
PPARγ Acetylation in Adipocytes Exacerbates BAT Whitening and Worsens Age-Associated Metabolic Dysfunction
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Studies
2.2. Western Blotting
2.3. Bone Processing and Analysis
2.4. Gene Expression
2.5. Hematoxylin and Eosin (H & E) Staining
2.6. Immunostaining of Ucp1
2.7. Statistics
2.8. Study Approval
3. Results
3.1. PPARγ Acetylation in Adipocytes Exacerbates the Age-Associated Metabolic Decline
3.2. PPARγ Acetylation Promotes Lipid Accumulation in BAT during Aging
3.3. Metabolic Improvements of Intermittent Fasting (IF) Are Dampened in Middle-Aged aKQ Mice
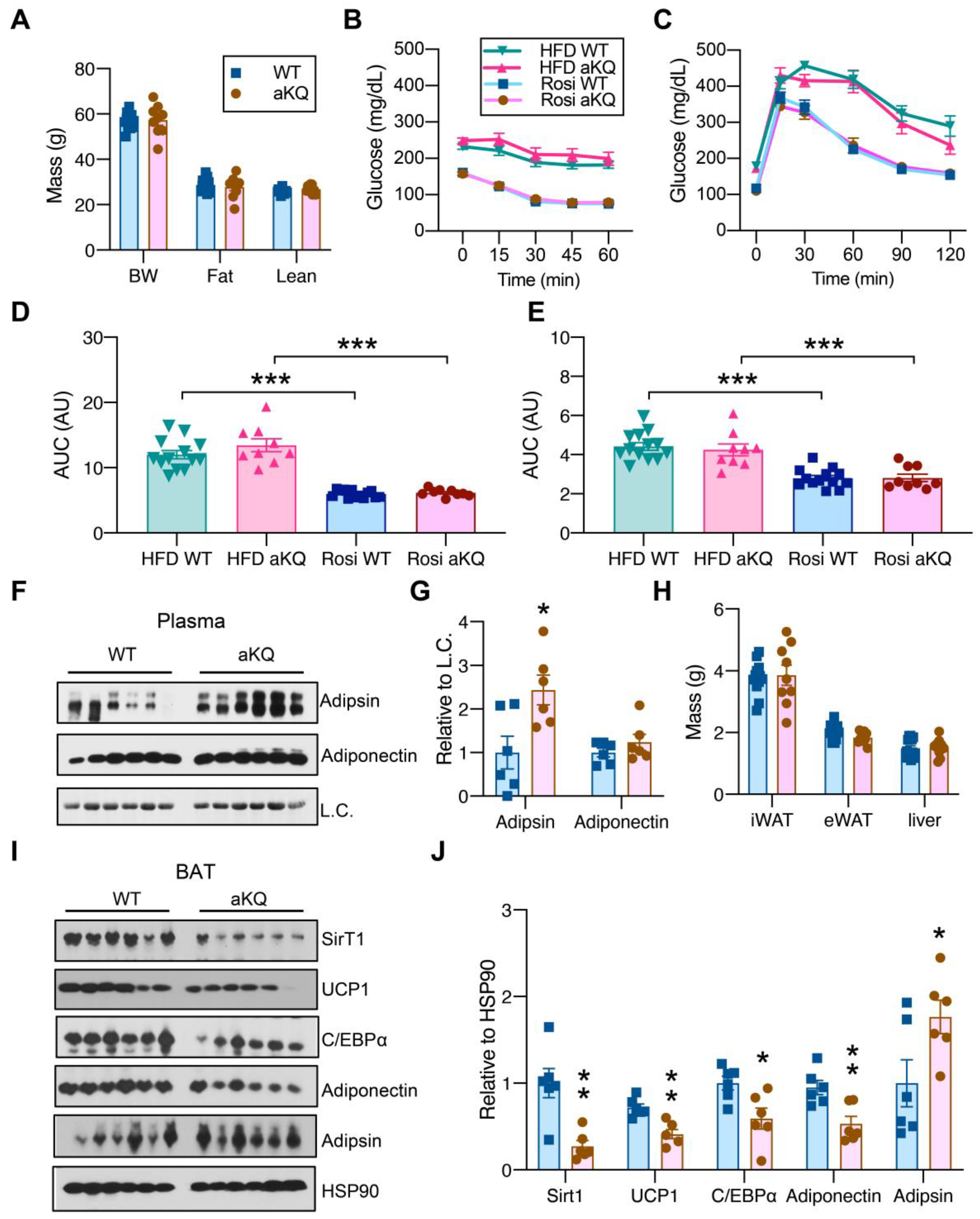
3.4. aKQ Mice Retain the Response to TZDs in Diet-Induced Obesity but with Impaired BAT Function
3.5. Activation of SirT1 Fails to Protect aKQ Mice from BAT Whitening
3.6. PPARγ Acetylation in Adipocytes Worsens TZD-Induced Bone Loss
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Santos, A.L.; Sinha, S. Obesity and aging: Molecular mechanisms and therapeutic approaches. Ageing Res. Rev. 2021, 67, 101268. [Google Scholar] [CrossRef] [PubMed]
- Bentley, R.A.; Ross, C.N.; O’Brien, M.J. Obesity, Metabolism, and Aging: A Multiscalar Approach. Prog. Mol. Biol. Transl. Sci. 2018, 155, 25–42. [Google Scholar] [CrossRef]
- Corbo, R.M.; Pinto, A.; Scacchi, R. Gender-specific association between FSHR and PPARG common variants and human longevity. Rejuvenation Res. 2013, 16, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Koutnikova, H.; Cock, T.A.; Watanabe, M.; Houten, S.M.; Champy, M.F.; Dierich, A.; Auwerx, J. Compensation by the muscle limits the metabolic consequences of lipodystrophy in PPAR gamma hypomorphic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 14457–14462. [Google Scholar] [CrossRef] [PubMed]
- Argmann, C.; Dobrin, R.; Heikkinen, S.; Auburtin, A.; Pouilly, L.; Cock, T.A.; Koutnikova, H.; Zhu, J.; Schadt, E.E.; Auwerx, J. Ppargamma2 is a key driver of longevity in the mouse. PLoS Genet. 2009, 5, e1000752. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Han, T.; Chu, X.; Lu, H.; Yang, X.; Zi, T.; Zhao, Y.; Wang, X.; Liu, Z.; Ruan, J.; et al. An isocaloric moderately high-fat diet extends lifespan in male rats and Drosophila. Cell Metab. 2021, 33, 581–597.e9. [Google Scholar] [CrossRef]
- Xu, L.; Ma, X.; Verma, N.; Perie, L.; Pendse, J.; Shamloo, S.; Josephson, A.M.; Wang, D.; Qiu, J.; Guo, M.; et al. PPARgamma agonists delay age-associated metabolic disease and extend longevity. Aging Cell 2020, 19, e13267. [Google Scholar] [CrossRef] [PubMed]
- Chandra, V.; Huang, P.; Hamuro, Y.; Raghuram, S.; Wang, Y.; Burris, T.P.; Rastinejad, F. Structure of the intact PPAR-gamma-RXR-α nuclear receptor complex on DNA. Nature 2008, 456, 350–356. [Google Scholar] [CrossRef]
- Lehrke, M.; Lazar, M.A. The many faces of PPARgamma. Cell 2005, 123, 993–999. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Uno, K. Peroxisome proliferator-activated receptor (PPAR alpha/gamma) agonists as a potential target to reduce cardiovascular risk in diabetes. Diabetes Vasc. Dis. Res. 2012, 9, 89–94. [Google Scholar] [CrossRef]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARgamma signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Mayoral, R.; Osborn, O.; McNelis, J.; Johnson, A.M.; Oh, D.Y.; Izquierdo, C.L.; Chung, H.; Li, P.; Traves, P.G.; Bandyopadhyay, G.; et al. Adipocyte SIRT1 knockout promotes PPARgamma activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Mol. Metab. 2015, 4, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Kim, J.B.; Sarraf, P.; Spiegelman, B.M. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science 1996, 274, 2100–2103. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Fan, L.; Chan, M.; Kraakman, M.J.; Yang, J.; Fan, Y.; Aaron, N.; Wan, Q.; Carrillo-Sepulveda, M.A.; Tall, A.R.; et al. PPARgamma Deacetylation Confers the Antiatherogenic Effect and Improves Endothelial Function in Diabetes Treatment. Diabetes 2020, 69, 1793–1803. [Google Scholar] [CrossRef]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 2005, 437, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wang, L.; Kon, N.; Zhao, W.; Lee, S.; Zhang, Y.; Rosenbaum, M.; Zhao, Y.; Gu, W.; Farmer, S.R.; et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell 2012, 150, 620–632. [Google Scholar] [CrossRef]
- He, Y.; Taub, A.B.; Yu, L.; Yao, Y.; Zhang, R.; Zahr, T.; Aaron, N.; LeSauter, J.; Fan, L.; Liu, L.; et al. PPARgamma Acetylation Orchestrates Adipose Plasticity and Metabolic Rhythms. Adv. Sci. 2023, 10, 2204190. [Google Scholar] [CrossRef]
- Kraakman, M.J.; Liu, Q.; Postigo-Fernandez, J.; Ji, R.; Kon, N.; Larrea, D.; Namwanje, M.; Fan, L.; Chan, M.; Area-Gomez, E.; et al. PPARgamma deacetylation dissociates thiazolidinedione’s metabolic benefits from its adverse effects. J. Clin. Investig. 2018, 128, 2600–2612. [Google Scholar] [CrossRef]
- Zahr, T.; Liu, L.; Chan, M.; Zhou, Q.; Cai, B.; He, Y.; Aaron, N.; Accili, D.; Sun, L.; Qiang, L. PPARgamma (Peroxisome Proliferator-Activated Receptor gamma) Deacetylation Suppresses Aging-Associated Atherosclerosis and Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 30–44. [Google Scholar] [CrossRef]
- Henderson, Y.O.; Bithi, N.; Link, C.; Yang, J.; Schugar, R.; Llarena, N.; Brown, J.M.; Hine, C. Late-life intermittent fasting decreases aging-related frailty and increases renal hydrogen sulfide production in a sexually dimorphic manner. Geroscience 2021, 43, 1527–1554. [Google Scholar] [CrossRef]
- Aaron, N.; Kraakman, M.J.; Zhou, Q.; Liu, Q.; Costa, S.; Yang, J.; Liu, L.; Yu, L.; Wang, L.; He, Y.; et al. Adipsin promotes bone marrow adiposity by priming mesenchymal stem cells. Elife 2021, 10, e69209. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Jiang, Y.; Wu, H.; Sun, F.; Li, Y.; He, H.; Wang, B.; Lu, Z.; Hu, Y.; Wei, X.; et al. Inhibition of Mitochondrial Calcium Overload by SIRT3 Prevents Obesity- or Age-Related Whitening of Brown Adipose Tissue. Diabetes 2020, 69, 165–180. [Google Scholar] [CrossRef] [PubMed]
- de Cabo, R.; Mattson, M.P. Effects of Intermittent Fasting on Health, Aging, and Disease. N. Engl. J. Med. 2019, 381, 2541–2551. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Longo, V.D.; Harvie, M. Impact of intermittent fasting on health and disease processes. Ageing Res. Rev. 2017, 39, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Xie, C.; Lu, S.; Nichols, R.G.; Tian, Y.; Li, L.; Patel, D.; Ma, Y.; Brocker, C.N.; Yan, T.; et al. Intermittent Fasting Promotes White Adipose Browning and Decreases Obesity by Shaping the Gut Microbiota. Cell Metab. 2017, 26, 801. [Google Scholar] [CrossRef]
- Fernandez, M.R.; Vilca, C.C.; Batista, L.O.; Figueiredo, L.S.; Ribeiro, R.A.; do Carmo, M.D.G.T.; Albuquerque, K.T. Fasting and refeeding cycles alter subcutaneous white depot growth dynamics and the morphology of brown adipose tissue in female rats. Br. J. Nutr. 2021, 126, 460–469. [Google Scholar] [CrossRef]
- Jarrar, M.H.; Baranova, A. PPARgamma activation by thiazolidinediones (TZDs) may modulate breast carcinoma outcome: The importance of interplay with TGFbeta signalling. J. Cell. Mol. Med. 2007, 11, 71–87. [Google Scholar] [CrossRef]
- Lebovitz, H.E. Thiazolidinediones: The Forgotten Diabetes Medications. Curr. Diabetes Rep. 2019, 19, 151. [Google Scholar] [CrossRef]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef]
- Konner, A.C.; Bruning, J.C. Selective insulin and leptin resistance in metabolic disorders. Cell Metab. 2012, 16, 144–152. [Google Scholar] [CrossRef]
- Shimizu, I.; Aprahamian, T.; Kikuchi, R.; Shimizu, A.; Papanicolaou, K.N.; MacLauchlan, S.; Maruyama, S.; Walsh, K. Vascular rarefaction mediates whitening of brown fat in obesity. J. Clin. Investig. 2014, 124, 2099–2112. [Google Scholar] [CrossRef] [PubMed]
- Stekovic, S.; Hofer, S.J.; Tripolt, N.; Aon, M.A.; Royer, P.; Pein, L.; Stadler, J.T.; Pendl, T.; Prietl, B.; Url, J.; et al. Alternate Day Fasting Improves Physiological and Molecular Markers of Aging in Healthy, Non-obese Humans. Cell Metab. 2020, 31, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Sutton, E.F.; Beyl, R.; Early, K.S.; Cefalu, W.T.; Ravussin, E.; Peterson, C.M. Early Time-Restricted Feeding Improves Insulin Sensitivity, Blood Pressure, and Oxidative Stress Even without Weight Loss in Men with Prediabetes. Cell Metab. 2018, 27, 1212–1221.e3. [Google Scholar] [CrossRef] [PubMed]
- Nowosad, K.; Sujka, M. Effect of Various Types of Intermittent Fasting (IF) on Weight Loss and Improvement of Diabetic Parameters in Human. Curr. Nutr. Rep. 2021, 10, 146–154. [Google Scholar] [CrossRef]
- Longo, V.D.; Panda, S. Fasting, Circadian Rhythms, and Time-Restricted Feeding in Healthy Lifespan. Cell Metab. 2016, 23, 1048–1059. [Google Scholar] [CrossRef]
- Ohno, H.; Shinoda, K.; Spiegelman, B.M.; Kajimura, S. PPARgamma agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metab. 2012, 15, 395–404. [Google Scholar] [CrossRef]
- Sass, F.; Schlein, C.; Jaeckstein, M.Y.; Pertzborn, P.; Schweizer, M.; Schinke, T.; Ballabio, A.; Scheja, L.; Heeren, J.; Fischer, A.W. TFEB deficiency attenuates mitochondrial degradation upon brown adipose tissue whitening at thermoneutrality. Mol. Metab. 2021, 47, 101173. [Google Scholar] [CrossRef]
- Bartelt, A.; Widenmaier, S.B.; Schlein, C.; Johann, K.; Goncalves, R.L.S.; Eguchi, K.; Fischer, A.W.; Parlakgul, G.; Snyder, N.A.; Nguyen, T.B.; et al. Brown adipose tissue thermogenic adaptation requires Nrf1-mediated proteasomal activity. Nat. Med. 2018, 24, 292–303. [Google Scholar] [CrossRef]
- Lazar, M.A. Reversing the curse on PPARgamma. J. Clin. Investig. 2018, 128, 2202–2204. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Z.; Ke, L.; Sun, Y.; Li, W.; Feng, X.; Zhu, W.; Chen, S. Resveratrol promotes white adipocytes browning and improves metabolic disorders in Sirt1-dependent manner in mice. FASEB J. 2020, 34, 4527–4539. [Google Scholar] [CrossRef]
- Wong, R.H.; Zaw, J.J.T.; Xian, C.J.; Howe, P.R. Regular Supplementation with Resveratrol Improves Bone Mineral Density in Postmenopausal Women: A Randomized, Placebo-Controlled Trial. J. Bone Miner. Res. 2020, 35, 2121–2131. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Zhang, R.; Yu, L.; Zahr, T.; Li, X.; Kim, T.-W.; Qiang, L. PPARγ Acetylation in Adipocytes Exacerbates BAT Whitening and Worsens Age-Associated Metabolic Dysfunction. Cells 2023, 12, 1424. https://doi.org/10.3390/cells12101424
He Y, Zhang R, Yu L, Zahr T, Li X, Kim T-W, Qiang L. PPARγ Acetylation in Adipocytes Exacerbates BAT Whitening and Worsens Age-Associated Metabolic Dysfunction. Cells. 2023; 12(10):1424. https://doi.org/10.3390/cells12101424
Chicago/Turabian StyleHe, Ying, Ruotong Zhang, Lexiang Yu, Tarik Zahr, Xueming Li, Tae-Wan Kim, and Li Qiang. 2023. "PPARγ Acetylation in Adipocytes Exacerbates BAT Whitening and Worsens Age-Associated Metabolic Dysfunction" Cells 12, no. 10: 1424. https://doi.org/10.3390/cells12101424