

The Structure, Function and Regulation of Protein Tyrosine Phosphatase Receptor Type J and Its Role in Diseases

 , ,
, ,
Abstract
:1. Introduction
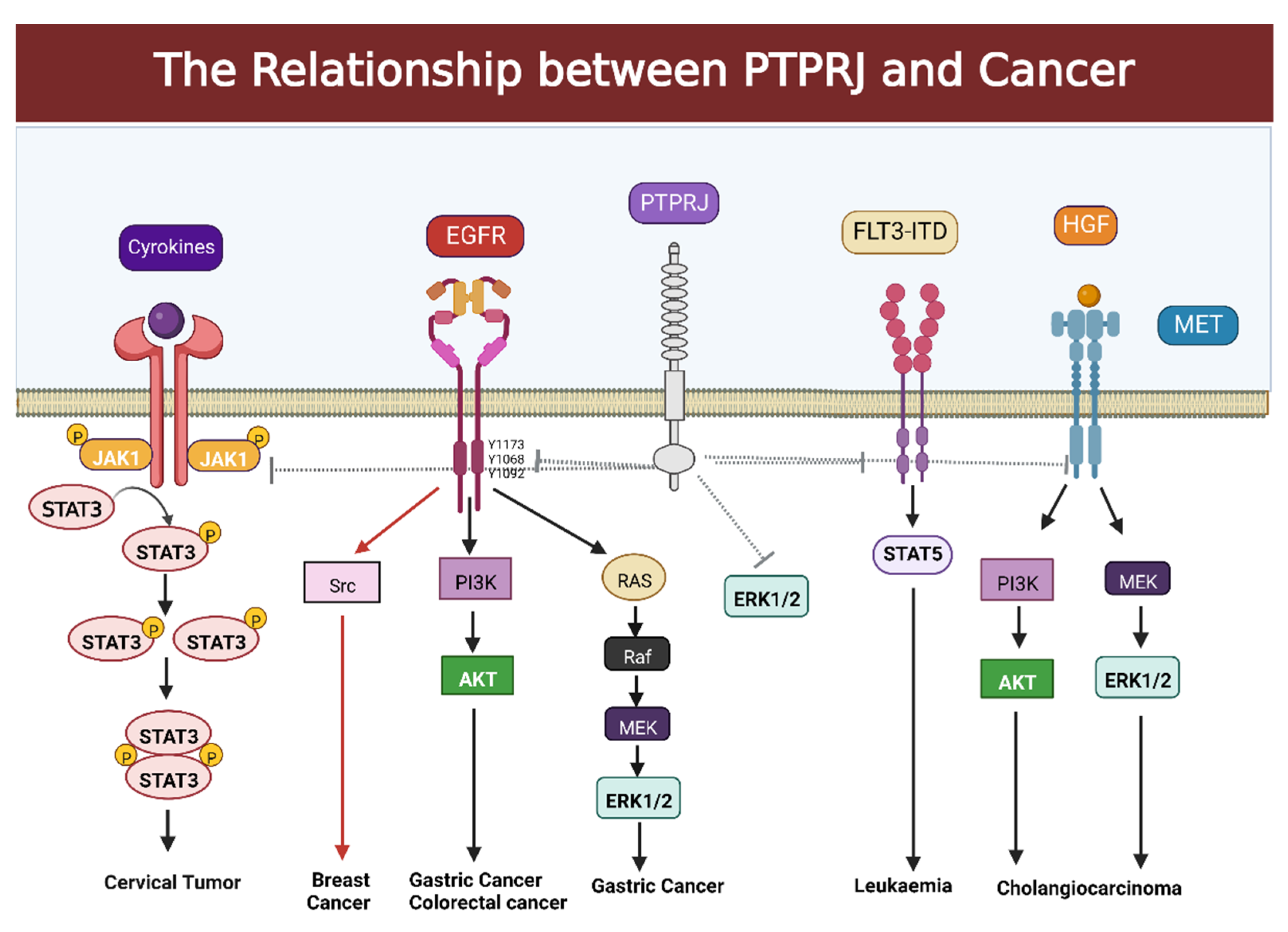
2. The Relationship between PTPRJ and Cancer
2.1. Association between PTPRJ and Gastric Cancer
2.2. Regulation of PTPRJ in Hepatocellular Carcinoma
2.3. PTPRJ and Its Relationship with Cholangiocarcinoma
2.4. Regulation of PTPRJ in Colorectal Cancer
2.5. PTPRJ and Its Relationship with Thyroid Cancer
2.6. PTPRJ and Its Relationship with Breast Cancer
2.7. PTPRJ and Its Relationship with Leukemia
2.8. PTPRJ and Its Relationship with Cervical Tumor
2.9. PTPRJ in Other Cancers
3. Contribution of PTPRJ to the Regulation of Metabolism
3.1. PTPRJ and Its Relationship with Insulin Resistance and Type 2 Diabetes
3.2. PTPRJ and Its Relationship with Obesity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
4. Contributions of PTPRJ to Axon Projection and Neurological Disorders
4.1. The Function of PTPRJ in Visual Topographic Map Formation
4.2. The Relationship between PTPRJ and Neurological Disorders
5. The Role of PTPRJ in Osteogenesis
6. The Relationship between PTPRJ and Platelets
7. The Relationship between PTPRJ and Immune Function
8. PTPRJ May Be a Potential Therapeutic Target
9. Concluding Remarks and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Waldt, N.; Scharnetzki, D.; Kesseler, C.; Kirches, E.; Stroscher, N.; Böhmer, F.-D.; Mawrin, C. Loss of PTPRJ/DEP-1 enhances NF2/Merlin-dependent meningioma development. J. Neurol. Sci. 2019, 408, 116553. [Google Scholar] [CrossRef]
- Spalinger, M.R.; McCole, D.F.; Rogler, G.; Scharl, M. Role of protein tyrosine phosphatases in regulating the immune system: Implications for chronic intestinal inflammation. Inflamm. Bowel Dis. 2015, 21, 645–655. [Google Scholar] [CrossRef]
- Bixby, J.L. Ligands and Signaling Through Receptor-Type Tyrosine Phosphatases. IUBMB Life 2001, 51, 157–163. [Google Scholar] [CrossRef]
- Bongard, R.D.; Lepley, M.; Thakur, K.; Talipov, M.R.; Nayak, J.; Lipinski, R.A.J.; Bohl, C.; Sweeney, N.; Ramchandran, R.; Rathore, R.; et al. Serendipitous discovery of light-induced (In Situ) formation of an Azo-bridged dimeric sulfonated naphthol as a potent PTP1B inhibitor. BMC Biochem. 2017, 18, 10. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Iglesias, A.; Schmoll, M. Protein phosphatases regulate growth, development, cellulases and secondary metabolism in Trichoderma reesei. Sci. Rep. 2019, 9, 10995. [Google Scholar] [CrossRef] [Green Version]
- Baek, M.; Kim, D.J. Protein Tyrosine Signaling and its Potential Therapeutic Implications in Carcinogenesis. Curr. Pharm. Des. 2017, 23, 4226–4246. [Google Scholar] [CrossRef]
- Ohtake, Y.; Saito, A.; Li, S. Diverse functions of protein tyrosine phosphatase σ in the nervous and immune systems. Exp. Neurol. 2018, 302, 196–204. [Google Scholar] [CrossRef]
- Takahashi, K.; Matafonov, A.; Sumarriva, K.; Ito, H.; Lauhan, C.; Zemel, D.; Tsuboi, N.; Chen, J.; Reynolds, A.; Takahashi, T. CD148 Tyrosine Phosphatase Promotes Cadherin Cell Adhesion. PLoS ONE 2014, 9, e112753. [Google Scholar] [CrossRef] [Green Version]
- Mohebiany, A.N.; Nikolaienko, R.M.; Bouyain, S.; Harroch, S. Receptor-type tyrosine phosphatase ligands: Looking for the needle in the haystack. FEBS J. 2012, 280, 388–400. [Google Scholar] [CrossRef] [Green Version]
- Shintani, T.; Higashi, S.; Suzuki, R.; Takeuchi, Y.; Ikaga, R.; Yamazaki, T.; Kobayashi, K.; Noda, M. PTPRJ Inhibits Leptin Signaling, and Induction of PTPRJ in the Hypothalamus Is a Cause of the Development of Leptin Resistance. Sci. Rep. 2017, 7, 11627. [Google Scholar] [CrossRef]
- Lu, Q.; Li, Q.; Lu, Q. Regulation of phagocytosis by TAM receptors and their ligands. Front. Biol. 2010, 5, 227–237. [Google Scholar] [CrossRef] [Green Version]
- Besco, J.A.; Frostholm, A.; Popesco, M.C.; Burghes, A.H.; Rotter, A. Genomic organization and alternative splicing of the human and mouse RPTPρ genes. BMC Genom. 2001, 2, 1. [Google Scholar] [CrossRef]
- Jeon, M.; Zinn, K. R3 receptor tyrosine phosphatases: Conserved regulators of receptor tyrosine kinase signaling and tubular organ development. Semin. Cell Dev. Biol. 2015, 37, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Kiyama, T.; Weber, N.; Whitaker, C.M.; Pan, P.; Badea, T.C.; Massey, S.C.; Mao, C. Characterization of Tbr2-expressing retinal ganglion cells. J. Comp. Neurol. 2021, 529, 3513–3532. [Google Scholar] [CrossRef]
- Kovalenko, M.; Denner, K.; Sandström, J.; Persson, C.; Groβ, S.; Jandt, E.; Vilella, R.; Böhmer, F.; Östman, A. Site-selective Dephosphorylation of the Platelet-derived Growth Factor β-Receptor by the Receptor-like Protein-tyrosine Phosphatase DEP-1. J. Biol. Chem. 2000, 275, 16219–16226. [Google Scholar] [CrossRef] [Green Version]
- Kappert, K.; Paulsson, J.; Sparwel, J.; Leppänen, O.; Hellberg, C.; Östman, A.; Micke, P. Dynamic changes in the expression of DEP-1 and other PDGF receptor-antagonizing PTPs during onset and termination of neointima formation. FASEB J. 2006, 21, 523–534. [Google Scholar] [CrossRef]
- Tarcic, G.; Boguslavsky, S.K.; Wakim, J.; Kiuchi, T.; Liu, A.; Reinitz, F.; Nathanson, D.; Takahashi, T.; Mischel, P.S.; Ng, T.; et al. An Unbiased Screen Identifies DEP-1 Tumor Suppressor as a Phosphatase Controlling EGFR Endocytosis. Curr. Biol. 2009, 19, 1788–1798. [Google Scholar] [CrossRef] [Green Version]
- Lampugnani, M.G.; Orsenigo, F.; Gagliani, M.C.; Tacchetti, C.; Dejana, E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J. Cell Biol. 2006, 174, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Keane, M.M.; Lowrey, G.A.; Ettenberg, S.A.; Dayton, M.A.; Lipkowitz, S. The protein tyrosine phosphatase DEP-1 is induced during differentiation and inhibits growth of breast cancer cells. Cancer Res. 1996, 56, 4236–4243. [Google Scholar]
- Laczmanska, I.; Sasiadek, M.M. Meta-analysis of association between Arg326Gln (rs1503185) and Gln276Pro (rs1566734) polymorphisms of PTPRJ gene and cancer risk. J. Appl. Genet. 2019, 60, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Aya-Bonilla, C.; Benton, M.; Keane, C.; Marlton, P.; Lea, R.; Gandhi, M.K.; Griffiths, L.R. High-resolution loss of heterozygosity screening implicates PTPRJ as a potential tumor suppressor gene that affects susceptibility to non-hodgkin’s lymphoma. Genes Chromosom. Cancer 2013, 52, 467–479. [Google Scholar] [CrossRef]
- Fournier, P.; Dussault, S.; Fusco, A.; Rivard, A.; Royal, I. Tyrosine Phosphatase PTPRJ/DEP-1 Is an Essential Promoter of Vascular Permeability, Angiogenesis, and Tumor Progression. Cancer Res. 2016, 76, 5080–5091. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Tu, R.; Li, K.; Ye, P.; Cui, X. Tumor Suppressor PTPRJ Is a Target of miR-155 in Colorectal Cancer. J. Cell. Biochem. 2017, 118, 3391–3400. [Google Scholar] [CrossRef]
- Zhao, S.; Sedwick, D.; Wang, Z. Genetic alterations of protein tyrosine phosphatases in human cancers. Oncogene 2014, 34, 3885–3894. [Google Scholar] [CrossRef] [Green Version]
- Bilotta, A.; Dattilo, V.; D’Agostino, S.; Belviso, S.; Scalise, S.; Bilotta, M.; Gaudio, E.; Paduano, F.; Perrotti, N.; Florio, T.; et al. A novel splice variant of the protein tyrosine phosphatase PTPRJ that encodes for a soluble protein involved in angiogenesis. Oncotarget 2016, 8, 10091–10102. [Google Scholar] [CrossRef] [Green Version]
- Borges, L.G.; Seifert, R.A.; Grant, F.J.; Hart, C.E.; Disteche, C.M.; Edelhoff, S.; Solca, F.F.; Lieberman, M.A.; Lindner, V.; Fischer, E.H.; et al. Cloning and Characterization of Rat Density-Enhanced Phosphatase-1, a Protein Tyrosine Phosphatase Expressed by Vascular Cells. Circ. Res. 1996, 79, 570–580. [Google Scholar] [CrossRef]
- Smart, C.E.; Amiri, M.E.A.; Wronski, A.; Dinger, M.E.; Crawford, J.; Ovchinnikov, D.A.; Vargas, A.C.; Reid, L.; Simpson, P.T.; Song, S.; et al. Expression and Function of the Protein Tyrosine Phosphatase Receptor J (PTPRJ) in Normal Mammary Epithelial Cells and Breast Tumors. PLoS ONE 2012, 7, e40742. [Google Scholar] [CrossRef] [Green Version]
- Mita, Y.; Yasuda, Y.; Sakai, A.; Yamamoto, H.; Toyooka, S.; Gunduz, M.; Tanabe, S.; Naomoto, Y.; Ouchida, M.; Shimizu, K. Missense polymorphisms of PTPRJ and PTPN13 genes affect susceptibility to a variety of human cancers. J. Cancer Res. Clin. Oncol. 2009, 136, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Sumarriva, K.; Kim, R.; Jiang, R.; Brantley-Sieders, D.M.; Chen, J.; Mernaugh, R.L.; Takahashi, T. Determination of the CD148-Interacting Region in Thrombospondin-1. PLoS ONE 2016, 11, e0154916. [Google Scholar] [CrossRef] [Green Version]
- Tsoyi, K.; Liang, X.; De Rossi, G.; Ryter, S.W.; Xiong, K.; Chu, S.G.; Liu, X.; Ith, B.; Celada, L.J.; Romero, F.; et al. CD148 Deficiency in Fibroblasts Promotes the Development of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 204, 312–325. [Google Scholar] [CrossRef]
- Takahashi, T.; Takahashi, K.; John, P.L.S.; Fleming, P.A.; Tomemori, T.; Watanabe, T.; Abrahamson, D.R.; Drake, C.J.; Shirasawa, T.; Daniel, T.O. A Mutant Receptor Tyrosine Phosphatase, CD148, Causes Defects in Vascular Development. Mol. Cell. Biol. 2003, 23, 1817–1831. [Google Scholar] [CrossRef]
- Krüger, J.; Brachs, S.; Trappiel, M.; Kintscher, U.; Meyborg, H.; Wellnhofer, E.; Thöne-Reineke, C.; Stawowy, P.; Östman, A.; Birkenfeld, A.L.; et al. Enhanced insulin signaling in density-enhanced phosphatase-1 (DEP-1) knockout mice. Mol. Metab. 2015, 4, 325–336. [Google Scholar] [CrossRef]
- Schneble, N.; Müller, J.; Kliche, S.; Bauer, R.; Wetzker, R.; Böhmer, F.-D.; Wang, Z.-Q.; Müller, J.P. The protein-tyrosine phosphatase DEP-1 promotes migration and phagocytic activity of microglial cells in part through negative regulation of fyn tyrosine kinase. Glia 2016, 65, 416–428. [Google Scholar] [CrossRef]
- Honda, H.; Inazawa, J.; Nishida, J.; Yazaki, Y.; Hirai, H. Molecular cloning, characterization, and chromosomal localization of a novel protein-tyrosine phosphatase, HPTP eta. Blood 1994, 84. [Google Scholar]
- Ostman, A.; Yang, Q.; Tonks, N.K. Expression of DEP-1, a receptor-like protein-tyrosine-phosphatase, is enhanced with increasing cell density. Proc. Natl. Acad. Sci. USA 1994, 91, 9680–9684. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Chen, S.; Wang, X.; Zhou, L.; Chen, L. miR-204-5p promotes preeclampsia serum-induced injury in human umbilical vein endothelial cells through regulation of the PTPRJ/Notch axis. Pregnancy Hypertens. Int. J. Women’s Cardiovasc. Heal. 2022, 28, 100–108. [Google Scholar] [CrossRef]
- Paduano, F.; Dattilo, V.; Narciso, D.; Bilotta, A.; Gaudio, E.; Menniti, M.; Agosti, V.; Palmieri, C.; Perrotti, N.; Fusco, A.; et al. Protein tyrosine phosphatase PTPRJ is negatively regulated by microRNA-328. FEBS J. 2012, 280, 401–412. [Google Scholar] [CrossRef]
- Östman, A.; Hellberg, C.; Böhmer, F.D. Protein-tyrosine phosphatases and cancer. Nat. Rev. Cancer 2006, 6, 307–320. [Google Scholar] [CrossRef]
- Ruivenkamp, C.A.; van Wezel, T.; Zanon, C.; Stassen, A.P.; Vlcek, C.; Csikós, T.; Klous, A.M.; Tripodis, N.; Perrakis, A.; Boerrigter, L.; et al. Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat. Genet. 2002, 31, 295–300. [Google Scholar] [CrossRef]
- Petermann, A.; Haase, D.; Wetzel, A.; Balavenkatraman, K.K.; Tenev, T.; Guehrs, K.-H.; Friedrich, S.; Nakamura, M.; Mawrin, C.; Böhmer, F.-D. Loss of the Protein-Tyrosine Phosphatase DEP-1/PTPRJ Drives Meningioma Cell Motility. Brain Pathol. 2010, 21, 405–418. [Google Scholar] [CrossRef]
- Godfrey, R.; Arora, D.; Bauer, R.; Stopp, S.; Müller, J.P.; Heinrich, T.; Böhmer, S.-A.; Dagnell, M.; Schnetzke, U.; Scholl, S.; et al. Cell transformation by FLT3 ITD in acute myeloid leukemia involves oxidative inactivation of the tumor suppressor protein-tyrosine phosphatase DEP-1/ PTPRJ. Blood 2012, 119, 4499–4511. [Google Scholar] [CrossRef] [Green Version]
- Palka, H.L.; Park, M.; Tonks, N.K. Hepatocyte Growth Factor Receptor Tyrosine Kinase Met Is a Substrate of the Receptor Protein-tyrosine Phosphatase DEP-1. J. Biol. Chem. 2003, 278, 5728–5735. [Google Scholar] [CrossRef]
- Sacco, F.; Tinti, M.; Palma, A.; Ferrari, E.; Nardozza, A.P.; van Huijsduijnen, R.H.; Takahashi, T.; Castagnoli, L.; Cesareni, G. Tumor Suppressor Density-enhanced Phosphatase-1 (DEP-1) Inhibits the RAS Pathway by Direct Dephosphorylation of ERK1/2 Kinases. J. Biol. Chem. 2009, 284, 22048–22058. [Google Scholar] [CrossRef] [Green Version]
- Bloch, E.; Sikorski, E.L.; Pontoriero, D.; Day, E.K.; Berger, B.W.; Lazzara, M.J.; Thévenin, D. Disrupting the transmembrane domain–mediated oligomerization of protein tyrosine phosphatase receptor J inhibits EGFR-driven cancer cell phenotypes. J. Biol. Chem. 2019, 294, 18796–18806. [Google Scholar] [CrossRef]
- Sun, Y.; Li, S.; Yu, W.; Chen, C.; Liu, T.; Li, L.; Zhang, D.; Zhao, Z.; Gao, J.; Wang, X.; et al. CD148 Serves as a Prognostic Marker of Gastric Cancer and Hinders Tumor Progression by Dephosphorylating EGFR. J. Cancer 2020, 11, 2667–2678. [Google Scholar] [CrossRef]
- Bupathi, M.; Kaseb, A.O.; Meric-Bernstam, F.; Naing, A. Hepatocellular carcinoma: Where there is unmet need. Mol. Oncol. 2015, 9, 1501–1509. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Luo, X.; Yang, S.; Zhou, C.; Pan, F.; Li, Q. MicroRNA-328 enhances cellular motility through posttranscriptional regulation of PTPRJ in human hepatocellular carcinoma. OncoTargets Ther. 2015, 8, 3159–3167. [Google Scholar] [CrossRef] [Green Version]
- Dai, R.; Li, J.; Fu, J.; Chen, Y.; Wang, R.; Zhao, X.; Luo, T.; Zhu, J.; Ren, Y.; Cao, J.; et al. The Tyrosine Kinase c-Met Contributes to the Pro-tumorigenic Function of the p38 Kinase in Human Bile Duct Cholangiocarcinoma Cells. J. Biol. Chem. 2012, 287, 39812–39823. [Google Scholar] [CrossRef] [Green Version]
- Ferracini, R.; Longati, P.; Naldini, L.; Vigna, E.; Comoglio, P.M. Identification of the major autophosphorylation site of the Met/hepatocyte growth factor receptor tyrosine kinase. J. Biol. Chem. 1991, 266. [Google Scholar]
- Iuliano, R.; Le Pera, I.; Cristofaro, C.; Baudi, F.; Arturi, F.; Pallante, P.; Martelli, M.L.; Trapasso, F.; Chiariotti, L.; Fusco, A. The tyrosine phosphatase PTPRJ/DEP-1 genotype affects thyroid carcinogenesis. Oncogene 2004, 23, 8432–8438. [Google Scholar] [CrossRef] [Green Version]
- Spring, K.; Fournier, P.; Lapointe, L.; Chabot, C.; Roussy, J.; Pommey, S.; Stagg, J.; Royal, I. The protein tyrosine phosphatase DEP-1/PTPRJ promotes breast cancer cell invasion and metastasis. Oncogene 2015, 34, 5536–5547. [Google Scholar] [CrossRef]
- Doshi, K.A.; Trotta, R.; Natarajan, K.; Rassool, F.V.; Tron, A.E.; Huszar, D.; Perrotti, D.; Baer, M.R. Pim kinase inhibition sensitizes FLT3-ITD acute myeloid leukemia cells to topoisomerase 2 inhibitors through increased DNA damage and oxidative stress. Oncotarget 2016, 7, 48280–48295. [Google Scholar] [CrossRef] [Green Version]
- Kawase, T.; Nakazawa, T.; Eguchi, T.; Tsuzuki, H.; Ueno, Y.; Amano, Y.; Mori, M.; Yoshida, T. Effect of Fms-like tyrosine kinase 3 (FLT3) ligand (FL) on antitumor activity of gilteritinib, a FLT3 inhibitor, in mice xenografted with FL-overexpressing cells. Oncotarget 2019, 10, 6111–6123. [Google Scholar] [CrossRef]
- Kresinsky, A.; Bauer, R.; Schnöder, T.M.; Berg, T.; Meyer, D.; Ast, V.; König, R.; Serve, H.; Heidel, F.H.; Böhmer, F.-D.; et al. Loss of DEP-1 (Ptprj) promotes myeloproliferative disease in FLT3-ITD acute myeloid leukemia. Haematologica 2018, 103, e505–e509. [Google Scholar] [CrossRef]
- Yan, C.-M.; Zhao, Y.-L.; Cai, H.-Y.; Miao, G.-Y.; Ma, W. Blockage of PTPRJ promotes cell growth and resistance to 5-FU through activation of JAK1/STAT3 in the cervical carcinoma cell line C33A. Oncol. Rep. 2015, 33, 1737–1744. [Google Scholar] [CrossRef] [Green Version]
- Scoles, D.R.; Nguyen, V.D.; Qin, Y.; Sun, C.-X.; Morrison, H.; Gutmann, D.H.; Pulst, S.-M. Neurofibromatosis 2 (NF2) tumor suppressor schwannomin and its interacting protein HRS regulate STAT signaling. Hum. Mol. Genet. 2002, 11, 3179–3189. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Zhang, Y. Integrated Analysis of Immunity- and Ferroptosis-Related Biomarker Signatures to Improve the Prognosis Prediction of Hepatocellular Carcinoma. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Paduano, F.; Ortuso, F.; Campiglia, P.; Raso, C.; Iaccino, E.; Gaspari, M.; Gaudio, E.; Mangone, G.; Carotenuto, A.; Bilotta, A.; et al. Isolation and Functional Characterization of Peptide Agonists of PTPRJ, a Tyrosine Phosphatase Receptor Endowed with Tumor Suppressor Activity. ACS Chem. Biol. 2012, 7, 1666–1676. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, M.; Carr, B.I. Involvement of receptor tyrosine phosphatase DEP-1 mediated PI3K-cofilin signaling pathway in Sorafenib-induced cytoskeletal rearrangement in hepatoma cells. J. Cell. Physiol. 2010, 224, 559–565. [Google Scholar] [CrossRef]
- Arora, D.; Stopp, S.; Böhmer, S.-A.; Schons, J.; Godfrey, R.; Masson, K.; Razumovskaya, E.; Rönnstrand, L.; Tänzer, S.; Bauer, R.; et al. Protein-tyrosine Phosphatase DEP-1 Controls Receptor Tyrosine Kinase FLT3 Signaling. J. Biol. Chem. 2011, 286, 10918–10929. [Google Scholar] [CrossRef] [Green Version]
- Boccaccio, C.; Comoglio, P.M. Invasive growth: A MET-driven genetic programme for cancer and stem cells. Nat. Cancer 2006, 6, 637–645. [Google Scholar] [CrossRef]
- Guo, X.; Liu, G.; Xie, X.; Li, J.; Hou, Z.; Gu, Y.; Yu, L. Expressions of CD23, IL-17 and MMP-9 in Patients with Colorectal Cancer. Iran. J. Public Health 2020. [Google Scholar] [CrossRef]
- Katsuyama, A.; Konno, T.; Shimoyama, S.; Kikuchi, H. The Mycotoxin Patulin Decreases Expression of Density-Enhanced Phosphatase-1 by Down-Regulating PPARγ in Human Colon Cancer Cells. Tohoku J. Exp. Med. 2014, 233, 265–274. [Google Scholar] [CrossRef]
- Balavenkatraman, K.K.; Jandt, E.; Friedrich, K.; Kautenburger, T.; Pool-Zobel, B.L.; Östman, A.; Böhmer, F.D. DEP-1 protein tyrosine phosphatase inhibits proliferation and migration of colon carcinoma cells and is upregulated by protective nutrients. Oncogene 2006, 25, 6319–6324. [Google Scholar] [CrossRef] [Green Version]
- Bertazza, L.; Barollo, S.; Radu, C.M.; Cavedon, E.; Simioni, P.; Faggian, D.; Plebani, M.; Pelizzo, M.R.; Rubin, B.; Boscaro, M.; et al. Synergistic antitumour activity of RAF265 and ZSTK474 on human TT medullary thyroid cancer cells. J. Cell. Mol. Med. 2015, 19, 2244–2252. [Google Scholar] [CrossRef]
- Trapasso, F.; Iuliano, R.; Boccia, A.; Stella, A.; Visconti, R.; Bruni, P.; Baldassarre, G.; Santoro, M.; Viglietto, G.; Fusco, A. Rat Protein Tyrosine Phosphatase η Suppresses the Neoplastic Phenotype of Retrovirally Transformed Thyroid Cells through the Stabilization of p27 Kip1. Mol. Cell. Biol. 2000, 20, 9236–9246. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, K.; Han, S.; Tian, Y.; Hu, P.; Xu, X.; He, Y.; Pan, W.; Gao, Y.; Zhang, Z.; et al. Chlorotoxin targets ERα/VASP signaling pathway to combat breast cancer. Cancer Med. 2019, 8, 1679–1693. [Google Scholar] [CrossRef] [Green Version]
- Lesueur, F.; Pharoah, P.D.; Laing, S.; Ahmed, S.; Jordan, C.; Smith, P.L.; Luben, R.; Wareham, N.J.; Easton, D.F.; Dunning, A.M.; et al. Allelic association of the human homologue of the mouse modifier Ptprj with breast cancer. Hum. Mol. Genet. 2005, 14, 2349–2356. [Google Scholar] [CrossRef] [Green Version]
- Huchko, M.; Adewumi, K.; Oketch, S.; Saduma, I.; Bukusi, E. ‘I’m here to save my life’: A qualitative study of experiences navigating a cryotherapy referral system for human papillomavirus-positive women in western Kenya. BMJ Open 2019, 9, e028669. [Google Scholar] [CrossRef] [Green Version]
- Roychowdhury, A.; Basu, M.; Pal, D.; Dutta, P.; Samadder, S.; Mondal, R.; Roy, A.K.; Roychoudhury, S.; Panda, C.K. PTPRJ is downregulated in cervical squamous cell carcinoma. J. Genet. 2022, 101, 29. [Google Scholar] [CrossRef]
- Áyen, Á.; Jimenez Martinez, Y.; Boulaiz, H. Targeted Gene Delivery Therapies for Cervical Cancer. Cancers 2020, 12, 1301. [Google Scholar] [CrossRef]
- D’Agostino, S.; Lanzillotta, D.; Varano, M.; Botta, C.; Baldrini, A.; Bilotta, A.; Scalise, S.; Dattilo, V.; Amato, R.; Gaudio, E.; et al. The receptor protein tyrosine phosphatase PTPRJ negatively modulates the CD98hc oncoprotein in lung cancer cells. Oncotarget 2018, 9, 23334–23348. [Google Scholar] [CrossRef] [Green Version]
- Aya-Bonilla, C.; Camilleri, E.; Haupt, L.M.; Lea, R.; Gandhi, M.K.; Griffiths, L.R. In silico analyses reveal common cellular pathways affected by loss of heterozygosity (LOH) events in the lymphomagenesis of Non-Hodgkin’s lymphoma (NHL). BMC Genom. 2014, 15, 390. [Google Scholar] [CrossRef]
- Stiefel, P.; Vallejo-Vaz, A.J.; Morillo, S.G.; Villar, J. Role of the Renin-Angiotensin System and Aldosterone on Cardiometabolic Syndrome. Int. J. Hypertens. 2011, 2011, 685238. [Google Scholar] [CrossRef] [Green Version]
- Syafruddin, S.E.; Nazarie, W.F.W.M.; Moidu, N.A.; Soon, B.H.; Mohtar, M.A. Integration of RNA-Seq and proteomics data identifies glioblastoma multiforme surfaceome signature. BMC Cancer 2021, 21, 850. [Google Scholar] [CrossRef]
- Sevillano, J.; Sánchez-Alonso, M.; Pizarro-Delgado, J.; Ramos-Álvarez, M. Role of Receptor Protein Tyrosine Phosphatases (RPTPs) in Insulin Signaling and Secretion. Int. J. Mol. Sci. 2021, 22, 5812. [Google Scholar] [CrossRef]
- Krüger, J.; Wellnhofer, E.; Meyborg, H.; Stawowy, P.; Östman, A.; Kintscher, U.; Kappert, K. Inhibition of Src homology 2 domain-containing phosphatase 1 increases insulin sensitivity in high-fat diet-induced insulin-resistant mice. FEBS Open Bio. 2016, 6, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Li, X.C.; Zhuo, J.L. Current Insights and New Perspectives on the Roles of Hyperglucagonemia in Non-Insulin–Dependent Type 2 Diabetes. Curr. Hypertens. Rep. 2013, 15, 522–530. [Google Scholar] [CrossRef] [Green Version]
- Shintani, T.; Higashi, S.; Takeuchi, Y.; Gaudio, E.; Trapasso, F.; Fusco, A.; Noda, M. The R3 receptor-like protein tyrosine phosphatase subfamily inhibits insulin signalling by dephosphorylating the insulin receptor at specific sites. J. Biochem. 2015, 158, 235–243. [Google Scholar] [CrossRef]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Soliman, A.T.; Yasin, M.; Kassem, A. Leptin in pediatrics: A hormone from adipocyte that wheels several functions in children. Indian J. Endocrinol. Metab. 2012, 16, S577–S587. [Google Scholar] [CrossRef]
- Bates, S.H.; Stearns, W.H.; Dundon, T.A.; Schubert, M.; Tso, A.W.K.; Wang, Y.; Banks, A.S.; Lavery, H.J.; Haq, A.K.; Maratos-Flier, E.; et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature 2003, 421, 856–859. [Google Scholar] [CrossRef]
- Terazawa, S.; Takada, M.; Sato, Y.; Nakajima, H.; Imokawa, G. The Attenuated Secretion of Hyaluronan by UVA-Exposed Human Fibroblasts Is Associated with Up- and Downregulation of HYBID and HAS2 Expression via Activated and Inactivated Signaling of the p38/ATF2 and JAK2/STAT3 Cascades. Int. J. Mol. Sci. 2021, 22, 2057. [Google Scholar] [CrossRef]
- Yu, Y.; Shintani, T.; Takeuchi, Y.; Shirasawa, T.; Noda, M. Protein Tyrosine Phosphatase Receptor Type J (PTPRJ) Regulates Retinal Axonal Projections by Inhibiting Eph and Abl Kinases in Mice. J. Neurosci. 2018, 38, 8345–8363. [Google Scholar] [CrossRef] [Green Version]
- Shintani, T.; Ihara, M.; Sakuta, H.; Takahashi, H.; Watakabe, I.; Noda, M. Eph receptors are negatively controlled by protein tyrosine phosphatase receptor type O. Nat. Neurosci. 2006, 9, 761–769. [Google Scholar] [CrossRef]
- Sakuraba, J.; Shintani, T.; Tani, S.; Noda, M. Substrate Specificity of R3 Receptor-like Protein-tyrosine Phosphatase Subfamily toward Receptor Protein-tyrosine Kinases. J. Biol. Chem. 2013, 288, 23421–23431. [Google Scholar] [CrossRef] [Green Version]
- Agarwala, S.; Ramachandra, N.B. Risk homozygous haplotype regions for autism identifies population-specific ten genes for numerous pathways. Egypt. J. Neurol. Psychiatry Neurosurg. 2021, 57, 69. [Google Scholar] [CrossRef]
- Rangaraju, S.; Raza, S.A.; Li, N.X.; Betarbet, R.; Dammer, E.B.; Duong, D.; Lah, J.J.; Seyfried, N.T.; Levey, A.I. Differential Phagocytic Properties of CD45low Microglia and CD45high Brain Mononuclear Phagocytes—Activation and Age-Related Effects. Front. Immunol. 2018, 9, 405. [Google Scholar] [CrossRef] [Green Version]
- Shalev, M.; Arman, E.; Stein, M.; Cohen-Sharir, Y.; Brumfeld, V.; Kapishnikov, S.; Royal, I.; Tuckermann, J.; Elson, A. PTPRJ promotes osteoclast maturation and activity by inhibiting Cbl-mediated ubiquitination of NFATc1 in late osteoclastogenesis. FEBS J. 2021, 288, 4702–4723. [Google Scholar] [CrossRef]
- Wen, R.; Wang, D. PTPRJ: A novel inherited thrombocytopenia gene. Blood 2019, 133, 1272–1274. [Google Scholar] [CrossRef]
- Senis, Y.A.; Tomlinson, M.G.; Ellison, S.; Mazharian, A.; Lim, J.; Zhao, Y.; Kornerup, K.N.; Auger, J.M.; Thomas, S.G.; Dhanjal, T.; et al. The tyrosine phosphatase CD148 is an essential positive regulator of platelet activation and thrombosis. Blood 2009, 113, 4942–4954. [Google Scholar] [CrossRef] [Green Version]
- Nagy, Z.; Mori, J.; Ivanova, V.-S.; Mazharian, A.; Senis, Y.A. Interplay between the tyrosine kinases Chk and Csk and phosphatase PTPRJ is critical for regulating platelets in mice. Blood 2020, 135, 1574–1587. [Google Scholar] [CrossRef]
- Mori, J.; Nagy, Z.; Di Nunzio, G.; Smith, C.W.; Geer, M.J.; Al Ghaithi, R.; Van Geffen, J.P.; Heising, S.; Boothman, L.; Tullemans, B.M.; et al. Maintenance of murine platelet homeostasis by the kinase Csk and phosphatase CD148. Blood J. Am. Soc. Hematol. 2018, 131, 1122–1144. [Google Scholar] [CrossRef]
- Nagy, Z.; Vögtle, T.; Geer, M.J.; Mori, J.; Heising, S.; Di Nunzio, G.; Gareus, R.; Tarakhovsky, A.; Weiss, A.; Neel, B.G.; et al. The Gp1ba-Cre transgenic mouse: A new model to delineate platelet and leukocyte functions. Blood 2019, 133, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Marconi, C.; Di Buduo, C.A.; Barozzi, S.; Palombo, F.; Pardini, S.; Zaninetti, C.; Pippucci, T.; Noris, P.; Balduini, A.; Seri, M.; et al. SLFN14-related thrombocytopenia: Identification within a large series of patients with inherited thrombocytopenia. Thromb. Haemost. 2016, 115, 1076–1079. [Google Scholar] [CrossRef]
- Johnson, B.; Lowe, G.C.; Futterer, J.; Lordkipanidze, M.; MacDonald, D.; Simpson, M.A.; Sanchez-Guiu, I.; Drake, S.; Bem, D.; Leo, V.; et al. Whole exome sequencing identifies genetic variants in inherited thrombocytopenia with secondary qualitative function defects. Haematologica 2016, 101, 1170–1179. [Google Scholar] [CrossRef] [Green Version]
- Marconi, C.; Di Buduo, C.A.; LeVine, K.; Barozzi, S.; Faleschini, M.; Bozzi, V.; Palombo, F.; McKinstry, S.; Lassandro, G.; Giordano, P.; et al. Loss-of-function mutations in PTPRJ cause a new form of inherited thrombocytopenia. Blood 2019, 133, 1346–1357. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.W.; Doan, K.; Park, J.; Chau, A.H.; Zhang, H.; Lowell, C.A.; Weiss, A. Receptor-like Tyrosine Phosphatases CD45 and CD148 Have Distinct Functions in Chemoattractant-Mediated Neutrophil Migration and Response to S. aureus. Immunity 2011, 35, 757–769. [Google Scholar] [CrossRef] [Green Version]
- Skrzypczynska, K.M.; Zhu, J.W.; Weiss, A. Positive Regulation of Lyn Kinase by CD148 Is Required for B Cell Receptor Signaling in B1 but Not B2 B Cells. Immunity 2016, 45, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Goob, G.; Adrian, J.; Cossu, C.; Hauck, C.R. Phagocytosis mediated by the human granulocyte receptor CEACAM3 is limited by the receptor-type protein tyrosine phosphatase PTPRJ. J. Biol. Chem. 2022. [Google Scholar] [CrossRef]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef]
- Kellie, S.; Craggs, G.; Bird, I.N.; Jones, G.E. The tyrosine phosphatase DEP-1 induces cytoskeletal rearrangements, aberrant cell-substratum interactions and a reduction in cell proliferation. J. Cell Sci. 2004, 117, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Trapasso, F.; Yendamuri, S.; Dumon, K.R.; Iuliano, R.; Cesari, R.; Feig, B.; Seto, R.; Infante, L.; Ishii, H.; Vecchione, A.; et al. Restoration of receptor-type protein tyrosine phosphatase function inhibits human pancreatic carcinoma cell growth in vitro and in vivo. Carcinogenesis 2004, 25, 2107–2114. [Google Scholar] [CrossRef]
- Takahashi, T.; Takahashi, K.; Mernaugh, R.L.; Tsuboi, N.; Liu, H.; Daniel, T.O. A monoclonal antibody against CD148, a receptor-like tyrosine phosphatase, inhibits endothelial-cell growth and angiogenesis. Blood 2006, 108, 1234–1242. [Google Scholar] [CrossRef] [Green Version]
- Le, H.T.T.; Cho, Y.-C.; Cho, A.S. Inhibition of protein tyrosine phosphatase non-receptor type 2 by PTP inhibitor XIX: Its role as a multiphosphatase inhibitor. BMB Rep. 2017, 50, 329–334. [Google Scholar] [CrossRef] [Green Version]
- Frankson, R.; Yu, Z.-H.; Bai, Y.; Li, Q.; Zhang, R.-Y.; Zhang, Z.-Y. Therapeutic Targeting of Oncogenic Tyrosine Phosphatases. Cancer Res. 2017, 77, 5701–5705. [Google Scholar] [CrossRef] [Green Version]
- Kostrzewa, T.; Styszko, J.; Gorska-Ponikowska, M.; Sledzinski, T.; Kuban-Jankowska, A. Inhibitors of Protein Tyrosine Phosphatase PTP1B With Anticancer Potential. Anticancer Res. 2019, 39, 3379–3384. [Google Scholar] [CrossRef]
- Sivaganesh, V.; Promi, N.; Maher, S.; Peethambaran, B. Emerging Immunotherapies against Novel Molecular Targets in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 2433. [Google Scholar] [CrossRef]
- Kostrzewa, T.; Sahu, K.K.; Gorska-Ponikowska, M.; Tuszynski, J.A.; Kuban-Jankowska, A. Synthesis of small peptide compounds, molecular docking, and inhibitory activity evaluation against phosphatases PTP1B and SHP2. Drug Des. Dev. Ther. 2018, 12, 4139–4147. [Google Scholar] [CrossRef]
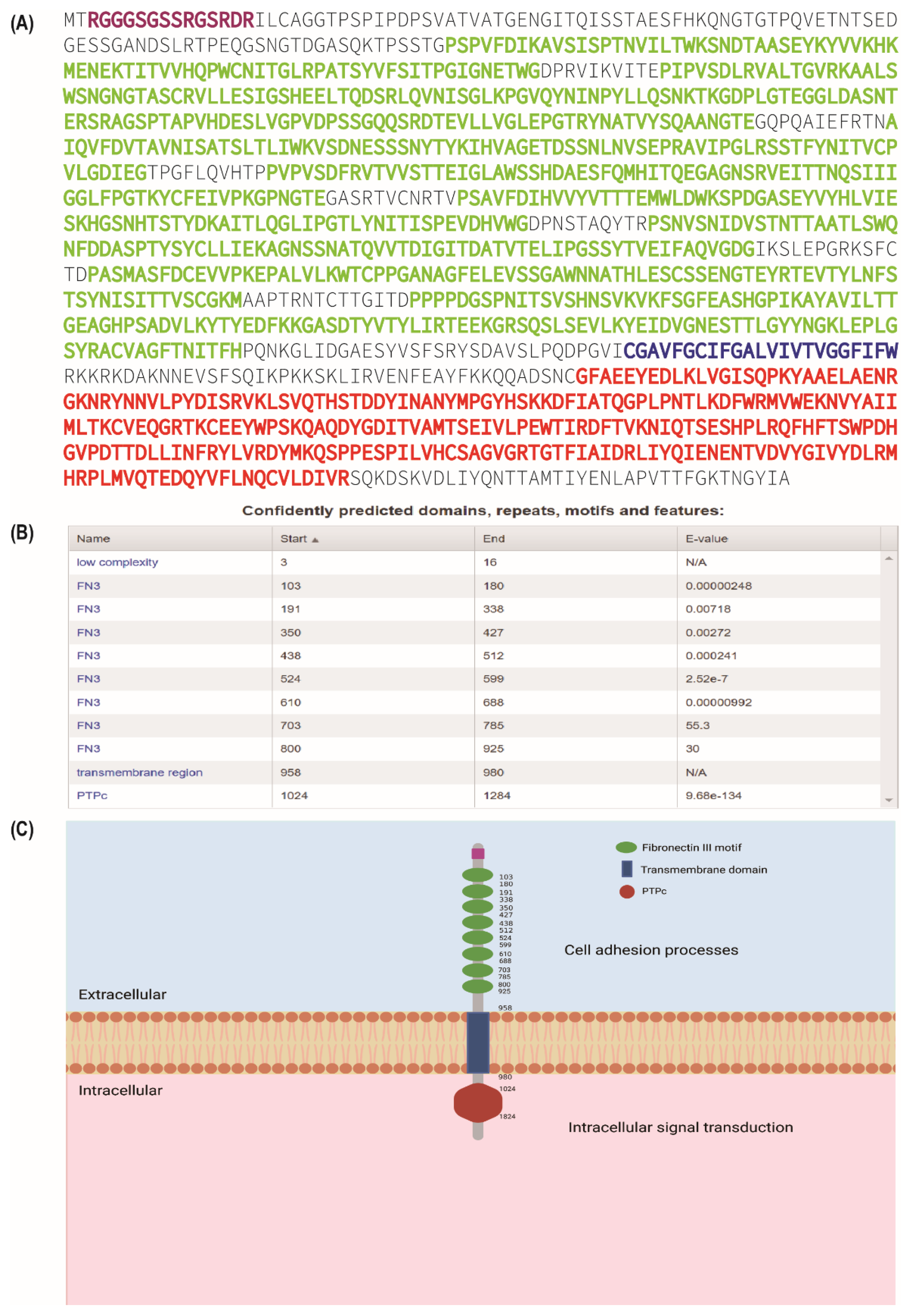


| Cancer | Mechanism | Cellular/Molecular Function | Reference |
|---|---|---|---|
| Gastric Cancer | Methylation in the 3′UTR region of the PTPRJ gene | EGFR, PI3K/AKT, and MEK/ERK pathways | [45] |
| Hepatocellular Carcinoma | MiR-328 acts directly on the 3′-UTR of PTPRJ | Not identified | [47] |
| Cholangiocarcinoma | c-Met dephosphorylation | PI3K/Akt and MEK/ERK | [42,48,49] |
| Colorectal Cancer | LOH of PTPRJ | AKT | [23] |
| Thyroid Cancer | LOH of PTPRJ | Not identified | [50] |
| Breast Cancer | Deletion and/or mutation | SRC, EGFP | [51] |
| Leukemia | PTPRJ oxidation | FLT3 | [52,53,54] |
| Cervical tumor tissue | Decreasing the phosphorylation levels of JAK1 and STAT3 | JAK1/STAT3 | [55] |
| Human Meningiomas | LOH of PTPRJ | PDGF | [56] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Zhang, P.; Liu, C.; Wang, Y.; Deng, Y.; Dong, W.; Yu, Y. The Structure, Function and Regulation of Protein Tyrosine Phosphatase Receptor Type J and Its Role in Diseases. Cells 2023, 12, 8. https://doi.org/10.3390/cells12010008
Li H, Zhang P, Liu C, Wang Y, Deng Y, Dong W, Yu Y. The Structure, Function and Regulation of Protein Tyrosine Phosphatase Receptor Type J and Its Role in Diseases. Cells. 2023; 12(1):8. https://doi.org/10.3390/cells12010008
Chicago/Turabian StyleLi, Huiting, Peng Zhang, Cencen Liu, Yiwei Wang, Yan Deng, Wei Dong, and Yang Yu. 2023. "The Structure, Function and Regulation of Protein Tyrosine Phosphatase Receptor Type J and Its Role in Diseases" Cells 12, no. 1: 8. https://doi.org/10.3390/cells12010008
APA StyleLi, H., Zhang, P., Liu, C., Wang, Y., Deng, Y., Dong, W., & Yu, Y. (2023). The Structure, Function and Regulation of Protein Tyrosine Phosphatase Receptor Type J and Its Role in Diseases. Cells, 12(1), 8. https://doi.org/10.3390/cells12010008