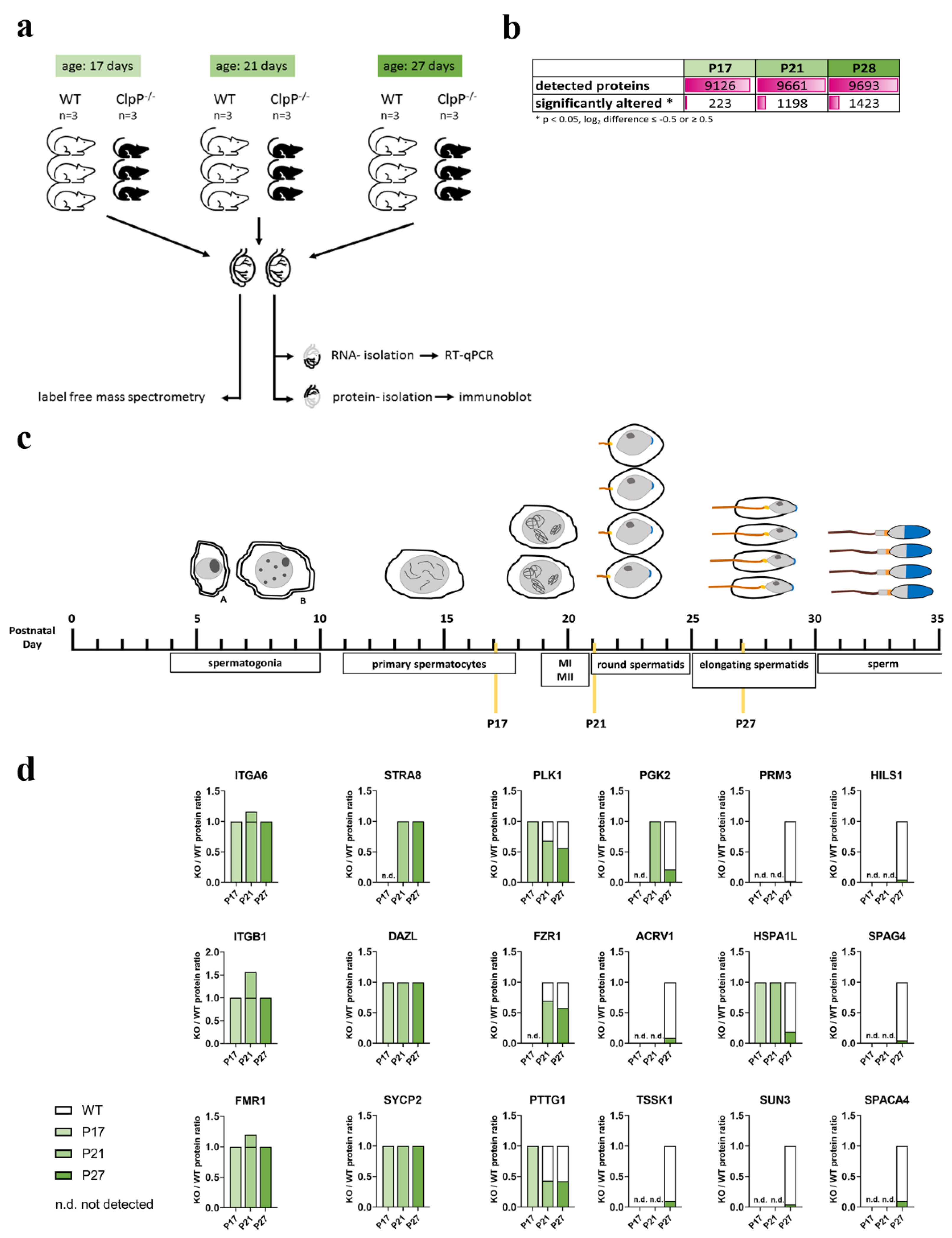
3.2. Global Proteome Data Analysis in Juveniles Recapitulates Lack of Progression to Metaphase-I in CLPP-Null Mice
3.3. Identification of Main Affected Pathways in Juvenile Clpp−/− Testes through Proteome and RT-qPCR Analyses
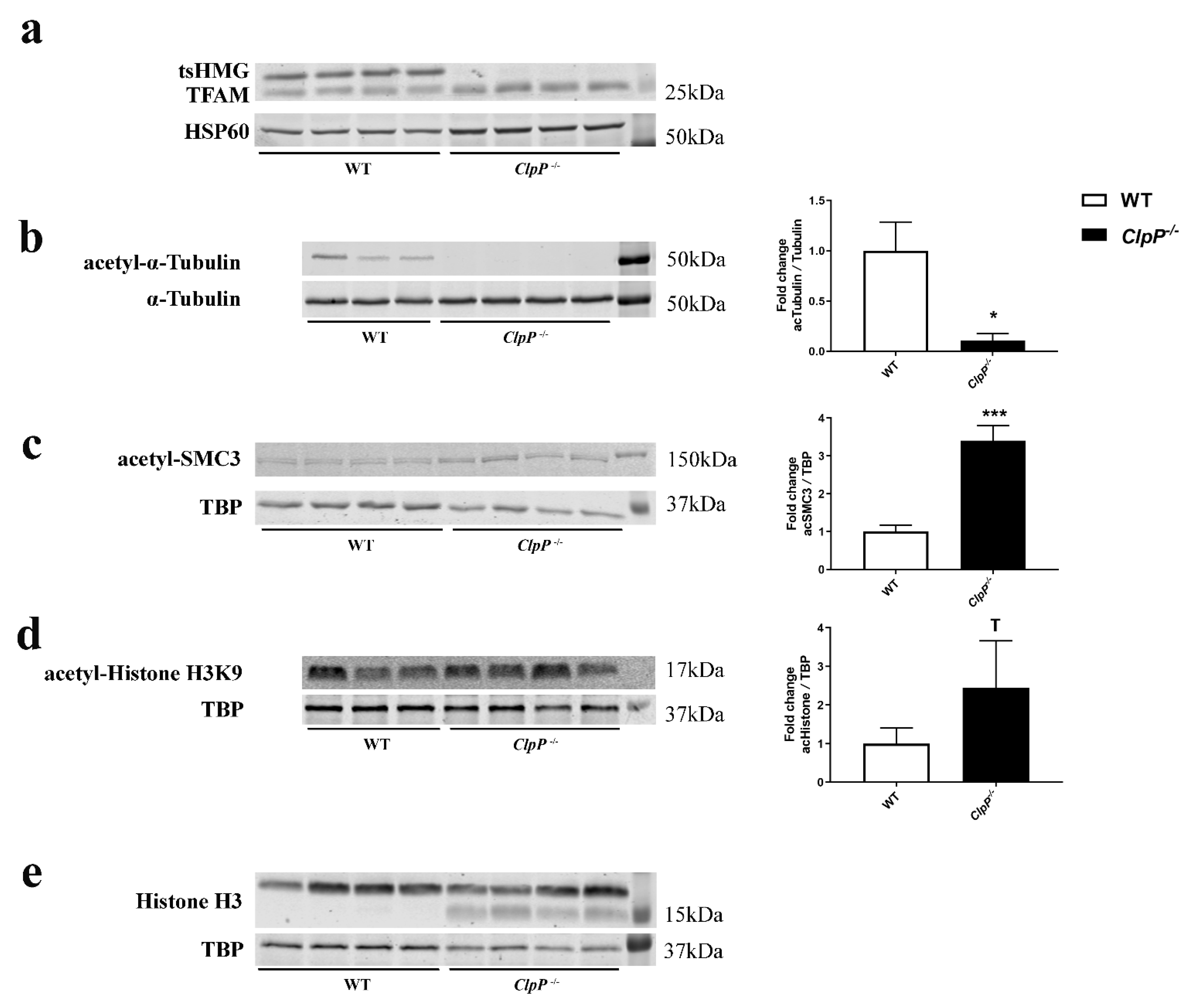
3.4. Microscopy and Quantitative Immunoblot Validation of Prominent Pathway Alterations Leading to Block
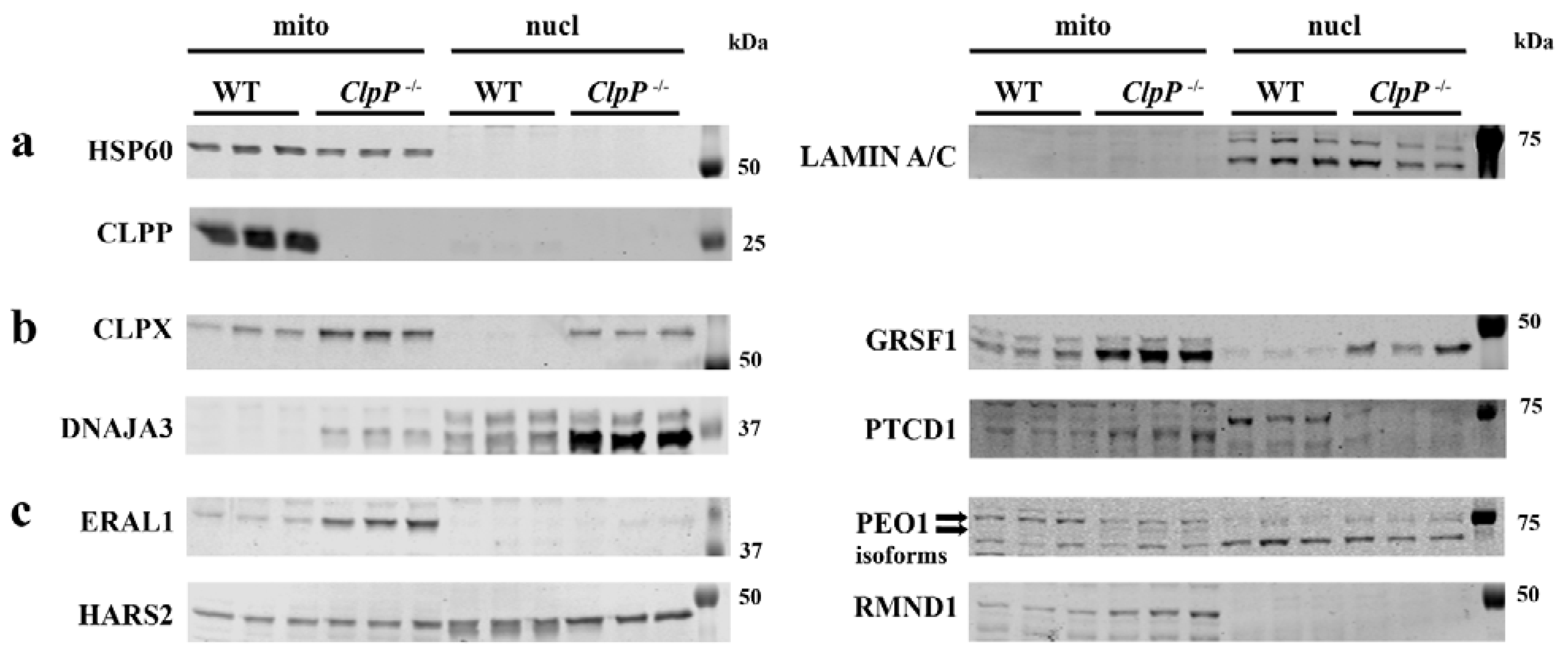
3.5. Redistribution of Excess Mitochondrial Proteins to the Nucleus Possibly Contributes to Pathway Alterations
4. Discussion
4.1. Exceptional Diplotene Arrest Phenotype in CLPP-Null Mice
4.2. Premature Desynapsis in CLPP-Null Mice
4.3. Early and Robust Depletion of Proteins Associated with Ciliopathies
4.4. The Innate Immune Response in CLPP-Null Spermatocytes
4.5. Abnormal Accumulation of Mitochondrial Proteins in Cytosol and Nucleus in Relation to the Meiotic Phenotype
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAA+ | ATPases associated with diverse cellular activities |
| ABC | Ammonium bicarbonate |
| ACN | Acetonitrile |
| ACRV1 | Acrosomal Vesicle Protein 1 |
| ACTB | Actin Beta |
| ADAM3 | ADAM Metallopeptidase Domain 3A |
| AGC | Automatic gain control |
| ALKBH7 | AlkB Homolog 7 |
| AURKA | Aurora Kinase A |
| AURKAIP1 | Aurora Kinase A Interacting Protein 1 |
| AURKC | Aurora Kinase C |
| BCA | Bicinchoninic acid assay |
| BET | Bromodomain and extraterminal domain |
| BIRC5 | Baculoviral IAP Repeat Containing 5 |
| BRDT | Bromodomain testis associated |
| BSA | Bovine serum albumin |
| CDCA8 | Cell Division Cycle Associated 8 |
| CENPE | Centromere Protein E |
| CENPU | Centromere Protein U |
| cGAMP | Cyclic guanosine monophosphate–adenosine monophosphate |
| cGAS | Cyclic GMP-AMP Synthase |
| CLPP | Caseinolytic Mitochondrial Matrix Peptidase Proteolytic Subunit |
| CLPX | Caseinolytic Mitochondrial Matrix Peptidase Chaperone Subunit X |
| CPC | Chromosome passenger complex |
| CSNK1D | Casein Kinase 1 Delta |
| DAZL | Deleted In Azoospermia Like |
| DCP1A | Decapping MRNA 1A |
| DMC1 | DNA Meiotic Recombinase 1 |
| DMRTC2 | Doublesex- and Mab-3-Rrelated transcription factor C2 |
| DNA | Deoxyribonucleid acid |
| DNAAF | Dynein Axonemal Assembly Factor 1 |
| DNAJA3 | DnaJ Heat Shock Protein Family (Hsp40) Member A3 |
| DNAJB13 | DnaJ Heat Shock Protein Family (Hsp40) Member B13 |
| DNAJB3 | DnaJ Heat Shock Protein Family (Hsp40) Member B3 |
| DNALI1 | Dynein Axonemal Light Intermediate Chain 1 |
| DSB | Double-strand break |
| ELISA | Enzyme-linked immunosorbent assay |
| EPPS | N-(2-Hydroxyethyl)piperazine-N′-(3-propanesulfonic acid) |
| ERAL1 | Era Like 12S Mitochondrial RRNA Chaperone 1 |
| ESPL1 | Extra Spindle Pole Bodies Like 1, Separase |
| FA | Formic acid |
| FZR1 | Fizzy And Cell Division Cycle 20 Related 1 |
| GAPDH | Glyceraldehyde-3-Phosphate Dehydrogenase |
| GFM1 | G Elongation Factor Mitochondrial 1 |
| γH2AX | Gamma histone H2A |
| GRSF1 | G-Rich RNA Sequence Binding Factor 1 |
| GTP | Guanosine triphosphate |
| H | Hour |
| H3S10p | Histone 3 Serine 10 phosphorylation |
| HARS2 | Histidyl-TRNA Synthetase 2, Mitochondrial |
| HCD | High-energy collision-induced dissociation |
| HILS1 | H1.9 Linker Histone, Pseudogene |
| hnRNP | Heterogeneous Nuclear Ribonucleoprotein |
| HPLC | High-performance liquid chromatography |
| HR | Homologous recombination |
| HSF5 | Heat Shock Transcription Factor 5 |
| HSP60 | Heat Shock Protein Family D (Hsp60) Member 1 |
| HSPA1L | Heat Shock Protein Family A (Hsp70) Member 1 Like |
| ID | Inner diameter |
| IFNAR | Interferon (alpha and beta) receptor 1 |
| INCENP | Inner Centromere Protein |
| kDA | kiloDalton |
| KO | Knockout |
| LARS2 | Leucyl-TRNA Synthetase 2, Mitochondrial |
| LC–MS | Liquid chromatography–mass spectrometry |
| LDHC | Lactate Dehydrogenase C |
| LRPPRC | Leucine Rich Pentatricopeptide Repeat Containing |
| MAD2L2 | Mitotic Arrest Deficient 2 Like 2 |
| MAVS | Mitochondrial antiviral signaling protein |
| MS | Mass spectrometry |
| MEFs | Mouse embryonic fibroblasts |
| Min | Minute |
| MLH1 | DNA Mismatch Repair Protein MutL Homolog 1 |
| MLH3 | MutL (E. Coli) Homolog 3 |
| mRNA | Messenger RNA |
| MRPL43 | Mitochondrial Ribosomal Protein L43 |
| MS | Mass spectrometry |
| mtDNA | Mitochondrial DNA |
| mtRNA | Mitochondrial RNA |
| n.d. | Not detected |
| NANOS3 | Nanos C2HC-Type Zinc Finger 3 |
| NCAPG2 | Non-SMC Condensin II Complex Subunit G2 |
| NCE | Normalized collision energy |
| NLR | Nod-like receptor |
| NMES1 | Normal mucosa of esophagus-specific gene 1 |
| NOS | Non-obstructive azoospermia |
| PAS | Periodic acid-Schiff |
| PBS | Phosphate-buffered saline |
| PCGF6 | Polycomb Group Ring Finger 6 |
| PD | Proteome Discoverer software |
| PEO1 | Progressive external ophthalmoplegia (=Twinkle) |
| PGK2 | Phosphoglycerate Kinase 2 |
| PLK1 | Polo Like Kinase 1 |
| POLDIP2 | DNA Polymerase Delta Interacting Protein 2 |
| PolG | Polymerase Gamma |
| PPP2R5C | Protein Phosphatase 2 Regulatory Subunit B’Gamma |
| PRLTS | Perrault syndrome |
| PRM3 | Protamine 3 |
| PRORP | Protein Only RNase P Catalytic Subunit |
| PSM | Peptide-spectrum matches |
| PTCD1 | Pentatricopeptide Repeat Domain |
| PTTG1 | PTTG1 Regulator Of Sister Chromatid Separation, Securin |
| qPCR | Quantitative polymerase chain reaction |
| RAD21L | RAD21 Cohesin Complex Component Like 1 |
| REC8 | REC8 Meiotic Recombination Protein |
| RF | Radio frequency |
| RHAU | RNA Helicase Associated With AU-Rich Element Protein |
| RIPA | Radioimmunoprecipitation assay buffer |
| RMND1 | Required For Meiotic Nuclear Division 1 Homolog |
| RNA | Ribonucleic acid |
| RPA | Replication Protein A |
| Rpm | Revolutions per minute |
| RT-qPCR | Reverse transcriptase quantitative polymerase chain reaction |
| s | Second |
| SAC | Spindle assembly checkpoint complex |
| SC | Synaptonemal complex |
| SDS | Sodium dodecylsulfate |
| SEM | Standard error of the mean |
| SGOL2 | Shugoshin 2 |
| SHCBP1L | SHC Binding And Spindle Associated 1 Like |
| SMC1B | Structural Maintenance Of Chromosomes 1B |
| SMC3 | Structural Maintenance Of Chromosomes 3 |
| SPACA4 | Sperm Acrosome Associated 4 |
| SPAG4 | Sperm Associated Antigen 4 |
| ssDNA | Single-strand DNA |
| STAG1/2/3 | Stromal Antigen 1/2/3 |
| STAT1 | Signal Transducer And Activator Of Transcription 1 |
| STING | Stimulator Of Interferon Response CGAMP Interactor 1 |
| STRA8 | Stimulated By Retinoic Acid 8 |
| STRING | Search tool for recurring instances of neighboring genes |
| SUN3 | Sad1 and UNC84 domain containing 3 |
| SYCE1L | Synaptonemal Complex Central Element Protein 1 Like |
| SYCP3 | Synaptonemal Complex Protein 3 |
| TBP | TATA-Box Binding Protein |
| TCEP | Tris(2-carboxyethyl)phosphin |
| TEX12 | Testis Expressed 12 |
| TFAM | Transcription Factor A, Mitochondrial |
| Th | Thomson (mass-to-charge ratio as mass spectrometry unit) |
| TID1S | Tumorous Imaginal Discs Protein Tid56 Homolog |
| TLR | Toll-like receptor |
| TMT | Tandem mass tag |
| TNRC6A | Trinucleotide Repeat Containing Adaptor 6A |
| TRIM33 | Tripartite Motif Containing 33 |
| tRNA | transfer RNA |
| tsHMG/TFAM | Testis-specific High-Mobility Group protein (TFAM testis isoform) |
| TSSK1B | Testis-Specific Serine Kinase 1B |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| VWA8 | Von Willebrand Factor A Domain Containing 8 |
| WT | Wild-type |
| XY body | Chromatin domain of the X and Y chromosomes |
| ZFE | Central animal facility |
References
- Gispert, S.; Parganlija, D.; Klinkenberg, M.; Drose, S.; Wittig, I.; Mittelbronn, M.; Grzmil, P.; Koob, S.; Hamann, A.; Walter, M.; et al. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum. Mol. Genet. 2013, 22, 4871–4887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkinson, E.M.; Rehman, A.U.; Walsh, T.; Clayton-Smith, J.; Lee, K.; Morell, R.J.; Drummond, M.C.; Khan, S.N.; Naeem, M.A.; Rauf, B.; et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 2013, 92, 605–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demain, L.A.; Urquhart, J.E.; O’Sullivan, J.; Williams, S.G.; Bhaskar, S.S.; Jenkinson, E.M.; Lourenco, C.M.; Heiberg, A.; Pearce, S.H.; Shalev, S.A.; et al. Expanding the genotypic spectrum of Perrault syndrome. Clin. Genet. 2017, 91, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Vertika, S.; Singh, K.K.; Rajender, S. Mitochondria, spermatogenesis, and male infertility—An update. Mitochondrion 2020, 54, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Franca, M.M.; Mendonca, B.B. Genetics of ovarian insufficiency and defects of folliculogenesis. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 36, 101594. [Google Scholar] [CrossRef] [PubMed]
- Tiosano, D.; Mears, J.A.; Buchner, D.A. Mitochondrial Dysfunction in Primary Ovarian Insufficiency. Endocrinology 2019, 160, 2353–2366. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Babayev, E.; Jiang, Z.; Li, G.; Zhang, M.; Esencan, E.; Horvath, T.; Seli, E. Mitochondrial unfolded protein response gene Clpp is required to maintain ovarian follicular reserve during aging, for oocyte competence, and development of pre-implantation embryos. Aging Cell 2018, 17, e12784. [Google Scholar] [CrossRef]
- Bhaskaran, S.; Pharaoh, G.; Ranjit, R.; Murphy, A.; Matsuzaki, S.; Nair, B.C.; Forbes, B.; Gispert, S.; Auburger, G.; Humphries, K.M.; et al. Loss of mitochondrial protease ClpP protects mice from diet-induced obesity and insulin resistance. EMBO Rep. 2018, 19, e45009. [Google Scholar] [CrossRef]
- Newman, W.G.; Friedman, T.B.; Conway, G.S.; Demain, L.A.M. Perrault Syndrome. In GeneReviews (R); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Faridi, R.; Rea, A.; Fenollar-Ferrer, C.; O’Keefe, R.T.; Gu, S.; Munir, Z.; Khan, A.A.; Riazuddin, S.; Hoa, M.; Naz, S.; et al. New insights into Perrault syndrome, a clinically and genetically heterogeneous disorder. Hum. Genet. 2021, 141, 805–819. [Google Scholar] [CrossRef]
- Hochberg, I.; Demain, L.A.M.; Richer, J.; Thompson, K.; Urquhart, J.E.; Rea, A.; Pagarkar, W.; Rodriguez-Palmero, A.; Schluter, A.; Verdura, E.; et al. Bi-allelic variants in the mitochondrial RNase P subunit PRORP cause mitochondrial tRNA processing defects and pleiotropic multisystem presentations. Am. J. Hum. Genet. 2021, 108, 2195–2204. [Google Scholar] [CrossRef]
- Auburger, G.; Key, J.; Gispert, S. The Bacterial ClpXP-ClpB Family Is Enriched with RNA-Binding Protein Complexes. Cells 2022, 11, 2370. [Google Scholar] [CrossRef] [PubMed]
- Wawrzynow, A.; Wojtkowiak, D.; Marszalek, J.; Banecki, B.; Jonsen, M.; Graves, B.; Georgopoulos, C.; Zylicz, M. The ClpX heat-shock protein of Escherichia coli, the ATP-dependent substrate specificity component of the ClpP-ClpX protease, is a novel molecular chaperone. EMBO J. 1995, 14, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.A.; Sauer, R.T. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta 2012, 1823, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Key, J.; Torres-Odio, S.; Bach, N.C.; Gispert, S.; Koepf, G.; Reichlmeir, M.; West, A.P.; Prokisch, H.; Freisinger, P.; Newman, W.G.; et al. Inactivity of Peptidase ClpP Causes Primary Accumulation of Mitochondrial Disaggregase ClpX with Its Interacting Nucleoid Proteins, and of mtDNA. Cells 2021, 10, 3354. [Google Scholar] [CrossRef] [PubMed]
- Torres-Odio, S.; Lei, Y.; Gispert, S.; Maletzko, A.; Key, J.; Menissy, S.S.; Wittig, I.; Auburger, G.; West, A.P. Loss of Mitochondrial Protease CLPP Activates Type I IFN Responses through the Mitochondrial DNA-cGAS-STING Signaling Axis. J. Immunol. 2021, 206, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Maletzko, A.; Key, J.; Wittig, I.; Gispert, S.; Koepf, G.; Canet-Pons, J.; Torres-Odio, S.; West, A.P.; Auburger, G. Increased presence of nuclear DNAJA3 and upregulation of cytosolic STAT1 and of nucleic acid sensors trigger innate immunity in the ClpP-null mouse. Neurogenetics 2021, 22, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Key, J.; Maletzko, A.; Kohli, A.; Gispert, S.; Torres-Odio, S.; Wittig, I.; Heidler, J.; Barcena, C.; Lopez-Otin, C.; Lei, Y.; et al. Loss of mitochondrial ClpP, Lonp1, and Tfam triggers transcriptional induction of Rnf213, a susceptibility factor for moyamoya disease. Neurogenetics 2020, 21, 187–203. [Google Scholar] [CrossRef]
- Granat, L.; Hunt, R.J.; Bateman, J.M. Mitochondrial retrograde signalling in neurological disease. Philos. Trans. R Soc. Lond. B Biol. Sci. 2020, 375, 20190415. [Google Scholar] [CrossRef]
- Monaghan, R.M.; Whitmarsh, A.J. Mitochondrial Proteins Moonlighting in the Nucleus. Trends Biochem. Sci. 2015, 40, 728–735. [Google Scholar] [CrossRef]
- Gewiss, R.; Topping, T.; Griswold, M.D. Cycles, waves, and pulses: Retinoic acid and the organization of spermatogenesis. Andrology 2020, 8, 892–897. [Google Scholar] [CrossRef]
- Grey, C.; de Massy, B. Chromosome Organization in Early Meiotic Prophase. Front. Cell Dev. Biol. 2021, 9, 688878. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.K.; Reeves, A.; Webb, L.M.; Ashley, T. Distribution of crossing over on mouse synaptonemal complexes using immunofluorescent localization of MLH1 protein. Genetics 1999, 151, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Chiba, T.; Sakai, J.; Hirose, K.; Yamamoto, M.; Hada, A.; Kuramoto, K.; Higuchi, K.; Mori, M. Mouse testis transcriptome revealed using serial analysis of gene expression. Mamm. Genome 2004, 15, 433–451. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.; Odom, D.T.; Kutter, C. The emergence of piRNAs against transposon invasion to preserve mammalian genome integrity. Nat. Commun. 2017, 8, 1411. [Google Scholar] [CrossRef] [Green Version]
- Lammers, J.H.; Offenberg, H.H.; van Aalderen, M.; Vink, A.C.; Dietrich, A.J.; Heyting, C. The gene encoding a major component of the lateral elements of synaptonemal complexes or the rat is related to X-linked lymphocyte-regulated genes. Mol. Cell Biol. 1994, 14, 1137–1146. [Google Scholar] [CrossRef]
- Wojtasz, L.; Daniel, K.; Roig, I.; Bolcun-Filas, E.; Xu, H.; Boonsanay, V.; Eckmann, C.R.; Cooke, H.J.; Jasin, M.; Keeney, S.; et al. Mouse HORMAD1 and HORMAD2, two conserved meiotic chromosomal proteins, are depleted from synapsed chromosome axes with the help of TRIP13 AAA-ATPase. PLoS Genet. 2009, 5, e1000702. [Google Scholar] [CrossRef] [Green Version]
- Schaarschmidt, D.; Ladenburger, E.M.; Keller, C.; Knippers, R. Human Mcm proteins at a replication origin during the G1 to S phase transition. Nucl. Acids Res. 2002, 30, 4176–4185. [Google Scholar] [CrossRef] [Green Version]
- Peters, A.H.; Plug, A.W.; van Vugt, M.J.; de Boer, P. A drying-down technique for the spreading of mammalian meiocytes from the male and female germline. Chromosom. Res. 1997, 5, 66–68. [Google Scholar] [CrossRef]
- Ghouil, R.; Miron, S.; Koornneef, L.; Veerman, J.; Paul, M.W.; Le Du, M.H.; Sleddens-Linkels, E.; van Rossum-Fikkert, S.E.; van Loon, Y.; Felipe-Medina, N.; et al. BRCA2 binding through a cryptic repeated motif to HSF2BP oligomers does not impact meiotic recombination. Nat. Commun. 2021, 12, 4605. [Google Scholar] [CrossRef] [PubMed]
- Klann, K.; Tascher, G.; Munch, C. Functional Translatome Proteomics Reveal Converging and Dose-Dependent Regulation by mTORC1 and eIF2alpha. Mol. Cell 2020, 77, 913–925.e914. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baghirova, S.; Hughes, B.G.; Hendzel, M.J.; Schulz, R. Sequential fractionation and isolation of subcellular proteins from tissue or cultured cells. MethodsX 2015, 2, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Song, N.; Liu, J.; An, S.; Nishino, T.; Hishikawa, Y.; Koji, T. Immunohistochemical Analysis of Histone H3 Modifications in Germ Cells during Mouse Spermatogenesis. Acta Histochem. Cytochem. 2011, 44, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisig, C.G.; Guiraldelli, M.F.; Kouznetsova, A.; Scherthan, H.; Hoog, C.; Dawson, D.S.; Pezza, R.J. Synaptonemal complex components persist at centromeres and are required for homologous centromere pairing in mouse spermatocytes. PLoS Genet. 2012, 8, e1002701. [Google Scholar] [CrossRef] [Green Version]
- Oakberg, E.F. Duration of spermatogenesis in the mouse and timing of stages of the cycle of the seminiferous epithelium. Am. J. Anat. 1956, 99, 507–516. [Google Scholar] [CrossRef]
- Shi, B.; Xue, J.; Yin, H.; Guo, R.; Luo, M.; Ye, L.; Shi, Q.; Huang, X.; Liu, M.; Sha, J.; et al. Dual functions for the ssDNA-binding protein RPA in meiotic recombination. PLoS Genet. 2019, 15, e1007952. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.M. Meiotic Silencing in Mammals. Annu. Rev. Genet. 2015, 49, 395–412. [Google Scholar] [CrossRef]
- Gispert, S.E.A. Available online: https://www.preprints.org/manuscript/202204.0245/v1 (accessed on 13 December 2022).
- Kluin, P.M.; Kramer, M.F.; de Rooij, D.G. Spermatogenesis in the immature mouse proceeds faster than in the adult. Int. J. Androl. 1982, 5, 282–294. [Google Scholar] [CrossRef]
- Bellve, A.R.; Cavicchia, J.C.; Millette, C.F.; O’Brien, D.A.; Bhatnagar, Y.M.; Dym, M. Spermatogenic cells of the prepuberal mouse. Isolation and morphological characterization. J. Cell Biol. 1977, 74, 68–85. [Google Scholar] [CrossRef]
- von Kopylow, K.; Spiess, A.N. Human spermatogonial markers. Stem. Cell Res. 2017, 25, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liang, Z.; Yang, J.; Wang, D.; Wang, H.; Zhu, M.; Geng, B.; Xu, E.Y. DAZL is a master translational regulator of murine spermatogenesis. Natl. Sci. Rev. 2019, 6, 455–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanno, N.; Kuninaka, S.; Fujimura, S.; Takemooto, K.; Okamura, K.; Takeda, N.; Araki, K.; Araki, M.; Saya, H.; Ishiguro, K.I. Phosphorylation of the Anaphase Promoting Complex activator FZR1/CDH1 is required for Meiosis II entry in mouse male germ cell. Sci. Rep. 2020, 10, 10094. [Google Scholar] [CrossRef] [PubMed]
- Nasa, I.; Kettenbach, A.N. Coordination of Protein Kinase and Phosphoprotein Phosphatase Activities in Mitosis. Front. Cell Dev. Biol. 2018, 6, 30. [Google Scholar] [CrossRef] [Green Version]
- Perez de Castro, I.; de Carcer, G.; Malumbres, M. A census of mitotic cancer genes: New insights into tumor cell biology and cancer therapy. Carcinogenesis 2007, 28, 899–912. [Google Scholar] [CrossRef] [Green Version]
- Nabti, I.; Grimes, R.; Sarna, H.; Marangos, P.; Carroll, J. Maternal age-dependent APC/C-mediated decrease in securin causes premature sister chromatid separation in meiosis II. Nat. Commun. 2017, 8, 15346. [Google Scholar] [CrossRef] [Green Version]
- Hermann, B.P.; Cheng, K.; Singh, A.; Roa-De La Cruz, L.; Mutoji, K.N.; Chen, I.C.; Gildersleeve, H.; Lehle, J.D.; Mayo, M.; Westernstroer, B.; et al. The Mammalian Spermatogenesis Single-Cell Transcriptome, from Spermatogonial Stem Cells to Spermatids. Cell Rep. 2018, 25, 1650–1667.e1658. [Google Scholar] [CrossRef] [Green Version]
- Green, C.D.; Ma, Q.; Manske, G.L.; Shami, A.N.; Zheng, X.; Marini, S.; Moritz, L.; Sultan, C.; Gurczynski, S.J.; Moore, B.B.; et al. A Comprehensive Roadmap of Murine Spermatogenesis Defined by Single-Cell RNA-Seq. Dev. Cell 2018, 46, 651–667.e610. [Google Scholar] [CrossRef] [Green Version]
- Clayton, S.A.; Daley, K.K.; MacDonald, L.; Fernandez-Vizarra, E.; Bottegoni, G.; O’Neil, J.D.; Major, T.; Griffin, D.; Zhuang, Q.; Adewoye, A.B.; et al. Inflammation causes remodeling of mitochondrial cytochrome c oxidase mediated by the bifunctional gene C15orf48. Sci. Adv. 2021, 7, eabl5182. [Google Scholar] [CrossRef]
- Lee, C.Q.E.; Kerouanton, B.; Chothani, S.; Zhang, S.; Chen, Y.; Mantri, C.K.; Hock, D.H.; Lim, R.; Nadkarni, R.; Huynh, V.T.; et al. Coding and non-coding roles of MOCCI (C15ORF48) coordinate to regulate host inflammation and immunity. Nat. Commun. 2021, 12, 2130. [Google Scholar] [CrossRef]
- Cahoon, C.K.; Hawley, R.S. Regulating the construction and demolition of the synaptonemal complex. Nat. Struct. Mol. Biol. 2016, 23, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Dix, D.J.; Allen, J.W.; Collins, B.W.; Poorman-Allen, P.; Mori, C.; Blizard, D.R.; Brown, P.R.; Goulding, E.H.; Strong, B.D.; Eddy, E.M. HSP70-2 is required for desynapsis of synaptonemal complexes during meiotic prophase in juvenile and adult mouse spermatocytes. Development 1997, 124, 4595–4603. [Google Scholar] [CrossRef] [PubMed]
- Dix, D.J.; Allen, J.W.; Collins, B.W.; Mori, C.; Nakamura, N.; Poorman-Allen, P.; Goulding, E.H.; Eddy, E.M. Targeted gene disruption of Hsp70-2 results in failed meiosis, germ cell apoptosis, and male infertility. Proc. Natl. Acad. Sci. USA 1996, 93, 3264–3268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saju, J.M.; Hossain, M.S.; Liew, W.C.; Pradhan, A.; Thevasagayam, N.M.; Tan, L.S.E.; Anand, A.; Olsson, P.E.; Orban, L. Heat Shock Factor 5 Is Essential for Spermatogenesis in Zebrafish. Cell Rep. 2018, 25, 3252–3261.e3254. [Google Scholar] [CrossRef] [Green Version]
- Zarkower, D.; Murphy, M.W. DMRT1: An Ancient Sexual Regulator Required for Human Gonadogenesis. Sex. Dev. 2022, 16, 112–125. [Google Scholar] [CrossRef]
- Kim, S.; Namekawa, S.H.; Niswander, L.M.; Ward, J.O.; Lee, J.T.; Bardwell, V.J.; Zarkower, D. A mammal-specific Doublesex homolog associates with male sex chromatin and is required for male meiosis. PLoS Genet. 2007, 3, e62. [Google Scholar] [CrossRef]
- Fouchecourt, S.; Picolo, F.; Elis, S.; Lecureuil, C.; Thelie, A.; Govoroun, M.; Bregeon, M.; Papillier, P.; Lareyre, J.J.; Monget, P. An evolutionary approach to recover genes predominantly expressed in the testes of the zebrafish, chicken and mouse. BMC Evol. Biol. 2019, 19, 137. [Google Scholar] [CrossRef] [Green Version]
- Miki, Y.; Devi, L.; Imai, Y.; Minami, N.; Koide, T.; Goel, S. Deletion of the PDZ-binding kinase (Pbk) gene does not affect male fertility in mice. Reprod. Fertil. Dev. 2020, 32, 893–902. [Google Scholar] [CrossRef]
- Loges, N.T.; Olbrich, H.; Becker-Heck, A.; Haffner, K.; Heer, A.; Reinhard, C.; Schmidts, M.; Kispert, A.; Zariwala, M.A.; Leigh, M.W.; et al. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009, 85, 883–889. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Chen, W.; Wang, L.; Qian, L. Clinical and Genetic Spectrum of Children with Primary Ciliary Dyskinesia in China. J. Pediatr. 2020, 225, 157–165.e155. [Google Scholar] [CrossRef]
- Horani, A.; Brody, S.L.; Ferkol, T.W.; Shoseyov, D.; Wasserman, M.G.; Ta-shma, A.; Wilson, K.S.; Bayly, P.V.; Amirav, I.; Cohen-Cymberknoh, M.; et al. CCDC65 mutation causes primary ciliary dyskinesia with normal ultrastructure and hyperkinetic cilia. PLoS ONE 2013, 8, e72299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merveille, A.C.; Davis, E.E.; Becker-Heck, A.; Legendre, M.; Amirav, I.; Bataille, G.; Belmont, J.; Beydon, N.; Billen, F.; Clement, A.; et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011, 43, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Shamseldin, H.E.; Shaheen, R.; Ewida, N.; Bubshait, D.K.; Alkuraya, H.; Almardawi, E.; Howaidi, A.; Sabr, Y.; Abdalla, E.M.; Alfaifi, A.Y.; et al. The morbid genome of ciliopathies: An update. Genet. Med. 2020, 22, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Parisi, S.; McKay, M.J.; Molnar, M.; Thompson, M.A.; van der Spek, P.J.; van Drunen-Schoenmaker, E.; Kanaar, R.; Lehmann, E.; Hoeijmakers, J.H.; Kohli, J. Rec8p, a meiotic recombination and sister chromatid cohesion phosphoprotein of the Rad21p family conserved from fission yeast to humans. Mol. Cell Biol. 1999, 19, 3515–3528. [Google Scholar] [CrossRef] [Green Version]
- Lester, W.C.; Johnson, T.; Hale, B.; Serra, N.; Elgart, B.; Wang, R.; Geyer, C.B.; Sperry, A.O. Aurora A Kinase (AURKA) is required for male germline maintenance and regulates sperm motility in the mouse. Biol. Reprod. 2021, 105, 1603–1616. [Google Scholar] [CrossRef]
- Kimmins, S.; Crosio, C.; Kotaja, N.; Hirayama, J.; Monaco, L.; Hoog, C.; van Duin, M.; Gossen, J.A.; Sassone-Corsi, P. Differential functions of the Aurora-B and Aurora-C kinases in mammalian spermatogenesis. Mol. Endocrinol. 2007, 21, 726–739. [Google Scholar] [CrossRef] [Green Version]
- Milenkovic, D.; Matic, S.; Kuhl, I.; Ruzzenente, B.; Freyer, C.; Jemt, E.; Park, C.B.; Falkenberg, M.; Larsson, N.G. TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D-loop strands and complete mtDNA replication. Hum. Mol. Genet. 2013, 22, 1983–1993. [Google Scholar] [CrossRef]
- Jemt, E.; Persson, O.; Shi, Y.; Mehmedovic, M.; Uhler, J.P.; Davila Lopez, M.; Freyer, C.; Gustafsson, C.M.; Samuelsson, T.; Falkenberg, M. Regulation of DNA replication at the end of the mitochondrial D-loop involves the helicase TWINKLE and a conserved sequence element. Nucleic Acids Res. 2015, 43, 9262–9275. [Google Scholar] [CrossRef] [Green Version]
- Dennerlein, S.; Rozanska, A.; Wydro, M.; Chrzanowska-Lightowlers, Z.M.; Lightowlers, R.N. Human ERAL1 is a mitochondrial RNA chaperone involved in the assembly of the 28S small mitochondrial ribosomal subunit. Biochem. J. 2010, 430, 551–558. [Google Scholar] [CrossRef] [Green Version]
- Tyynismaa, H.; Sembongi, H.; Bokori-Brown, M.; Granycome, C.; Ashley, N.; Poulton, J.; Jalanko, A.; Spelbrink, J.N.; Holt, I.J.; Suomalainen, A. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum. Mol. Genet. 2004, 13, 3219–3227. [Google Scholar] [CrossRef]
- Meersseman, C.; Lejard, V.; Rebours, E.; Boussaha, M.; Maftah, A.; Petit, D.; Rocha, D. Bovine TWINKLE and mitochondrial ribosomal protein L43 genes are regulated by an evolutionary conserved bidirectional promoter. Gene 2014, 537, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Pierce, S.B.; Chisholm, K.M.; Lynch, E.D.; Lee, M.K.; Walsh, T.; Opitz, J.M.; Li, W.; Klevit, R.E.; King, M.C. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 6543–6548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossmanith, W. Processing of human mitochondrial tRNA(Ser(AGY))GCU: A novel pathway in tRNA biosynthesis. J. Mol. Biol. 1997, 265, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Karasik, A.; Fierke, C.A.; Koutmos, M. Interplay between substrate recognition, 5′ end tRNA processing and methylation activity of human mitochondrial RNase P. RNA 2019, 25, 1646–1660. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G.; Garman, J.D.; Oldfors, A.; Barsh, G.S.; Clayton, D.A. A single mouse gene encodes the mitochondrial transcription factor A and a testis-specific nuclear HMG-box protein. Nat. Genet. 1996, 13, 296–302. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, X.; Li, Y.; Kim, B.J.; Jia, J.; Huang, Z.; Yang, T.; Fu, X.; Jung, S.Y.; Wang, Y.; et al. Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol. Cell 2008, 31, 143–151. [Google Scholar] [CrossRef]
- Matilainen, O.; Quiros, P.M.; Auwerx, J. Mitochondria and Epigenetics–Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef]
- Nikkanen, J.; Forsstrom, S.; Euro, L.; Paetau, I.; Kohnz, R.A.; Wang, L.; Chilov, D.; Viinamaki, J.; Roivainen, A.; Marjamaki, P.; et al. Mitochondrial DNA Replication Defects Disturb Cellular dNTP Pools and Remodel One-Carbon Metabolism. Cell Metab. 2016, 23, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Kota, S.K.; Feil, R. Epigenetic transitions in germ cell development and meiosis. Dev. Cell 2010, 19, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Yi, S.J.; Kim, K. Histone tail cleavage as a novel epigenetic regulatory mechanism for gene expression. BMB Rep. 2018, 51, 211–218. [Google Scholar] [CrossRef]
- Zhou, P.; Wu, E.; Alam, H.B.; Li, Y. Histone cleavage as a mechanism for epigenetic regulation: Current insights and perspectives. Curr. Mol. Med. 2014, 14, 1164–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, L.; Juszkiewicz, S.; Blears, D.; Bajpe, P.K.; Han, Z.; Faull, P.; Mitter, R.; Stewart, A.; Snijders, A.P.; Hegde, R.S.; et al. Translation stress and collided ribosomes are co-activators of cGAS. Mol. Cell 2021, 81, 2808–2822.e2810. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; He, Y.; Tang, H.; Chen, X.; Liu, S.; Tao, Y. cGAS/STING: Novel perspectives of the classic pathway. Mol. Biomed. 2020, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Xu, P.; Rowlett, C.M.; Jing, T.; Shinde, O.; Lei, Y.; West, A.P.; Liu, W.R.; Li, P. The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature 2020, 587, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Langberg, K.; Abbas, S.; Odermatt, E.; Yerramothu, P.; Volaric, M.; Reidenbach, M.A.; Krentz, K.J.; Rubinstein, C.D.; Brautigan, D.L.; et al. A non-canonical, interferon-independent signaling activity of cGAMP triggers DNA damage response signaling. Nat. Commun. 2021, 12, 6207. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Lu, B.; Garrido, N.; Spelbrink, J.N.; Suzuki, C.K. Tid1 isoforms are mitochondrial DnaJ-like chaperones with unique carboxyl termini that determine cytosolic fate. J. Biol. Chem. 2006, 281, 13150–13158. [Google Scholar] [CrossRef] [Green Version]
- Rackham, O.; Davies, S.M.; Shearwood, A.M.; Hamilton, K.L.; Whelan, J.; Filipovska, A. Pentatricopeptide repeat domain protein 1 lowers the levels of mitochondrial leucine tRNAs in cells. Nucleic Acids Res. 2009, 37, 5859–5867. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, M.I.; Mercer, T.R.; Davies, S.M.; Shearwood, A.M.; Nygard, K.K.; Richman, T.R.; Mattick, J.S.; Rackham, O.; Filipovska, A. RNA processing in human mitochondria. Cell Cycle 2011, 10, 2904–2916. [Google Scholar] [CrossRef]
- Uittenbogaard, M.; Chiaramello, A. Novel subcellular localization of the DNA helicase Twinkle at the kinetochore complex during mitosis in neuronal-like progenitor cells. Histochem. Cell Biol. 2016, 145, 275–286. [Google Scholar] [CrossRef]
- Nikali, K.; Suomalainen, A.; Saharinen, J.; Kuokkanen, M.; Spelbrink, J.N.; Lonnqvist, T.; Peltonen, L. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum. Mol. Genet. 2005, 14, 2981–2990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, K.; Sato, A.; Yoshida, K.; Morita, T.; Tanaka, H.; Inoue, S.; Yonekawa, H.; Hayashi, J. Mitochondria-related male infertility. Proc. Natl. Acad. Sci. USA 2006, 103, 15148–15153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Kauppila, T.E.S.; Motori, E.; Li, X.; Atanassov, I.; Folz-Donahue, K.; Bonekamp, N.A.; Albarran-Gutierrez, S.; Stewart, J.B.; Larsson, N.G. Increased Total mtDNA Copy Number Cures Male Infertility Despite Unaltered mtDNA Mutation Load. Cell Metab. 2017, 26, 429–436.e424. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Bai, X.; Zhang, D.; Han, C.; Yuan, J.; Liu, W.; Cao, X.; Chen, Z.; Shangguan, F.; Zhu, Z.; et al. Mammalian elongation factor 4 regulates mitochondrial translation essential for spermatogenesis. Nat. Struct. Mol. Biol. 2016, 23, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Varuzhanyan, G.; Rojansky, R.; Sweredoski, M.J.; Graham, R.L.J.; Hess, S.; Ladinsky, M.S.; Chan, D.C. Mitochondrial fusion is required for spermatogonial differentiation and meiosis. Elife 2019, 8, e51601. [Google Scholar] [CrossRef]
- Zhang, L.F.; Tan-Tai, W.J.; Li, X.H.; Liu, M.F.; Shi, H.J.; Martin-DeLeon, P.A.; O, W.S.; Chen, H. PHB regulates meiotic recombination via JAK2-mediated histone modifications in spermatogenesis. Nucleic Acids Res. 2020, 48, 4780–4796. [Google Scholar] [CrossRef]
- MacQueen, A.J.; Hochwagen, A. Checkpoint mechanisms: The puppet masters of meiotic prophase. Trends Cell Biol. 2011, 21, 393–400. [Google Scholar] [CrossRef]
- Lane, S.; Kauppi, L. Meiotic spindle assembly checkpoint and aneuploidy in males versus females. Cell Mol. Life Sci. 2019, 76, 1135–1150. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Zhu, G.; Yan, A.; He, J.; Liu, Y.; Li, L.; Yang, X.; Dong, C.; Kee, K. IGSF11 is required for pericentric heterochromatin dissociation during meiotic diplotene. PLoS Genet. 2021, 17, e1009778. [Google Scholar] [CrossRef]
- Zhu, D.; Dix, D.J.; Eddy, E.M. HSP70-2 is required for CDC2 kinase activity in meiosis I of mouse spermatocytes. Development 1997, 124, 3007–3014. [Google Scholar] [CrossRef]
- Rogon, C.; Ulbricht, A.; Hesse, M.; Alberti, S.; Vijayaraj, P.; Best, D.; Adams, I.R.; Magin, T.M.; Fleischmann, B.K.; Hohfeld, J. HSP70-binding protein HSPBP1 regulates chaperone expression at a posttranslational level and is essential for spermatogenesis. Mol. Biol. Cell 2014, 25, 2260–2271. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Palmer, K.; Handel, M.A. Mutation of Eif4g3, encoding a eukaryotic translation initiation factor, causes male infertility and meiotic arrest of mouse spermatocytes. Development 2010, 137, 1699–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopinathan, L.; Szmyd, R.; Low, D.; Diril, M.K.; Chang, H.Y.; Coppola, V.; Liu, K.; Tessarollo, L.; Guccione, E.; van Pelt, A.M.M.; et al. Emi2 Is Essential for Mouse Spermatogenesis. Cell Rep. 2017, 20, 697–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Leu, N.A.; Ma, J.; Chmatal, L.; Ruthel, G.; Bloom, J.C.; Lampson, M.A.; Schimenti, J.C.; Luo, M.; Wang, P.J. SKP1 drives the prophase I to metaphase I transition during male meiosis. Sci. Adv. 2020, 6, eaaz2129. [Google Scholar] [CrossRef] [Green Version]
- Nickerson, H.D.; Joshi, A.; Wolgemuth, D.J. Cyclin A1-deficient mice lack histone H3 serine 10 phosphorylation and exhibit altered aurora B dynamics in late prophase of male meiosis. Dev. Biol. 2007, 306, 725–735. [Google Scholar] [CrossRef] [Green Version]
- Chotiner, J.Y.; Wolgemuth, D.J.; Wang, P.J. Functions of cyclins and CDKs in mammalian gametogenesisdagger. Biol. Reprod. 2019, 101, 591–601. [Google Scholar] [CrossRef]
- Xu, J.; Gao, J.; Liu, J.; Huang, X.; Zhang, H.; Ma, A.; Ye, J.; Zhang, X.; Li, Y.; Yang, G.; et al. ZFP541 maintains the repression of pre-pachytene transcriptional programs and promotes male meiosis progression. Cell Rep. 2022, 38, 110540. [Google Scholar] [CrossRef]
- Ehrmann, I.; Crichton, J.H.; Gazzara, M.R.; James, K.; Liu, Y.; Grellscheid, S.N.; Curk, T.; de Rooij, D.; Steyn, J.S.; Cockell, S.; et al. An ancient germ cell-specific RNA-binding protein protects the germline from cryptic splice site poisoning. Elife 2019, 8, e39304. [Google Scholar] [CrossRef]
- Galán-Martínez, J.; Berenguer, I.; Del Carmen Maza, M.; Stamatakis, K.; Gironès, N.; Fresno, M. TCFL5 deficiency impairs the pachytene to diplotene transition during spermatogenesis in the mouse. Sci. Rep. 2022, 12, 10956. [Google Scholar] [CrossRef]
- Li, Y.; Meng, R.; Li, S.; Gu, B.; Xu, X.; Zhang, H.; Tan, X.; Shao, T.; Wang, J.; Xu, D.; et al. The ZFP541-KCTD19 complex is essential for pachytene progression by activating meiotic genes during mouse spermatogenesis. J. Genet. Genom. 2022, 49, 1029–1041. [Google Scholar] [CrossRef]
- Horisawa-Takada, Y.; Kodera, C.; Takemoto, K.; Sakashita, A.; Horisawa, K.; Maeda, R.; Shimada, R.; Usuki, S.; Fujimura, S.; Tani, N.; et al. Meiosis-specific ZFP541 repressor complex promotes developmental progression of meiotic prophase towards completion during mouse spermatogenesis. Nat. Commun. 2021, 12, 3184. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, K.; Biasini, A.; Yu, T.; Säflund, M.; Mou, H.; Arif, A.; Eghbali, A.; Colpan, C.; Gainetdinov, I.; de Rooij, D.G.; et al. The transcription factor TCFL5 responds to A MYB to elaborate the male meiotic program in mice. Reproduction 2022, REP-22-0355. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Biasini, A.; Cecchini, K.; Saflund, M.; Mou, H.; Arif, A.; Eghbali, A.; de Rooij, D.; Weng, Z.; Zamore, P.D.; et al. A-MYB/TCFL5 regulatory architecture ensures the production of pachytene piRNAs in placental mammals. RNA 2022, 29, 30–43, 079472.122. [Google Scholar] [CrossRef]
- Siep, M.; Sleddens-Linkels, E.; Mulders, S.; van Eenennaam, H.; Wassenaar, E.; Van Cappellen, W.A.; Hoogerbrugge, J.; Grootegoed, J.A.; Baarends, W.M. Basic helix-loop-helix transcription factor Tcfl5 interacts with the Calmegin gene promoter in mouse spermatogenesis. Nucleic Acids Res. 2004, 32, 6425–6436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Shi, X.; Bi, Y.; Qi, L.; Guo, X.; Wang, L.; Zhou, Z.; Sha, J. SHCBP1L, a conserved protein in mammals, is predominantly expressed in male germ cells and maintains spindle stability during meiosis in testis. Mol. Hum. Reprod. 2014, 20, 463–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiat, L.S.; Hui, K.M.; Gopalan, G. Aurora-A kinase interacting protein (AIP), a novel negative regulator of human Aurora-A kinase. J. Biol. Chem. 2002, 277, 45558–45565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koc, E.C.; Cimen, H.; Kumcuoglu, B.; Abu, N.; Akpinar, G.; Haque, M.E.; Spremulli, L.L.; Koc, H. Identification and characterization of CHCHD1, AURKAIP1, and CRIF1 as new members of the mammalian mitochondrial ribosome. Front. Physiol. 2013, 4, 183. [Google Scholar] [CrossRef] [Green Version]
- Katayama, H.; Sasai, K.; Czerniak, B.A.; Carter, J.L.; Sen, S. Aurora-A kinase phosphorylation of Aurora-A kinase interacting protein (AIP) and stabilization of the enzyme-substrate complex. J. Cell Biochem. 2007, 102, 1318–1331. [Google Scholar] [CrossRef]
- Nguyen, A.L.; Drutovic, D.; Vazquez, B.N.; El Yakoubi, W.; Gentilello, A.S.; Malumbres, M.; Solc, P.; Schindler, K. Genetic Interactions between the Aurora Kinases Reveal New Requirements for AURKB and AURKC during Oocyte Meiosis. Curr. Biol. 2018, 28, 3458–3468.e3455. [Google Scholar] [CrossRef] [Green Version]
- Musacchio, A.; Salmon, E.D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393. [Google Scholar] [CrossRef]
- Enguita-Marruedo, A.; Martín-Ruiz, M.; García, E.; Gil-Fernández, A.; Parra, M.T.; Viera, A.; Rufas, J.S.; Page, J. Transition from a meiotic to a somatic-like DNA damage response during the pachytene stage in mouse meiosis. PLoS Genet. 2019, 15, e1007439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, K.; Lange, J.; Hached, K.; Fu, J.; Anastassiadis, K.; Roig, I.; Cooke, H.J.; Stewart, A.F.; Wassmann, K.; Jasin, M.; et al. Meiotic homologue alignment and its quality surveillance are controlled by mouse HORMAD1. Nat. Cell Biol. 2011, 13, 599–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcon, E.; Moens, P. MLH1p and MLH3p localize to precociously induced chiasmata of okadaic-acid-treated mouse spermatocytes. Genetics 2003, 165, 2283–2287. [Google Scholar] [CrossRef] [PubMed]
- Moens, P.B.; Kolas, N.K.; Tarsounas, M.; Marcon, E.; Cohen, P.E.; Spyropoulos, B. The time course and chromosomal localization of recombination-related proteins at meiosis in the mouse are compatible with models that can resolve the early DNA-DNA interactions without reciprocal recombination. J. Cell Sci. 2002, 115, 1611–1622. [Google Scholar] [CrossRef]
- Ashley, T.; Walpita, D.; de Rooij, D.G. Localization of two mammalian cyclin dependent kinases during mammalian meiosis. J. Cell Sci. 2001, 114, 685–693. [Google Scholar] [CrossRef]
- Santucci-Darmanin, S.; Walpita, D.; Lespinasse, F.; Desnuelle, C.; Ashley, T.; Paquis-Flucklinger, V. MSH4 acts in conjunction with MLH1 during mammalian meiosis. FASEB J. 2000, 14, 1539–1547. [Google Scholar] [CrossRef]
- Plug, A.W.; Peters, A.H.; Keegan, K.S.; Hoekstra, M.F.; de Boer, P.; Ashley, T. Changes in protein composition of meiotic nodules during mammalian meiosis. J. Cell Sci. 1998, 111, 413–423. [Google Scholar] [CrossRef]
- Baker, S.M.; Plug, A.W.; Prolla, T.A.; Bronner, C.E.; Harris, A.C.; Yao, X.; Christie, D.M.; Monell, C.; Arnheim, N.; Bradley, A.; et al. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat. Genet. 1996, 13, 336–342. [Google Scholar] [CrossRef]
- Kolas, N.K.; Svetlanov, A.; Lenzi, M.L.; Macaluso, F.P.; Lipkin, S.M.; Liskay, R.M.; Greally, J.; Edelmann, W.; Cohen, P.E. Localization of MMR proteins on meiotic chromosomes in mice indicates distinct functions during prophase I. J. Cell Biol. 2005, 171, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Adelman, C.A.; Petrini, J.H. ZIP4H (TEX11) deficiency in the mouse impairs meiotic double strand break repair and the regulation of crossing over. PLoS Genet. 2008, 4, e1000042. [Google Scholar] [CrossRef]
- Liu, Y.; Tarsounas, M.; O’regan, P.; West, S.C. Role of RAD51C and XRCC3 in genetic recombination and DNA repair. J. Biol. Chem. 2007, 282, 1973–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, N.; Talib, S.Z.A.; Singh, P.; Goh, C.M.F.; Liu, K.; Schimenti, J.C.; Kaldis, P. A novel function for CDK2 activity at meiotic crossover sites. PLoS Biol. 2020, 18, e3000903. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Lin, C.; Kim, S.T.; Roig, I.; Chen, H.; Liu, L.; Veith, G.M.; Jin, R.U.; Keeney, S.; Jasin, M.; et al. The E3 ubiquitin ligase Cullin 4A regulates meiotic progression in mouse spermatogenesis. Dev. Biol. 2011, 356, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellard, S.R.; Skinner, M.W.; Zhao, X.; Shults, C.; Jordan, P.W. PLK1 depletion alters homologous recombination and synaptonemal complex disassembly events during mammalian spermatogenesis. Mol. Biol. Cell 2022, 33, ar37. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Wang, X.; Jin, M.; Li, L.; Zhu, J.; Kang, Y.; Chen, Z.; Sun, Y.; Zha, C. Cilia regulate meiotic recombination in zebrafish. J. Mol. Cell Biol. 2022, 14, mjac049. [Google Scholar] [CrossRef]
- López-Jiménez, P.; Pérez-Martín, S.; Hidalgo, I.; Garcia-Gonzalo, F.R.; Page, J.; Gómez, R. The Male Mouse Meiotic Cilium Emanates from the Mother Centriole at Zygotene Prior to Centrosome Duplication. Available online: https://www.biorxiv.org/content/10.1101/2022.10.20.512932v1 (accessed on 13 December 2022).
- Chaudhry, B.; Henderson, D.J. Cilia, mitochondria, and cardiac development. J. Clin. Investig. 2019, 129, 2666–2668. [Google Scholar] [CrossRef] [Green Version]
- Burkhalter, M.D.; Sridhar, A.; Sampaio, P.; Jacinto, R.; Burczyk, M.S.; Donow, C.; Angenendt, M.; Competence Network for Congenital Heart Defects, I.; Hempel, M.; Walther, P.; et al. Imbalanced mitochondrial function provokes heterotaxy via aberrant ciliogenesis. J. Clin. Investig. 2019, 129, 2841–2855. [Google Scholar] [CrossRef] [Green Version]
- O’Toole, J.F.; Liu, Y.; Davis, E.E.; Westlake, C.J.; Attanasio, M.; Otto, E.A.; Seelow, D.; Nurnberg, G.; Becker, C.; Nuutinen, M.; et al. Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronophthisis-like nephropathy. J. Clin. Investig. 2010, 120, 791–802. [Google Scholar] [CrossRef]
- Hu, J.; Barr, M.M. ATP-2 interacts with the PLAT domain of LOV-1 and is involved in Caenorhabditis elegans polycystin signaling. Mol. Biol. Cell 2005, 16, 458–469. [Google Scholar] [CrossRef] [Green Version]
- Widlak, W.; Vydra, N. The Role of Heat Shock Factors in Mammalian Spermatogenesis. Adv. Anat. Embryol. Cell Biol. 2017, 222, 45–65. [Google Scholar] [CrossRef]
- Enguita-Marruedo, A.; Sleddens-Linkels, E.; Ooms, M.; de Geus, V.; Wilke, M.; Blom, E.; Dohle, G.R.; Looijenga, L.H.J.; van Cappellen, W.; Baart, E.B.; et al. Meiotic arrest occurs most frequently at metaphase and is often incomplete in azoospermic men. Fertil. Steril. 2019, 112, 1059–1070.e1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Q.; Lau, R.K.; Mathews, I.T.; Birkholz, E.A.; Watrous, J.D.; Azimi, C.S.; Pogliano, J.; Jain, M.; Corbett, K.D. HORMA Domain Proteins and a Trip13-like ATPase Regulate Bacterial cGAS-like Enzymes to Mediate Bacteriophage Immunity. Mol. Cell 2020, 77, 709–722.e7. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; Garcia-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange consortium in 2020: Enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 2020, 48, D1145–D1152. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Key, J.; Gispert, S.; Koornneef, L.; Sleddens-Linkels, E.; Kohli, A.; Torres-Odio, S.; Koepf, G.; Amr, S.; Reichlmeir, M.; Harter, P.N.; et al. CLPP Depletion Causes Diplotene Arrest; Underlying Testis Mitochondrial Dysfunction Occurs with Accumulation of Perrault Proteins ERAL1, PEO1, and HARS2. Cells 2023, 12, 52. https://doi.org/10.3390/cells12010052
Key J, Gispert S, Koornneef L, Sleddens-Linkels E, Kohli A, Torres-Odio S, Koepf G, Amr S, Reichlmeir M, Harter PN, et al. CLPP Depletion Causes Diplotene Arrest; Underlying Testis Mitochondrial Dysfunction Occurs with Accumulation of Perrault Proteins ERAL1, PEO1, and HARS2. Cells. 2023; 12(1):52. https://doi.org/10.3390/cells12010052
Chicago/Turabian StyleKey, Jana, Suzana Gispert, Lieke Koornneef, Esther Sleddens-Linkels, Aneesha Kohli, Sylvia Torres-Odio, Gabriele Koepf, Shady Amr, Marina Reichlmeir, Patrick N. Harter, and et al. 2023. "CLPP Depletion Causes Diplotene Arrest; Underlying Testis Mitochondrial Dysfunction Occurs with Accumulation of Perrault Proteins ERAL1, PEO1, and HARS2" Cells 12, no. 1: 52. https://doi.org/10.3390/cells12010052
APA StyleKey, J., Gispert, S., Koornneef, L., Sleddens-Linkels, E., Kohli, A., Torres-Odio, S., Koepf, G., Amr, S., Reichlmeir, M., Harter, P. N., West, A. P., Münch, C., Baarends, W. M., & Auburger, G. (2023). CLPP Depletion Causes Diplotene Arrest; Underlying Testis Mitochondrial Dysfunction Occurs with Accumulation of Perrault Proteins ERAL1, PEO1, and HARS2. Cells, 12(1), 52. https://doi.org/10.3390/cells12010052