
Dasatinib and Trametinib Promote Anti-Tumor Metabolic Activity
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Metabolomics
2.3. Cellular Assays
2.4. Statistical Analysis
3. Results
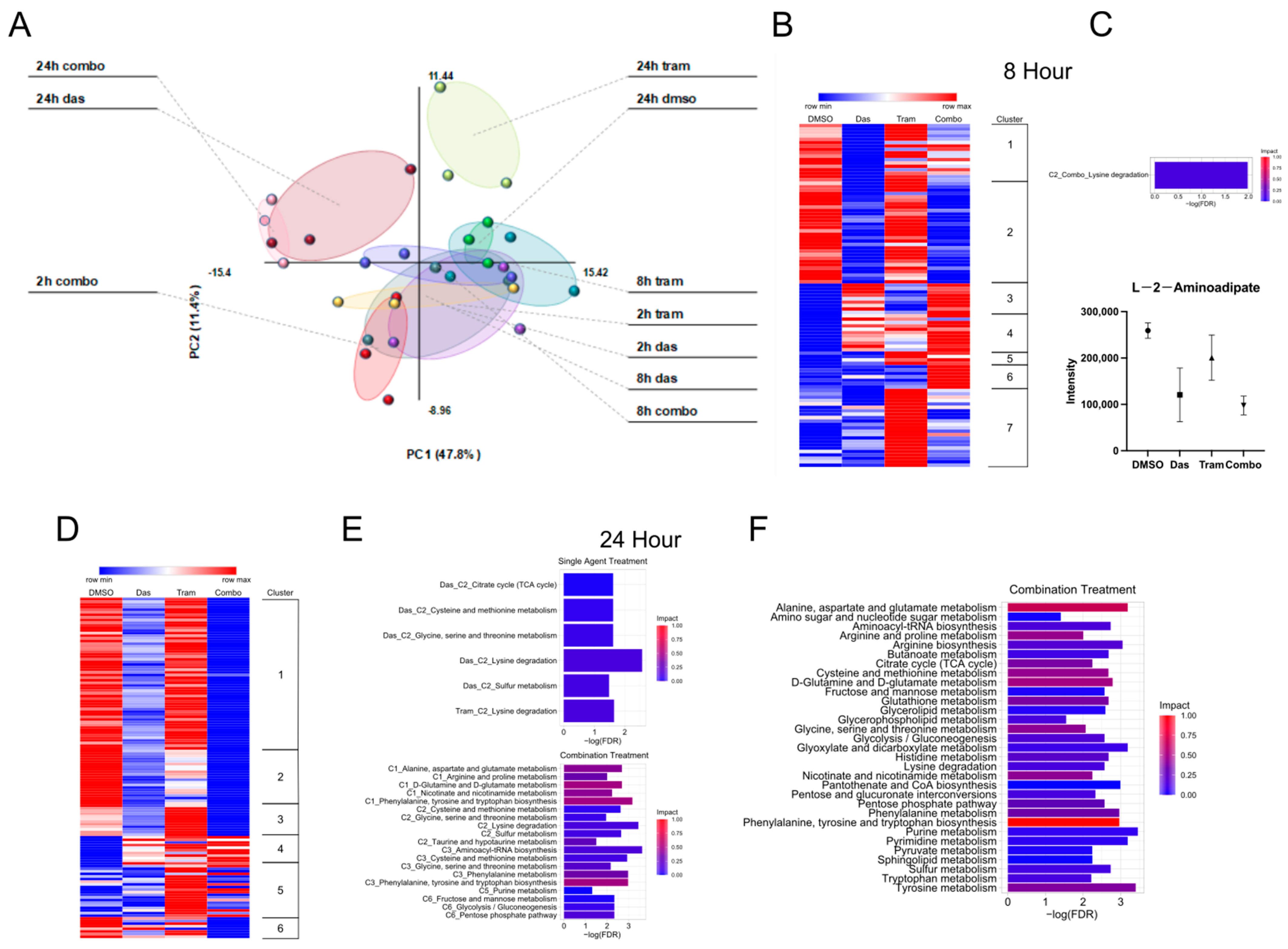
3.1. Effects of Dasatinib and Trametinib on Metabolic Activity in ATC Cells
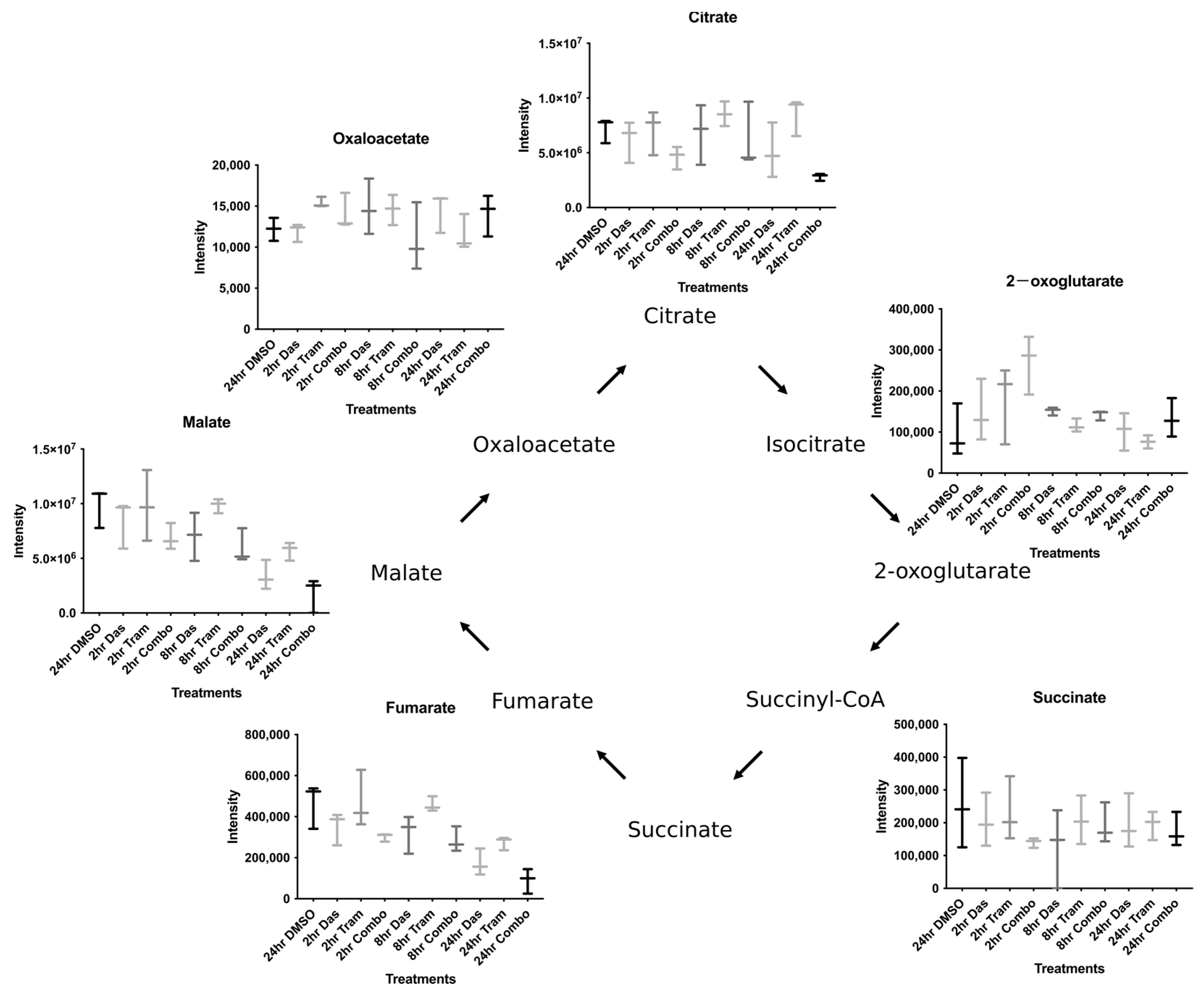
3.2. Combined Dasatinib and Trametinib Treatment Increases 2-oxoglutarate
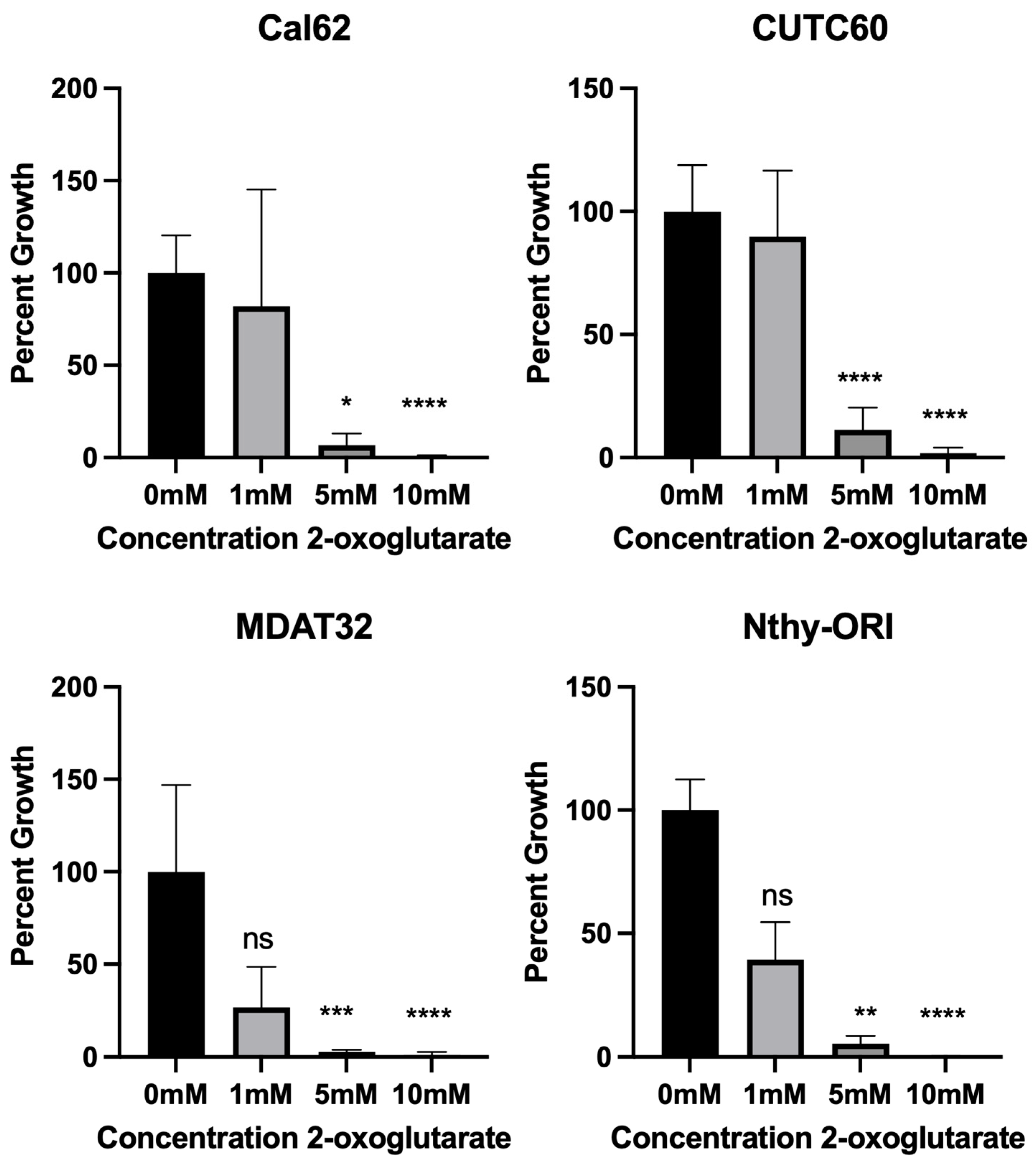
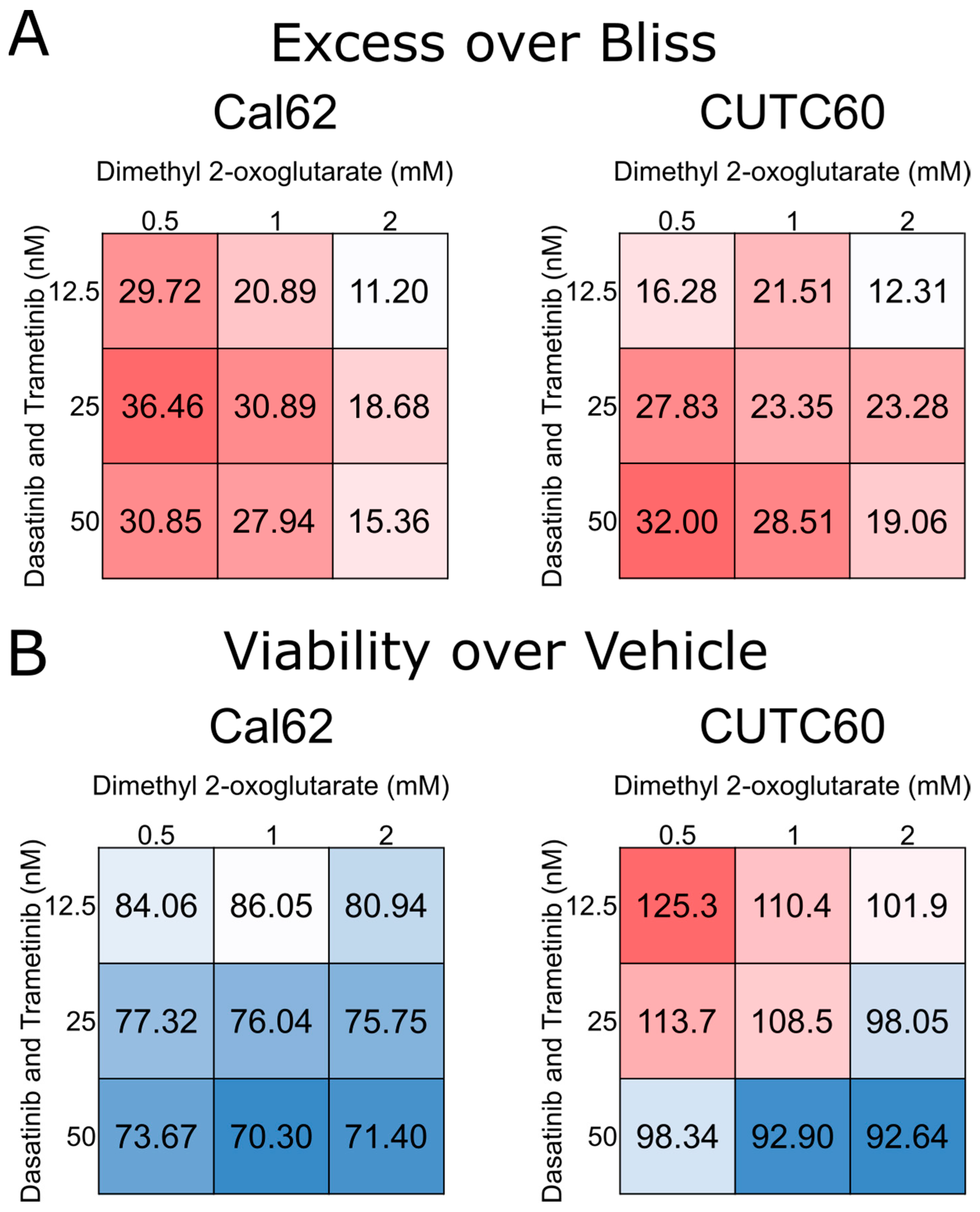
3.3. 2-oxoglutarate Synergizes with Dasatinib and Trametinib to Inhibit Proliferation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davies, L.; Welch, H.G. Current Thyroid Cancer Trends in the United States. JAMA Otolaryngol. Head Neck Surg. 2014, 140, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Nixon, I.J.; Whitcher, M.M.; Palmer, F.L.; Tuttle, R.M.; Shaha, A.R.; Shah, J.P.; Patel, S.G.; Ganly, I. The Impact of Distant Metastases at Presentation on Prognosis in Patients with Differentiated Carcinoma of the Thyroid Gland. Thyroid 2012, 22, 884–889. [Google Scholar] [CrossRef]
- Durante, C.; Haddy, N.; Baudin, E.; Leboulleux, S.; Hartl, D.; Travagli, J.P.; Caillou, B.; Ricard, M.; Lumbroso, J.D.; De Vathaire, F.; et al. Long-Term Outcome of 444 Patients with Distant Metastases from Papillary and Follicular Thyroid Carcinoma: Benefits and Limits of Radioiodine Therapy. J. Clin. Endocrinol. Metab. 2006, 91, 2892–2899. [Google Scholar] [CrossRef]
- Molinaro, E.; Romei, C.; Biagini, A.; Sabini, E.; Agate, L.; Mazzeo, S.; Materazzi, G.; Sellari-Franceschini, S.; Ribechini, A.; Torregrossa, L.; et al. Anaplastic Thyroid Carcinoma: From Clinicopathology to Genetics and Advanced Therapies. Nat. Rev. Endocrinol. 2017, 13, 644. [Google Scholar] [CrossRef]
- Xing, M. Molecular Pathogenesis and Mechanisms of Thyroid Cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Kang, H.B.; Fan, J.; Lin, R.; Elf, S.; Ji, Q.; Zhao, L.; Jin, L.; Seo, J.H.; Shan, C.; Arbiser, J.L.; et al. Metabolic Rewiring by Oncogenic Braf V600e Links Ketogenesis Pathway to Braf-Mek1 Signaling. Mol. Cell 2015, 59, 345–358. [Google Scholar] [CrossRef]
- Kimmelman, A.C. Metabolic Dependencies in Ras-Driven Cancers. Clin. Cancer Res. 2015, 21, 1828–1834. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Borkhuu, O.; Bao, W.; Yang, Y.T. Signaling Pathways in Thyroid Cancer and Their Therapeutic Implications. J. Clin. Med. Res. 2016, 8, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Michailidi, C.; Giaginis, C.; Stolakis, V.; Alexandrou, P.; Klijanienko, J.; Delladetsima, I.; Chatzizacharias, N.; Tsourouflis, G.; Theocharis, S. Evaluation of Fak and Src Expression in Human Benign and Malignant Thyroid Lesions. Pathol. Oncol. Res. 2010, 16, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.M.; Jing, X.; Pike, L.A.; Zhou, Q.; Lim, D.J.; Sams, S.B.; Lund, G.S.; Sharma, V.; Haugen, B.R.; Schweppe, R.E. Targeted Inhibition of Src Kinase with Dasatinib Blocks Thyroid Cancer Growth and Metastasis. Clin. Cancer Res. 2012, 18, 3580–3591. [Google Scholar] [CrossRef] [PubMed]
- Beadnell, T.C.; Nassar, K.W.; Rose, M.M.; Clark, E.G.; Danysh, B.P.; Hofmann, M.-C.; Pozdeyev, N.; Schweppe, R.E. Src-Mediated Regulation of the Pi3k Pathway in Advanced Papillary and Anaplastic Thyroid Cancer. Oncogenesis 2018, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Cai, Q.; Shenoy, A.K.; Lim, S.; Zhang, Y.; Charles, S.; Tarrash, M.; Fu, X.; Kamarajugadda, S.; Trevino, J.G.; et al. Src Drives the Warburg Effect and Therapy Resistance by Inactivating Pyruvate Dehydrogenase through Tyrosine-289 Phosphorylation. Oncotarget 2016, 7, 25113–25124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, S.; Jiang, B.; Huang, L.; Ji, Z.; Li, X.; Zhou, H.; Han, A.; Chen, A.; Wu, Y.; et al. C-Src Phosphorylation and Activation of Hexokinase Promotes Tumorigenesis and Metastasis. Nat. Commun. 2017, 8, 13732. [Google Scholar] [CrossRef] [PubMed]
- Montero-Conde, C.; Ruiz-Llorente, S.; Dominguez, J.M.; Knauf, J.A.; Viale, A.; Sherman, E.J.; Ryder, M.; Ghossein, R.A.; Rosen, N.; Fagin, J.A. Relief of Feedback Inhibition of Her3 Transcription by Raf and Mek Inhibitors Attenuates Their Antitumor Effects in Braf-Mutant Thyroid Carcinomas. Cancer Discov. 2013, 3, 520–533. [Google Scholar] [CrossRef]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G.; et al. Dabrafenib and Trametinib Treatment in Patients with Locally Advanced or Metastatic Braf V600-Mutant Anaplastic Thyroid Cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H. Chronic Myeloid Leukemia: 2022 Update on Diagnosis, Therapy, and Monitoring. Am. J. Hematol. 2022, 97, 1236–1256. [Google Scholar] [CrossRef]
- Landa, I.; Pozdeyev, N.; Korch, C.; Marlow, L.A.; Smallridge, R.C.; Copland, J.A.; Henderson, Y.C.; Lai, S.Y.; Clayman, G.L.; Onoda, N.; et al. Comprehensive Genetic Characterization of Human Thyroid Cancer Cell Lines: A Validated Panel for Preclinical Studies. Clin. Cancer Res. 2019, 25, 3141–3151. [Google Scholar] [CrossRef] [PubMed]
- Henderson, Y.C.; Ahn, S.H.; Ryu, J.; Chen, Y.; Williams, M.D.; El-Naggar, A.K.; Gagea, M.; Schweppe, R.E.; Haugen, B.R.; Lai, S.Y.; et al. Development and Characterization of Six New Human Papillary Thyroid Carcinoma Cell Lines. J. Clin. Endocrinol. Metab. 2015, 100, E243–E252. [Google Scholar] [CrossRef] [PubMed]
- Nemkov, T.; Hansen, K.C.; D’Alessandro, A. A Three-Minute Method for High-Throughput Quantitative Metabolomics and Quantitative Tracing Experiments of Central Carbon and Nitrogen Pathways. Rapid Commun. Mass Spectrom. 2017, 31, 663–673. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Amelio, I.; Berkers, C.R.; Antonov, A.; Vousden, K.H.; Melino, G.; Zolla, L. Metabolic Effect of Tap63α: Enhanced Glycolysis and Pentose Phosphate Pathway, Resulting in Increased Antioxidant Defense. Oncotarget 2014, 5, 7722–7733. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Wishart, D.S.; Xia, J. Using Metaboanalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]
- Bliss, C.I. The Toxicity of Poisons Applied Jointly1. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Tennant, D.A.; Gottlieb, E. Hif Prolyl Hydroxylase-3 Mediates Alpha-Ketoglutarate-Induced Apoptosis and Tumor Suppression. J. Mol. Med. 2010, 88, 839–849. [Google Scholar] [CrossRef]
- Sica, V.; Pedro, J.M.B.-S.; Izzo, V.; Pol, J.; Pierredon, S.; Enot, D.; Durand, S.; Bossut, N.; Chery, A.; Souquere, S.; et al. Lethal Poisoning of Cancer Cells by Respiratory Chain Inhibition Plus Dimethyl A-Ketoglutarate. Cell Rep. 2019, 27, 820–834.e9. [Google Scholar] [CrossRef]
- Sasanakietkul, T.; Murtha, T.D.; Javid, M.; Korah, R.; Carling, T. Epigenetic Modifications in Poorly Differentiated and Anaplastic Thyroid Cancer. Mol. Cell. Endocrinol. 2018, 469, 23–37. [Google Scholar] [CrossRef]
- Faam, B.; Ghaffari, M.A.; Ghadiri, A.; Azizi, F. Epigenetic Modifications in Human Thyroid Cancer. Biomed. Rep. 2015, 3, 3–8. [Google Scholar] [CrossRef]
- Morris, J.P.; Yashinskie, J.J.; Koche, R.; Chandwani, R.; Tian, S.; Chen, C.C.; Baslan, T.; Marinkovic, Z.S.; Sánchez-Rivera, F.J.; Leach, S.D.; et al. A-Ketoglutarate Links P53 to Cell Fate During Tumour Suppression. Nature 2019, 573, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Baracco, E.E.; Castoldi, F.; Durand, S.; Enot, D.P.; Tadic, J.; Kainz, K.; Madeo, F.; Chery, A.; Izzo, V.; Maiuri, M.C.; et al. A-Ketoglutarate Inhibits Autophagy. Aging 2019, 11, 3418–3431. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Park, S.; Hall, M.N. Mtor Signalling and Cellular Metabolism Are Mutual Determinants in Cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Haga, Y.; Higashisaka, K.; Yang, L.; Sekine, N.; Lin, Y.; Tsujino, H.; Nagano, K.; Tsutsumi, Y. Inhibition of Akt/Mtor Pathway Overcomes Intrinsic Resistance to Dasatinib in Triple-Negative Breast Cancer. Biochem. Biophys. Res. Commun. 2020, 533, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.; Wankell, M.; Ho, V.; White, R.; Deo, N.; Devine, C.; Dewdney, B.; Bhathal, P.; Govaere, O.; Roskams, T.; et al. Targeting Mtor and Src Restricts Hepatocellular Carcinoma Growth in a Novel Murine Liver Cancer Model. PLoS ONE 2019, 14, e0212860. [Google Scholar] [CrossRef]
- Skoko, J.; Rožanc, J.; Charles, E.M.; Alexopoulos, L.G.; Rehm, M. Post-Treatment De-Phosphorylation of P53 Correlates with Dasatinib Responsiveness in Malignant Melanoma. BMC Cell Biol. 2018, 19, 28. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolf, E.L.; Beadnell, T.C.; Rose, M.M.; D’Alessandro, A.; Nemkov, T.; Hansen, K.C.; Schweppe, R.E. Dasatinib and Trametinib Promote Anti-Tumor Metabolic Activity. Cells 2023, 12, 1374. https://doi.org/10.3390/cells12101374
Bolf EL, Beadnell TC, Rose MM, D’Alessandro A, Nemkov T, Hansen KC, Schweppe RE. Dasatinib and Trametinib Promote Anti-Tumor Metabolic Activity. Cells. 2023; 12(10):1374. https://doi.org/10.3390/cells12101374
Chicago/Turabian StyleBolf, Eric L., Thomas C. Beadnell, Madison M. Rose, Angelo D’Alessandro, Travis Nemkov, Kirk C. Hansen, and Rebecca E. Schweppe. 2023. "Dasatinib and Trametinib Promote Anti-Tumor Metabolic Activity" Cells 12, no. 10: 1374. https://doi.org/10.3390/cells12101374