
Beneficial Effect of ACI-24 Vaccination on Aβ Plaque Pathology and Microglial Phenotypes in an Amyloidosis Mouse Model

 , , ,
, , ,
Abstract
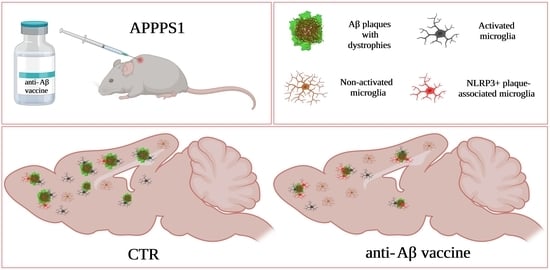
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Animal Treatment
2.3. Blood Collection
2.4. Quantification of Antigen-Specific Antibodies
2.5. Generation of the ApoE Antibody 26C11
2.6. Immunohistochemistry
2.7. Image Acquisition, Analysis and Quantification
2.8. Biochemical Analysis of the Brain
2.9. Brain Gene Expression Profiling
2.10. Isolation of Primary Microglia
2.11. Sample Preparation for Proteomics
2.12. LC-MS/MS Analysis
3. Results
3.1. ACI-24 Triggers Substantial Immune Response
3.2. ACI-24 Reduces Aβ Load in APPPS1 Mice
3.3. ACI-24 More Effectively Reduces Aβ Pathology in the Offspring Generated by the Transgenic Mother
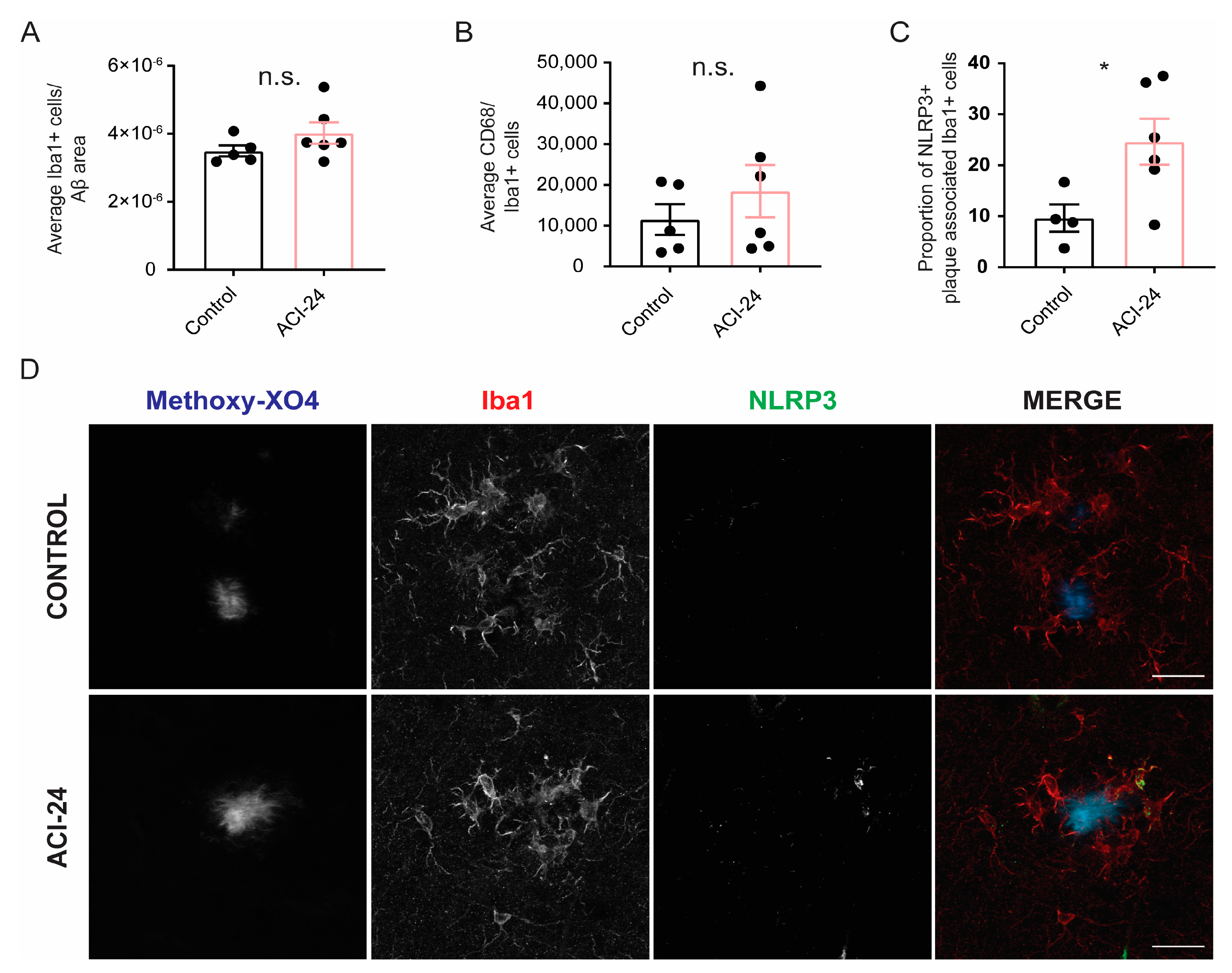
3.4. Microglia at the Plaque Are Changed Following ACI-24 Vaccination
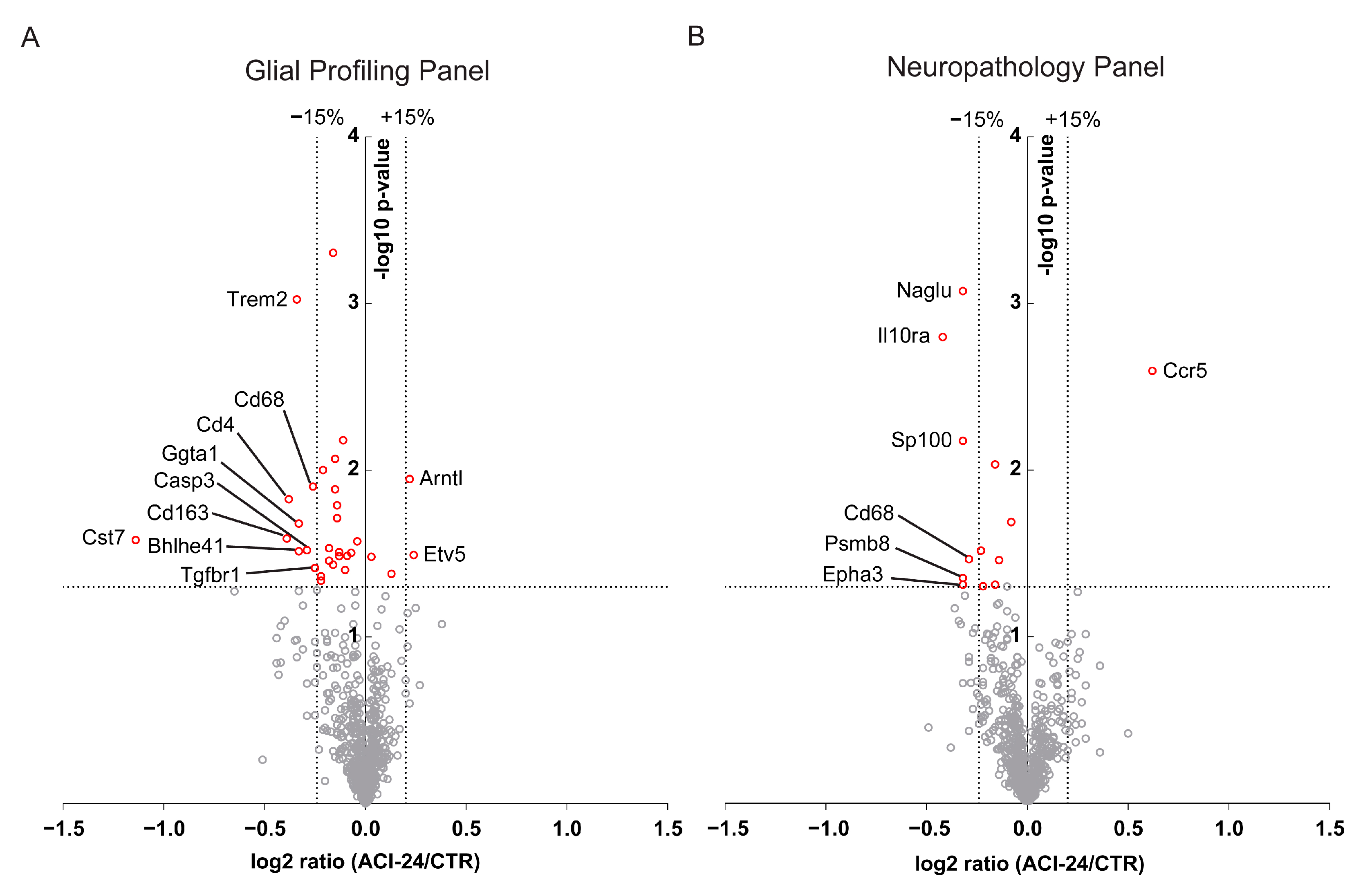
3.5. Global Beneficial Microglial Effect of the ACI-24 Vaccination
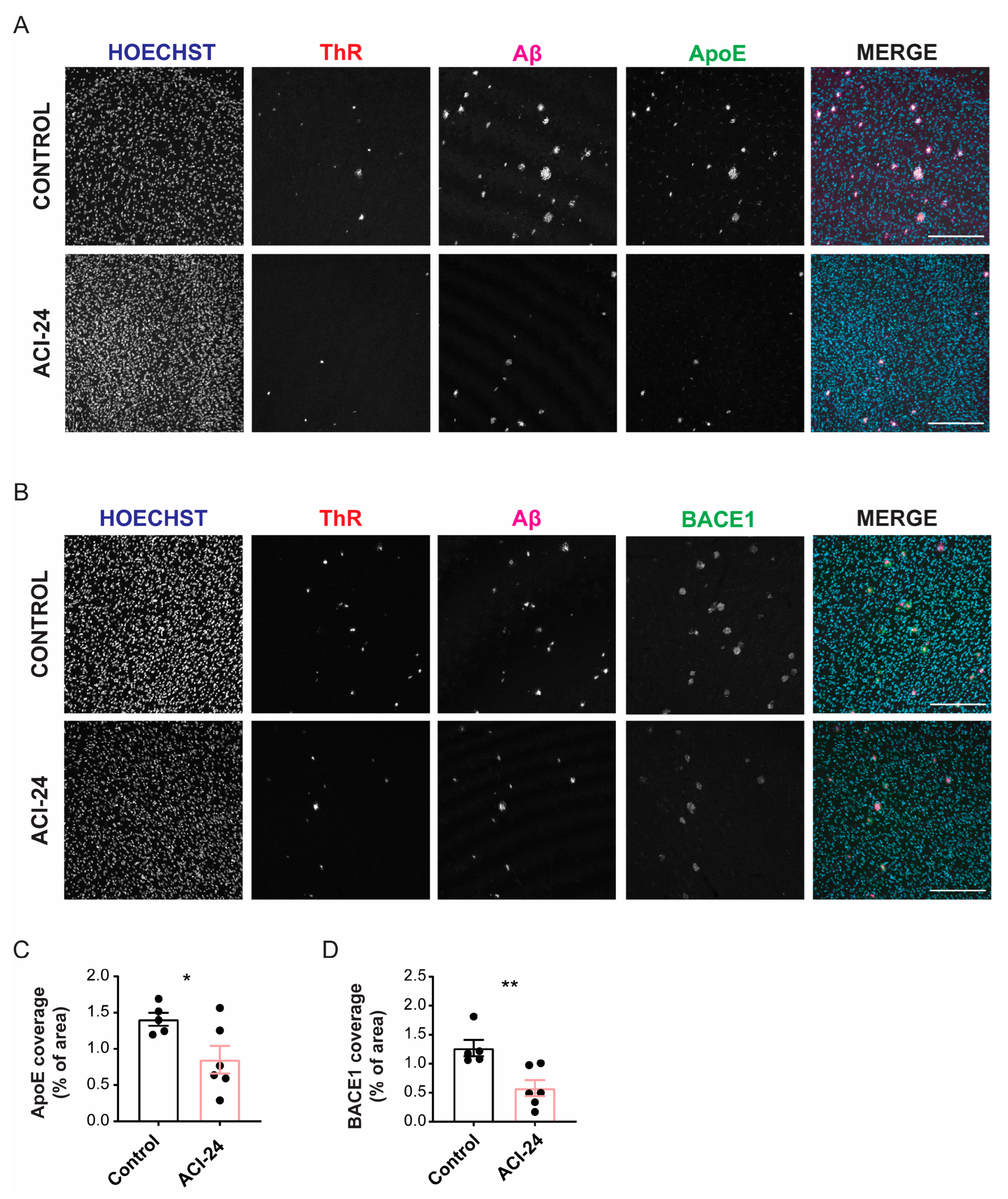
3.6. ACI-24 Reduces ApoE Protein Levels and Neuronal Injury
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s Disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Strohmeyer, R.; Kovelowski, C.J.; Li, R. Microglia and inflammatory mechanisms in the clearance of amyloid beta peptide. GLIA 2002, 40, 260–269. [Google Scholar] [CrossRef]
- Lee, C.Y.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Condello, C.; Schain, A.; Harb, R.; Grutzendler, J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. J. Neurosci. 2010, 30, 17091–17101. [Google Scholar] [CrossRef] [Green Version]
- Njie, E.G.; Boelen, E.; Stassen, F.R.; Steinbusch, H.W.; Borchelt, D.R.; Streit, W.J. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol. Aging 2012, 33, 195.e1–195.e12. [Google Scholar] [CrossRef] [Green Version]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 2013, 8, e60921. [Google Scholar] [CrossRef]
- Daria, A.; Colombo, A.; Llovera, G.; Hampel, H.; Willem, M.; Liesz, A.; Haass, C.; Tahirovic, S. Young microglia restore amyloid plaque clearance of aged microglia. EMBO J. 2017, 36, 583–603. [Google Scholar]
- Sebastian Monasor, L.; Müller, S.A.; Colombo, A.V.; Tanrioever, G.; König, J.; Roth, S.; Liesz, A.; Berghofer, A.; Piechotta, A.; Prestel, M.; et al. Fibrillar Aβ triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. Elife 2020, 9, 1–33. [Google Scholar] [CrossRef]
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Podleśny-Drabiniok, A.; Marcora, E.; Goate, A.M. Microglial phagocytosis: A disease-associated process emerging from Alzheimer’s disease genetics. Trends Neurosci. 2020, 43, 965–979. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol. 2015, 77, 75–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A unique microglia type associated with restricting development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Raj, D.; Saiepour, N.; Van Dam, D.; Brouwer, N.; Holtman, I.R.; Eggen, B.J.L.; Möller, T.; Tamm, J.A.; Abdourahman, A.; et al. Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer’s disease. Neurobiol. Aging 2017, 55, 115–122. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017, 47, 566–581.e569. [Google Scholar] [CrossRef] [Green Version]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Song, C.; Shi, J.; Zhang, P.; Zhang, Y.; Xu, J.; Zhao, L.; Zhang, R.; Wang, H.; Chen, H. Immunotherapy for Alzheimer’s disease: Targeting β-amyloid and beyond. Transl. Neurodegener. 2022, 11, 18. [Google Scholar] [CrossRef]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Gilman, S.; Koller, M.; Black, R.S.; Jenkins, L.; Griffith, S.G.; Fox, N.C.; Eisner, L.; Kirby, L.; Rovira, M.B.; Forette, F.; et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005, 64, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Hock, C.; Nitsch, R.M. Clinical observations with AN-1792 using TAPIR analyses. Neuro-Degener. Dis. 2005, 2, 273–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orgogozo, J.M.; Gilman, S.; Dartigues, J.F.; Laurent, B.; Puel, M.; Kirby, L.C.; Jouanny, P.; Dubois, B.; Eisner, L.; Flitman, S.; et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003, 61, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Monsonego, A.; Imitola, J.; Petrovic, S.; Zota, V.; Nemirovsky, A.; Baron, R.; Fisher, Y.; Owens, T.; Weiner, H.L. Abeta-induced meningoencephalitis is IFN-gamma-dependent and is associated with T cell-dependent clearance of Abeta in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5048–5053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabira, T. Immunization therapy for Alzheimer disease: A comprehensive review of active immunization strategies. Tohoku J. Exp. Med. 2010, 220, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Delrieu, J.; Ousset, P.J.; Caillaud, C.; Vellas, B. “Clinical trials in Alzheimer’s disease”: Immunotherapy approaches. J. Neurochem. 2012, 120 (Suppl. 1), 186–193. [Google Scholar] [CrossRef]
- Lowe, S.L.; Willis, B.A.; Hawdon, A.; Natanegara, F.; Chua, L.; Foster, J.; Shcherbinin, S.; Ardayfio, P.; Sims, J.R. Donanemab (LY3002813) dose-escalation study in Alzheimer’s disease. Alzheimers Dement. 2021, 7, e12112. [Google Scholar] [CrossRef]
- Fillit, H.; Green, A. Aducanumab and the FDA—Where are we now? Nat. Rev. Neurol. 2021, 17, 129–130. [Google Scholar] [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in early Alzheimer’s disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Farlow, M.; McDade, E.; Clifford, D.B.; Wang, G.; Llibre-Guerra, J.J.; Hitchcock, J.M.; Mills, S.L.; Santacruz, A.M.; Aschenbrenner, A.J.; et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat. Med. 2021, 27, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, R.E.; Rother, C.; Rasmussen, J.; Schelle, J.; Bergmann, C.; Ullrich Gavilanes, E.M.; Fritschi, S.K.; Buehler, A.; Baumann, F.; Skodras, A.; et al. Acute targeting of pre-amyloid seeds in transgenic mice reduces Alzheimer-like pathology later in life. Nat. Neurosci. 2020, 23, 1580–1588. [Google Scholar] [CrossRef]
- Kwan, P.; Konno, H.; Chan, K.Y.; Baum, L. Rationale for the development of an Alzheimer’s disease vaccine. Hum. Vaccines Immunother. 2020, 16, 645–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salloway, S.; Sperling, R.; Brashear, H.R. Phase 3 trials of solanezumab and bapineuzumab for Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farlow, M.R.; Andreasen, N.; Riviere, M.E.; Vostiar, I.; Vitaliti, A.; Sovago, J.; Caputo, A.; Winblad, B.; Graf, A. Long-term treatment with active Aβ immunotherapy with CAD106 in mild Alzheimer’s disease. Alzheimers Res. Ther. 2015, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, F.; Sadowsky, C.; Holstein, A.; Leterme Gle, P.; Peng, Y.; Jackson, N.; Fox, N.C.; Ketter, N.; Liu, E.; Ryan, J.M. Two Phase 2 Multiple Ascending-Dose Studies of Vanutide Cridificar (ACC-001) and QS-21 Adjuvant in Mild-to-Moderate Alzheimer’s Disease. J. Alzheimers Dis. 2016, 51, 1131–1143. [Google Scholar] [CrossRef]
- Hull, M.; Sadowsky, C.; Arai, H.; Le Prince Leterme, G.; Holstein, A.; Booth, K.; Peng, Y.; Yoshiyama, T.; Suzuki, H.; Ketter, N.; et al. Long-Term Extensions of Randomized Vaccination Trials of ACC-001 and QS-21 in Mild to Moderate Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 696–708. [Google Scholar] [CrossRef]
- Vandenberghe, R.; Riviere, M.E.; Caputo, A.; Sovago, J.; Maguire, R.P.; Farlow, M.; Marotta, G.; Sanchez-Valle, R.; Scheltens, P.; Ryan, J.M.; et al. Active Aβ immunotherapy CAD106 in Alzheimer’s disease: A phase 2b study. Alzheimers Dement. 2017, 3, 10–22. [Google Scholar] [CrossRef]
- Lacosta, A.M.; Pascual-Lucas, M.; Pesini, P.; Casabona, D.; Pérez-Grijalba, V.; Marcos-Campos, I.; Sarasa, L.; Canudas, J.; Badi, H.; Monleón, I.; et al. Safety, tolerability and immunogenicity of an active anti-Aβ(40) vaccine (ABvac40) in patients with Alzheimer’s disease: A randomised, double-blind, placebo-controlled, phase I trial. Alzheimers Res. Ther. 2018, 10, 12. [Google Scholar] [CrossRef]
- Muhs, A.; Hickman, D.T.; Pihlgren, M.; Chuard, N.; Giriens, V.; Meerschman, C.; van der Auwera, I.; van Leuven, F.; Sugawara, M.; Weingertner, M.C.; et al. Liposomal vaccines with conformation-specific amyloid peptide antigens define immune response and efficacy in APP transgenic mice. Proc. Natl. Acad. Sci. USA 2007, 104, 9810–9815. [Google Scholar] [CrossRef] [PubMed]
- Belichenko, P.V.; Madani, R.; Rey-Bellet, L.; Pihlgren, M.; Becker, A.; Plassard, A.; Vuillermot, S.; Giriens, V.; Nosheny, R.L.; Kleschevnikov, A.M.; et al. An anti-beta-amyloid vaccine for treating cognitive deficits in a mouse model of down syndrome. PLoS ONE 2016, 11, e0152471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickman, D.T.; Lopez-Deber, M.P.; Ndao, D.M.; Silva, A.B.; Nand, D.; Pihlgren, M.; Giriens, V.; Madani, R.; St-Pierre, A.; Karastaneva, H.; et al. Sequence-independent control of peptide conformation in liposomal vaccines for targeting protein misfolding diseases. J. Biol. Chem. 2011, 286, 13966–13976. [Google Scholar] [CrossRef] [Green Version]
- Saido, T.C.; Iwatsubo, T.; Mann, D.M.; Shimada, H.; Ihara, Y.; Kawashima, S. Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques. Neuron 1995, 14, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Pike, C.J.; Overman, M.J.; Cotman, C.W. Amino-terminal deletions enhance aggregation of beta-amyloid peptides in vitro. J. Biol. Chem. 1995, 270, 23895–23898. [Google Scholar] [CrossRef] [Green Version]
- Vukicevic, M.; Fiorini, E.; Siegert, S.; Carpintero, R.; Rincon-Restrepo, M.; Lopez-Deber, P.; Piot, N.; Ayer, M.; Rentero, I.; Babolin, C.; et al. An amyloid beta vaccine that safely drives immunity to a key pathological species in Alzheimer’s disease: Pyroglutamate amyloid beta. Brain Commun. 2022, 4, fcac022. [Google Scholar] [CrossRef] [PubMed]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jaggi, F.; Wolburg, H.; Gengler, S.; et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piedrahita, J.A.; Zhang, S.H.; Hagaman, J.R.; Oliver, P.M.; Maeda, N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc. Natl. Acad. Sci. USA 1992, 89, 4471–4475. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, A.; Eimer, S.; Okochi, M.; Smialowska, A.; Kaether, C.; Baumeister, R.; Haass, C.; Steiner, H. The GxGD motif of presenilin contributes to catalytic function and substrate identification of gamma-secretase. J. Neurosci. 2006, 26, 3821–3828. [Google Scholar] [CrossRef] [Green Version]
- Heindl, S.; Gesierich, B.; Benakis, C.; Llovera, G.; Duering, M.; Liesz, A. Automated morphological analysis of microglia after stroke. Front. Cell Neurosci. 2018, 12, 106. [Google Scholar] [CrossRef]
- Colombo, A.; Dinkel, L.; Müller, S.A.; Sebastian Monasor, L.; Schifferer, M.; Cantuti-Castelvetri, L.; König, J.; Vidatic, L.; Bremova-Ertl, T.; Lieberman, A.P.; et al. Loss of NPC1 enhances phagocytic uptake and impairs lipid trafficking in microglia. Nat. Commun. 2021, 12, 1158. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Ma, J.; Yee, A.; Brewer, H.B.; Jr Das, S.; Potter, H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature 1994, 372, 92–94. [Google Scholar] [CrossRef]
- Huynh, T.V.; Liao, F.; Francis, C.M.; Robinson, G.O.; Serrano, J.R.; Jiang, H.; Roh, J.; Finn, M.B.; Sullivan, P.M.; Esparza, T.J.; et al. Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of β-amyloidosis. Neuron 2017, 96, 1013–1023.e1014. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.C.; Zhao, N.; Fu, Y.; Wang, N.; Linares, C.; Tsai, C.W.; Bu, G. ApoE4 accelerates early seeding of amyloid pathology. Neuron 2017, 96, 1024–1032.e1023. [Google Scholar] [CrossRef] [Green Version]
- Kandalepas, P.C.; Sadleir, K.R.; Eimer, W.A.; Zhao, J.; Nicholson, D.A.; Vassar, R. The Alzheimer’s beta-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol. 2013, 126, 329–352. [Google Scholar] [CrossRef] [Green Version]
- Viña, J.; Lloret, A. Why women have more Alzheimer’s disease than men: Gender and mitochondrial toxicity of amyloid-beta peptide. J. Alzheimers Dis. 2010, 20 (Suppl. S2), S527–S533. [Google Scholar] [CrossRef] [Green Version]
- Illouz, T.; Nicola, R.; Ben-Shushan, L.; Madar, R.; Biragyn, A.; Okun, E. Maternal antibodies facilitate Amyloid-β clearance by activating Fc-receptor-Syk-mediated phagocytosis. Commun. Biol. 2021, 4, 329. [Google Scholar] [CrossRef]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.T.; Wang, K.; Hu, G.; Wang, X.; Miao, Z.; Azevedo, J.A.; Suh, E.; Van Deerlin, V.M.; Choi, D.; Roeder, K.; et al. APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol. 2020, 140, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Das, B.; Zhou, J.; Hu, X.; Yan, R. Targeted BACE-1 inhibition in microglia enhances amyloid clearance and improved cognitive performance. Sci. Adv. 2022, 8, eabo3610. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Ferrucci, L.; Kapogiannis, D. Effects of monoclonal antibodies against amyloid-β on clinical and biomarker outcomes and adverse event risks: A systematic review and meta-analysis of phase III RCTs in Alzheimer’s disease. Ageing Res. Rev. 2021, 68, 101339. [Google Scholar] [CrossRef]
- Chakrabarty, P.; Li, A.; Ceballos-Diaz, C.; Eddy, J.A.; Funk, C.C.; Moore, B.; DiNunno, N.; Rosario, A.M.; Cruz, P.E.; Verbeeck, C.; et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron 2015, 85, 519–533. [Google Scholar] [CrossRef] [Green Version]
- Lewcock, J.W.; Schlepckow, K.; Di Paolo, G.; Tahirovic, S.; Monroe, K.M.; Haass, C. Emerging microglia biology defines novel therapeutic approaches for Alzheimer’s Disease. Neuron 2020, 108, 801–821. [Google Scholar] [CrossRef]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 2000, 6, 916–919. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rudan Njavro, J.; Vukicevic, M.; Fiorini, E.; Dinkel, L.; Müller, S.A.; Berghofer, A.; Bordier, C.; Kozlov, S.; Halle, A.; Buschmann, K.; et al. Beneficial Effect of ACI-24 Vaccination on Aβ Plaque Pathology and Microglial Phenotypes in an Amyloidosis Mouse Model. Cells 2023, 12, 79. https://doi.org/10.3390/cells12010079
Rudan Njavro J, Vukicevic M, Fiorini E, Dinkel L, Müller SA, Berghofer A, Bordier C, Kozlov S, Halle A, Buschmann K, et al. Beneficial Effect of ACI-24 Vaccination on Aβ Plaque Pathology and Microglial Phenotypes in an Amyloidosis Mouse Model. Cells. 2023; 12(1):79. https://doi.org/10.3390/cells12010079
Chicago/Turabian StyleRudan Njavro, Jasenka, Marija Vukicevic, Emma Fiorini, Lina Dinkel, Stephan A. Müller, Anna Berghofer, Chiara Bordier, Stanislav Kozlov, Annett Halle, Katrin Buschmann, and et al. 2023. "Beneficial Effect of ACI-24 Vaccination on Aβ Plaque Pathology and Microglial Phenotypes in an Amyloidosis Mouse Model" Cells 12, no. 1: 79. https://doi.org/10.3390/cells12010079
APA StyleRudan Njavro, J., Vukicevic, M., Fiorini, E., Dinkel, L., Müller, S. A., Berghofer, A., Bordier, C., Kozlov, S., Halle, A., Buschmann, K., Capell, A., Giudici, C., Willem, M., Feederle, R., Lichtenthaler, S. F., Babolin, C., Montanari, P., Pfeifer, A., Kosco-Vilbois, M., & Tahirovic, S. (2023). Beneficial Effect of ACI-24 Vaccination on Aβ Plaque Pathology and Microglial Phenotypes in an Amyloidosis Mouse Model. Cells, 12(1), 79. https://doi.org/10.3390/cells12010079