

Ginsentide TP1 Protects Hypoxia-Induced Dysfunction and ER Stress-Linked Apoptosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Extraction and Purification of TP1 and Other Ginsentides
2.3. Cell Culture
2.4. Confocal Microscopic-Based Cellular Uptake Analyses
2.5. Pulsed SILAC Labeling
2.6. Protein Extraction and Tryptic Digestion
2.7. Reverse-Phase Chromatography Fractionation and Liquid Chromatography with Tandem Mass Spectrometry
2.8. Mass Spectrometric Data Analysis and Bioinformatics
2.9. LDH Assay
2.10. MTT Assay
2.11. Monocyte–Epithelium Adhesion Assay
2.12. NO Detection
2.13. Western Blot Analysis
2.14. Cell Cycle Analysis
2.15. qRT-PCR
2.16. Apoptosis Detection
2.17. ROS Detection
2.18. Immunostaining
2.19. Aggregated Protein Isolation
2.20. Statistical Analyses
3. Results
3.1. TP1 Is a Hyperstable Nontoxic and Non-Membranolytic Cell-Penetrating Peptide
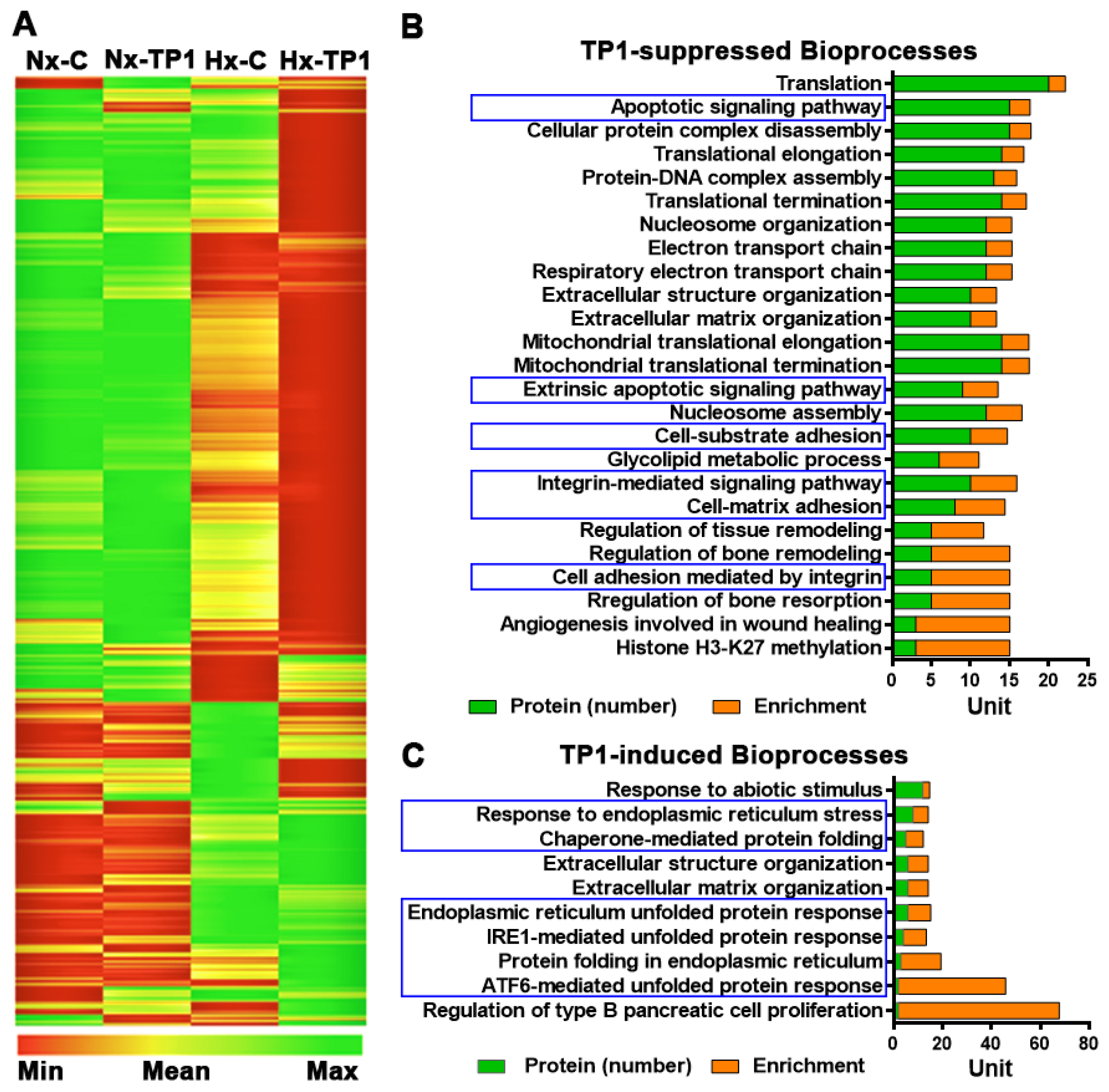
3.2. TP1 Alters Hypoxia-Modulated Endothelial Proteome
3.3. TP1 Limits Hypoxia-Induced Endothelial Activation and THP-1 Cell Adhesion
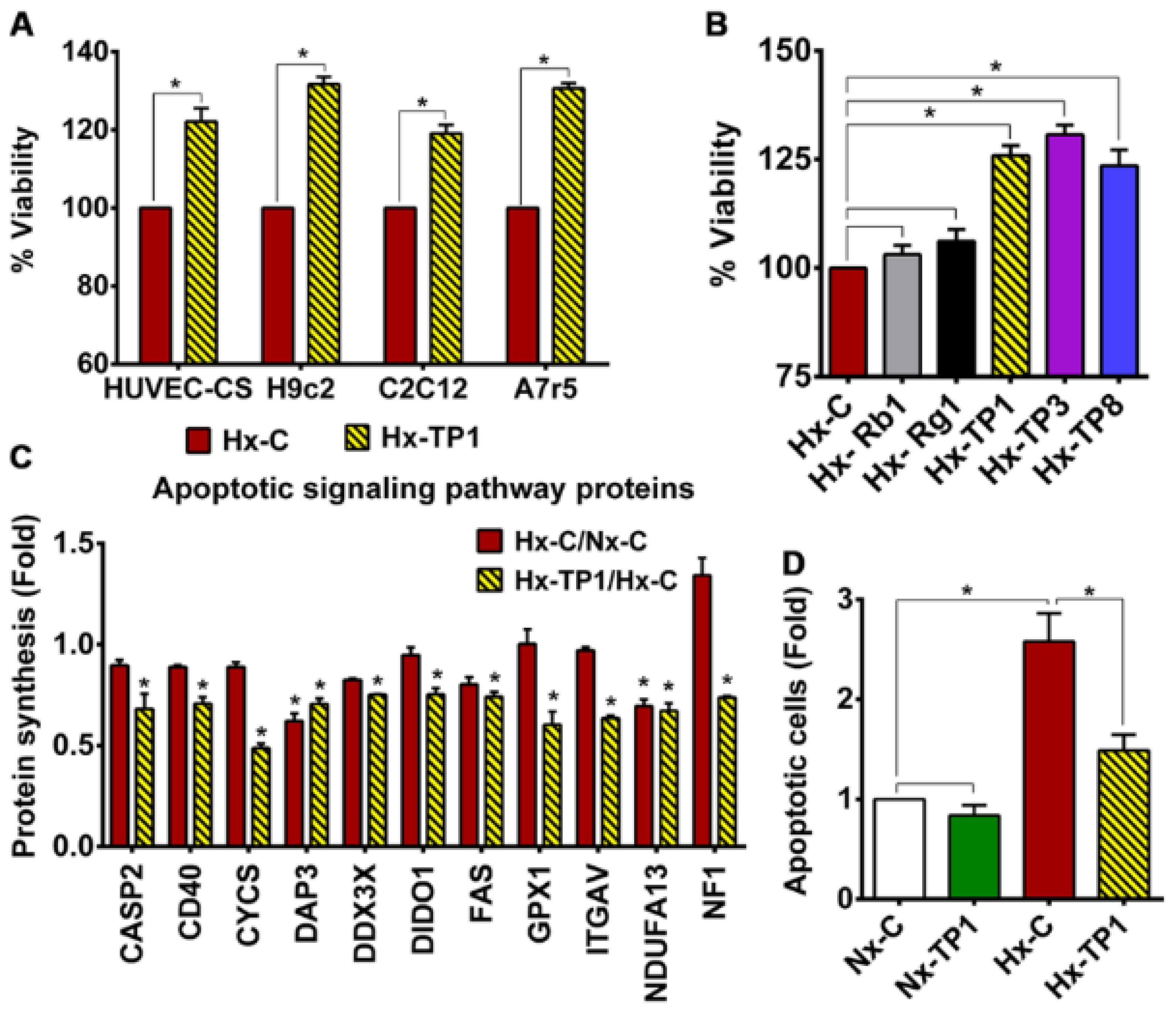
3.4. TP1 Promotes EC Survival by Antagonizing Apoptotic Signaling during Hypoxia
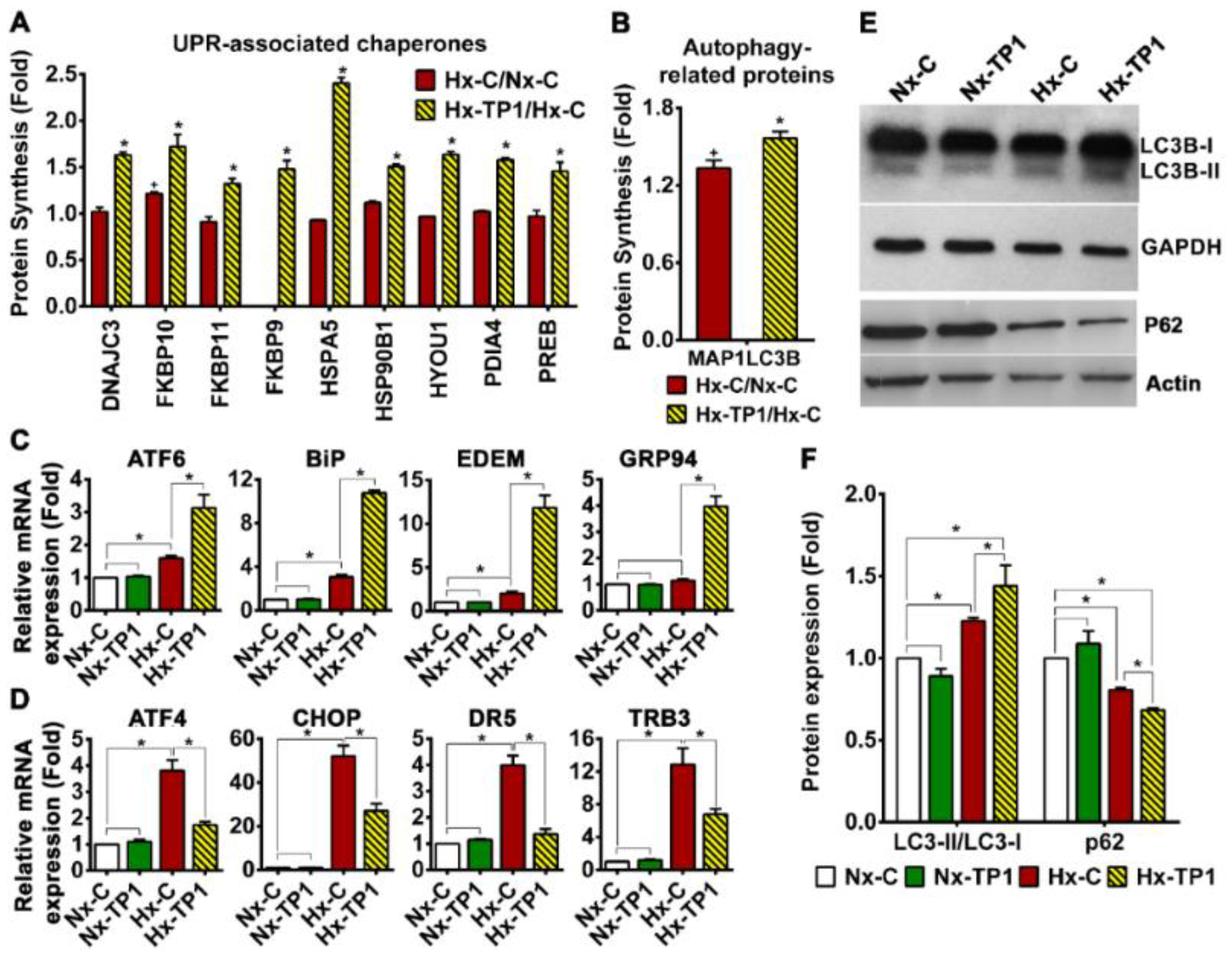
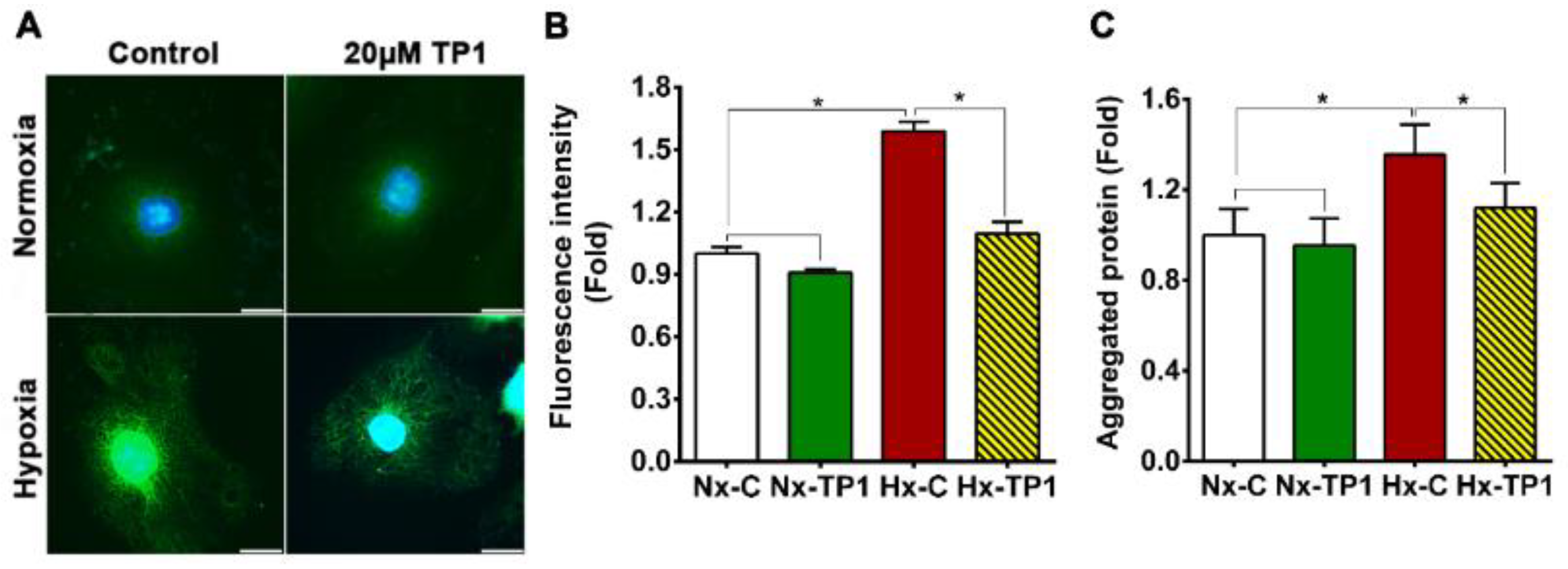
3.5. TP1 Treatment Rescues ECs from Hypoxia-Induced ER Stress
3.6. TP1 Restores NO Production and Reduces Reactive Oxygen Species Generation in Hypoxic ECs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoyos, C.M.; Melehan, K.L.; Liu, P.Y.; Grunstein, R.R.; Phillips, C.L. Does obstructive sleep apnea cause endothelial dysfunction? A critical review of the literature. Sleep Med. Rev. 2015, 20, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Ghofrani, H.A.; Weissmann, N.; Aldashev, A.; Zhao, L. Pathophysiology and treatment of high-altitude pulmonary vascular disease. Circulation 2015, 131, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.R.; Khayat, R.; Ponikowski, P.; Augostini, R.; Stellbrink, C.; Mianulli, M.; Abraham, W.T. Mechanisms and clinical consequences of untreated central sleep apnea in heart failure. J. Am. Coll. Cardiol. 2015, 65, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lin, S.; Zeng, Y. An Update on Obstructive Sleep Apnea for Atherosclerosis: Mechanism, Diagnosis, and Treatment. Front. Cardiovasc. Med. 2021, 8, 647071. [Google Scholar] [CrossRef]
- Haranczyk, M.; Konieczynska, M.; Plazak, W. Endothelial dysfunction in obstructive sleep apnea patients. Sleep Breath 2022, 26, 231–242. [Google Scholar] [CrossRef]
- Peracaula, M.; Torres, D.; Poyatos, P.; Luque, N.; Rojas, E.; Obrador, A.; Orriols, R.; Tura-Ceide, O. Endothelial Dysfunction and Cardiovascular Risk in Obstructive Sleep Apnea: A Review Article. Life 2022, 12, 537. [Google Scholar] [CrossRef]
- Wang, L.; Yin, J.; Nickles, H.T.; Ranke, H.; Tabuchi, A.; Hoffmann, J.; Tabeling, C.; Barbosa-Sicard, E.; Chanson, M.; Kwak, B.R.; et al. Hypoxic pulmonary vasoconstriction requires connexin 40-mediated endothelial signal conduction. J. Clin. Investig. 2012, 122, 4218–4230. [Google Scholar] [CrossRef]
- Burnstock, G.; Ralevic, V. Purinergic signaling and blood vessels in health and disease. Pharmacol. Rev. 2014, 66, 102–192. [Google Scholar] [CrossRef]
- Erlinge, D.; Burnstock, G. P2 receptors in cardiovascular regulation and disease. Purinergic Signal. 2008, 4, 1–20. [Google Scholar] [CrossRef]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef]
- Pober, J.S. Endothelial activation: Intracellular signaling pathways. Arthritis Res. Ther. 2002, 4 (Suppl. S3), S109. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. Responses to reductive stress in the cardiovascular system. Free. Radic. Biol. Med. 2017, 109, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Szocs, K. Endothelial dysfunction and reactive oxygen species production in ischemia/reperfusion and nitrate tolerance. Gen. Physiol. Biophys. 2004, 23, 265–295. [Google Scholar] [PubMed]
- Bourdier, G.; Flore, P.; Sanchez, H.; Pepin, J.L.; Belaidi, E.; Arnaud, C. High-intensity training reduces intermittent hypoxia-induced ER stress and myocardial infarct size. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H279–H289. [Google Scholar] [CrossRef]
- Battson, M.L.; Lee, D.M.; Gentile, C.L. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H355–H367. [Google Scholar] [CrossRef]
- Choi, Y.Y.; Kim, S.; Han, J.H.; Nam, D.H.; Park, K.M.; Kim, S.Y.; Woo, C.H. CHOP deficiency inhibits methylglyoxal-induced endothelial dysfunction. Biochem. Biophys. Res. Commun. 2016, 480, 362–368. [Google Scholar] [CrossRef]
- Wee, J.J.; Mee Park, K.; Chung, A.S. Biological Activities of Ginseng and Its Application to Human Health. In Herbal Medicine: Biomolecular and Clinical Aspects, 2nd ed.; Benzie, I.F.F., Wachtel-Galor, S., Eds.; CRC Press/Taylor & Francis LLC: Boca Raton, FL, USA, 2011. [Google Scholar]
- Wang, H.; Peng, D.; Xie, J. Ginseng leaf-stem: Bioactive constituents and pharmacological functions. Chin. Med. 2009, 4, 20. [Google Scholar] [CrossRef]
- Tam, J.P.; Nguyen, G.K.T.; Loo, S.; Wang, S.; Yang, D.; Kam, A. Ginsentides: Cysteine and Glycine-rich Peptides from the Ginseng Family with Unusual Disulfide Connectivity. Sci. Rep. 2018, 8, 16201. [Google Scholar] [CrossRef]
- Dutta, B.; Huang, J.; To, J.; Tam, J.P. LIR Motif-Containing Hyperdisulfide beta-Ginkgotide is Cytoprotective, Adaptogenic, and Scaffold-Ready. Molecules 2019, 24, 2417. [Google Scholar] [CrossRef]
- Loo, S.; Kam, A.; Li, B.B.; Feng, N.; Wang, X.; Tam, J.P. Discovery of Hyperstable Noncanonical Plant-Derived Epidermal Growth Factor Receptor Agonist and Analogs. J. Med. Chem. 2021, 64, 7746–7759. [Google Scholar] [CrossRef]
- Tam, J.P.; Wang, S.; Wong, K.H.; Tan, W.L. Antimicrobial Peptides from Plants. Pharmaceuticals 2015, 8, 711–757. [Google Scholar] [CrossRef] [PubMed]
- Loo, S.; Tay, S.V.; Kam, A.; Lee, W.; Tam, J.P. Hololectin Interdomain Linker Determines Asparaginyl Endopeptidase-Mediated Maturation of Antifungal Hevein-Like Peptides in Oats. Front. Plant Sci. 2022, 13, 899740. [Google Scholar] [CrossRef] [PubMed]
- Loo, S.; Tay, S.V.; Kam, A.; Tang, F.; Fan, J.S.; Yang, D.; Tam, J.P. Anti-Fungal Hevein-like Peptides Biosynthesized from Quinoa Cleavable Hololectins. Molecules 2021, 26, 5909. [Google Scholar] [CrossRef]
- Loo, S.; Kam, A.; Dutta, B.; Zhang, X.; Feng, N.; Sze, S.K.; Liu, C.-F.; Wang, X.; Tam, J.P. Decoding the Cure-all Effects of Ginseng. bioRxiv 2023. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzym. 2011, 490, 71–92. [Google Scholar] [CrossRef]
- Kam, A.; Loo, S.; Dutta, B.; Sze, S.K.; Tam, J.P. Plant-derived mitochondria-targeting cysteine-rich peptide modulates cellular bioenergetics. J. Biol. Chem. 2019, 294, 4000–4011. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Tse, S.W.; Xue, G.; Assisi, C.; Maqueda, A.S.; Ramon, G.P.X.; Low, J.K.; Kon, O.L.; Tay, C.Y.; Tam, J.P.; et al. Pulsed SILAC-based proteomic analysis unveils hypoxia- and serum starvation-induced de novo protein synthesis with PHD finger protein 14 (PHF14) as a hypoxia sensitive epigenetic regulator in cell cycle progression. Oncotarget 2019, 10, 2136–2150. [Google Scholar] [CrossRef]
- Finney, A.C.; Stokes, K.Y.; Pattillo, C.B.; Orr, A.W. Integrin signaling in atherosclerosis. Cell. Mol. Life Sci. CMLS 2017, 74, 2263–2282. [Google Scholar] [CrossRef]
- Chen, J.; Green, J.; Yurdagul, A., Jr.; Albert, P.; McInnis, M.C.; Orr, A.W. alphavbeta3 Integrins Mediate Flow-Induced NF-kappaB Activation, Proinflammatory Gene Expression, and Early Atherogenic Inflammation. Am. J. Pathol. 2015, 185, 2575–2589. [Google Scholar] [CrossRef]
- Akbar, N.; Digby, J.E.; Cahill, T.J.; Tavare, A.N.; Corbin, A.L.; Saluja, S.; Dawkins, S.; Edgar, L.; Rawlings, N.; Ziberna, K.; et al. Endothelium-derived extracellular vesicles promote splenic monocyte mobilization in myocardial infarction. JCI Insight 2017, 2, e93344. [Google Scholar] [CrossRef] [PubMed]
- Roebuck, K.A.; Finnegan, A. Regulation of intercellular adhesion molecule-1 (CD54) gene expression. J. Leukoc. Biol. 1999, 66, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Luo, Y.X.; Chen, H.Z.; Liu, D.P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5, 175. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.M.; Tentolouris, C.; Papageorgiou, N.; Stefanadis, C. The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species and endothelial function-role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharmacol. Toxicol. 2012, 110, 87–94. [Google Scholar] [CrossRef]
- Norouzirad, R.; Gonzalez-Muniesa, P.; Ghasemi, A. Hypoxia in Obesity and Diabetes: Potential Therapeutic Effects of Hyperoxia and Nitrate. Oxid. Med. Cell. Longev. 2017, 2017, 5350267. [Google Scholar] [CrossRef]
- Michell, B.J.; Griffiths, J.E.; Mitchelhill, K.I.; Rodriguez-Crespo, I.; Tiganis, T.; Bozinovski, S.; de Montellano, P.R.; Kemp, B.E.; Pearson, R.B. The Akt kinase signals directly to endothelial nitric oxide synthase. Curr. Biol. 1999, 9, 845–848. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Siamwala, J.H.; Chatterjee, S. eNOS phosphorylation in health and disease. Biochimie 2010, 92, 1186–1198. [Google Scholar] [CrossRef]
- Mohanan, P.; Subramaniyam, S.; Mathiyalagan, R.; Yang, D.C. Molecular signaling of ginsenosides Rb1, Rg1, and Rg3 and their mode of actions. J. Ginseng Res. 2018, 42, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Ma, Q.; Fan, C.; Chen, X.; Zhang, H.; Tang, M. Ginsenosides Rb1 and Rg1 Protect Primary Cultured Astrocytes against Oxygen-Glucose Deprivation/Reoxygenation-Induced Injury via Improving Mitochondrial Function. Int. J. Mol. Sci. 2019, 20, 6086. [Google Scholar] [CrossRef] [PubMed]
- Rajput, S.A.; Shaukat, A.; Rajput, I.R.; Kamboh, A.A.; Iqbal, Z.; Saeed, M.; Akhtar, R.W.; Shah, S.A.H.; Raza, M.A.; El Askary, A.; et al. Ginsenoside Rb1 prevents deoxynivalenol-induced immune injury via alleviating oxidative stress and apoptosis in mice. Ecotoxicol. Environ. Saf. 2021, 220, 112333. [Google Scholar] [CrossRef] [PubMed]
- Ratan, Z.A.; Haidere, M.F.; Hong, Y.H.; Park, S.H.; Lee, J.O.; Lee, J.; Cho, J.Y. Pharmacological potential of ginseng and its major component ginsenosides. J. Ginseng Res. 2021, 45, 199–210. [Google Scholar] [CrossRef]
- Choi, S.H.; Jung, S.W.; Lee, B.H.; Kim, H.J.; Hwang, S.H.; Kim, H.K.; Nah, S.Y. Ginseng pharmacology: A new paradigm based on gintonin-lysophosphatidic acid receptor interactions. Front. Pharmacol. 2015, 6, 245. [Google Scholar] [CrossRef]
- Leung, K.W.; Wong, A.S. Pharmacology of ginsenosides: A literature review. Chin. Med. 2010, 5, 20. [Google Scholar] [CrossRef]
- Schaefer, A.; van Duijn, T.J.; Majolee, J.; Burridge, K.; Hordijk, P.L. Endothelial CD2AP Binds the Receptor ICAM-1 To Control Mechanosignaling, Leukocyte Adhesion, and the Route of Leukocyte Diapedesis In Vitro. J. Immunol. 2017, 198, 4823–4836. [Google Scholar] [CrossRef]
- Anbarasan, C.; Bavanilatha, M.; Latchumanadhas, K.; Ajit Mullasari, S. ICAM-1 molecular mechanism and genome wide SNP’s association studies. Indian Heart J. 2015, 67, 282–287. [Google Scholar] [CrossRef]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A., Jr.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68. [Google Scholar] [CrossRef]
- Li, S.; Xu, J.; Yao, W.; Li, H.; Liu, Q.; Xiao, F.; Irwin, M.G.; Xia, Z.; Ruan, W. Sevoflurane pretreatment attenuates TNF-alpha-induced human endothelial cell dysfunction through activating eNOS/NO pathway. Biochem. Biophys. Res. Commun. 2015, 460, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Badiola, N.; Penas, C.; Minano-Molina, A.; Barneda-Zahonero, B.; Fado, R.; Sanchez-Opazo, G.; Comella, J.X.; Sabria, J.; Zhu, C.; Blomgren, K.; et al. Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis. 2011, 2, e149. [Google Scholar] [CrossRef] [PubMed]
- Thuerauf, D.J.; Marcinko, M.; Gude, N.; Rubio, M.; Sussman, M.A.; Glembotski, C.C. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ. Res. 2006, 99, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Kitakaze, M. ER stress in cardiovascular disease. J. Mol. Cell. Cardiol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Cimellaro, A.; Perticone, M.; Fiorentino, T.V.; Sciacqua, A.; Hribal, M.L. Role of endoplasmic reticulum stress in endothelial dysfunction. Nutr. Metab. Cardiovasc. Dis. NMCD 2016, 26, 863–871. [Google Scholar] [CrossRef]
- Takaki, A.; Morikawa, K.; Tsutsui, M.; Murayama, Y.; Tekes, E.; Yamagishi, H.; Ohashi, J.; Yada, T.; Yanagihara, N.; Shimokawa, H. Crucial role of nitric oxide synthases system in endothelium-dependent hyperpolarization in mice. J. Exp. Med. 2008, 205, 2053–2063. [Google Scholar] [CrossRef]
- Wu, S.; Gao, X.; Yang, S.; Meng, M.; Yang, X.; Ge, B. The role of endoplasmic reticulum stress in endothelial dysfunction induced by homocysteine thiolactone. Fundam. Clin. Pharmacol. 2015, 29, 252–259. [Google Scholar] [CrossRef]
- Oslowski, C.M.; Urano, F. The binary switch between life and death of endoplasmic reticulum-stressed beta cells. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 107–112. [Google Scholar] [CrossRef]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- De Pascali, F.; Hemann, C.; Samons, K.; Chen, C.A.; Zweier, J.L. Hypoxia and reoxygenation induce endothelial nitric oxide synthase uncoupling in endothelial cells through tetrahydrobiopterin depletion and S-glutathionylation. Biochemistry 2014, 53, 3679–3688. [Google Scholar] [CrossRef]
- Fish, J.E.; Yan, M.S.; Matouk, C.C.; St Bernard, R.; Ho, J.J.; Gavryushova, A.; Srivastava, D.; Marsden, P.A. Hypoxic repression of endothelial nitric-oxide synthase transcription is coupled with eviction of promoter histones. J. Biol. Chem. 2010, 285, 810–826. [Google Scholar] [CrossRef] [PubMed]
- Widmer, R.J.; Lerman, A. Endothelial dysfunction and cardiovascular disease. Glob. Cardiol. Sci. Pract. 2014, 2014, 291–308. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Watanabe, Y.; Moriya, Y.; Kawano, S.; Yamamoto, T.; Matsumoto, M.; Takami, T.; Kobayashi, D.; Araki, N.; Yoshizawa, A.C.; et al. jPOSTrepo: An international standard data repository for proteomes. Nucleic Acids Res. 2017, 45, D1107–D1111. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dutta, B.; Loo, S.; Kam, A.; Sze, S.K.; Tam, J.P. Ginsentide TP1 Protects Hypoxia-Induced Dysfunction and ER Stress-Linked Apoptosis. Cells 2023, 12, 1401. https://doi.org/10.3390/cells12101401
Dutta B, Loo S, Kam A, Sze SK, Tam JP. Ginsentide TP1 Protects Hypoxia-Induced Dysfunction and ER Stress-Linked Apoptosis. Cells. 2023; 12(10):1401. https://doi.org/10.3390/cells12101401
Chicago/Turabian StyleDutta, Bamaprasad, Shining Loo, Antony Kam, Siu Kwan Sze, and James P. Tam. 2023. "Ginsentide TP1 Protects Hypoxia-Induced Dysfunction and ER Stress-Linked Apoptosis" Cells 12, no. 10: 1401. https://doi.org/10.3390/cells12101401