

Evaluation of RAS Mutational Status in Liquid Biopsy to Monitor Disease Progression in Metastatic Colorectal Cancer Patients
 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Study Design, Population, and Setting
2.2. Patients’ Assessment and Follow-Up
2.3. Sample Collection
2.4. ctDNA Extraction and Purification
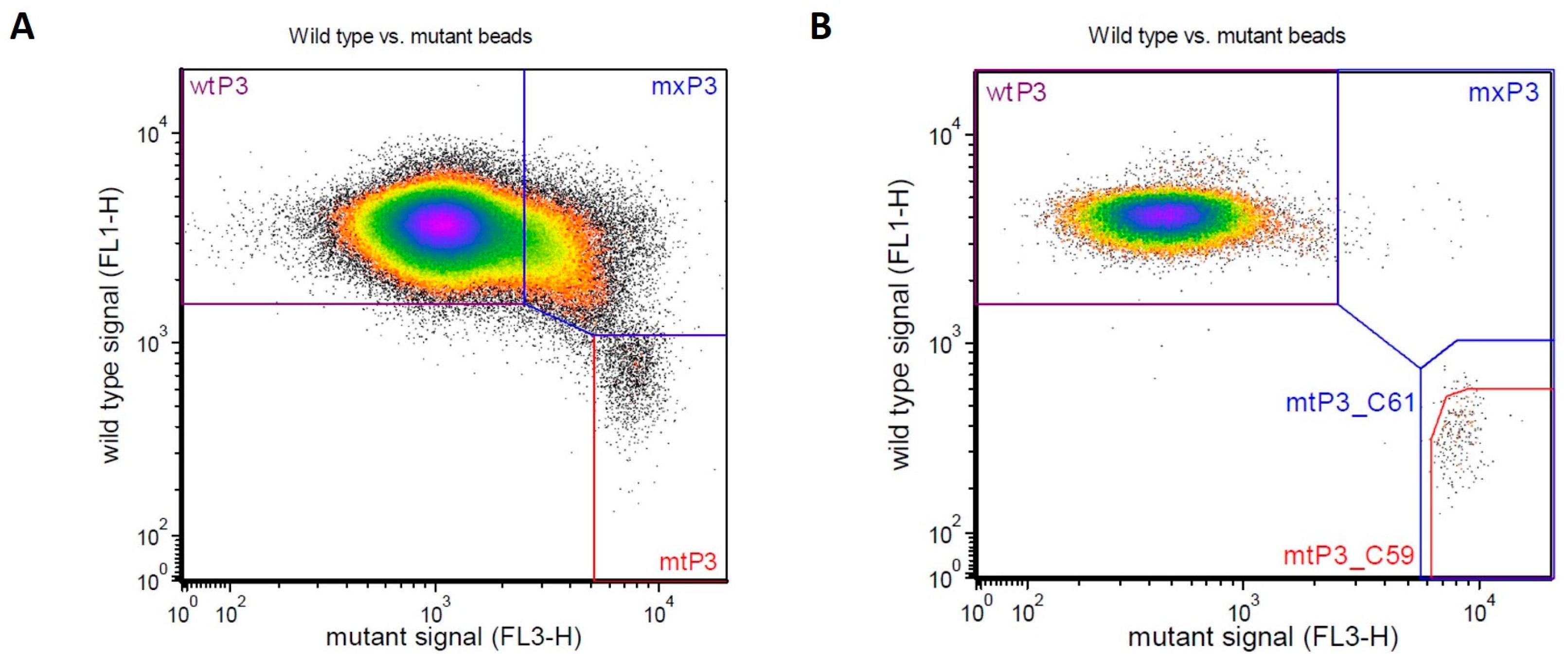
2.5. BEAMing
2.6. Statistical Analysis
3. Results
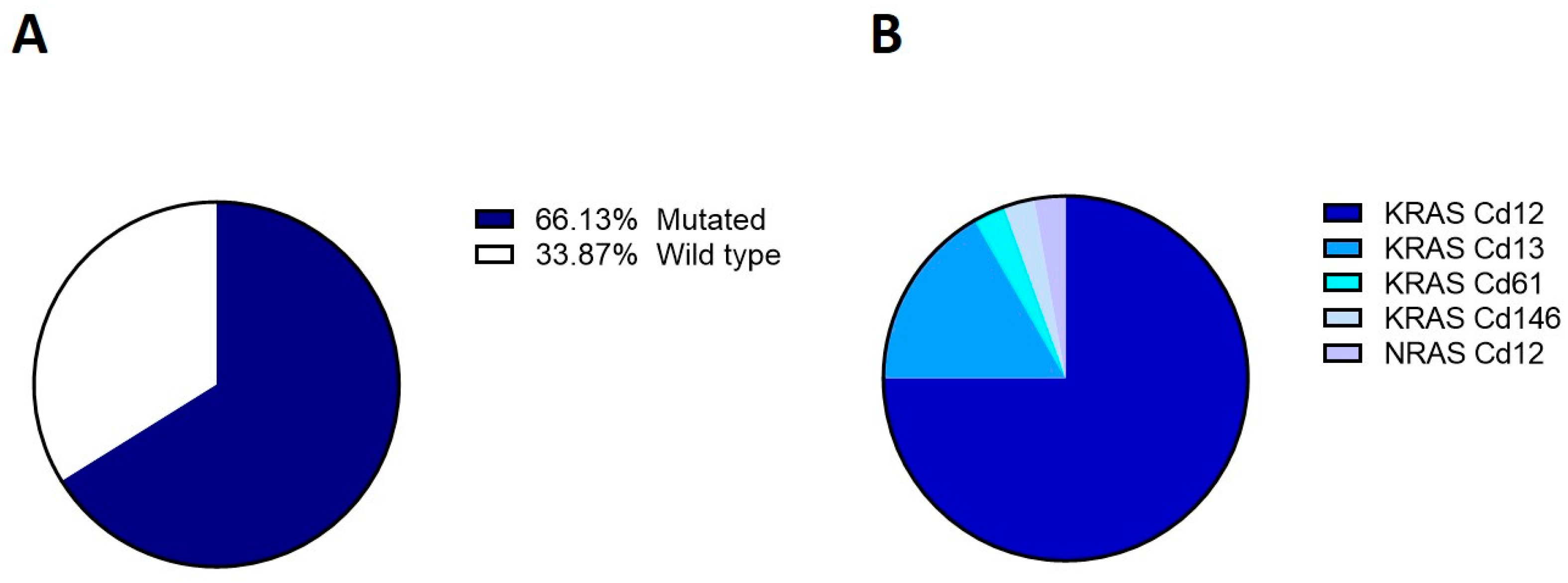
3.1. KRAS and NRAS Mutational Status in Tissue Samples
3.2. KRAS and NRAS Mutational Status Evaluation by BEAMing
Diagnostic Performance and Concordance between Tissue and Plasma KRAS and NRAS
3.3. KRAS and NRAS Status and Clinical Outcomes
3.4. Assessment of KRAS and NRAS Mutant Allele Fraction
3.5. Monitoring of KRAS and NRAS Mutational Status over Time
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [PubMed]
- Messersmith, W.A. Systemic Management of Colorectal Cancer. J. Natl. Compr. Cancer Netw. 2017, 15, 699–702. [Google Scholar] [CrossRef]
- Li, Z.N.; Zhao, L.; Yu, L.F.; Wei, M.J. BRAF and KRAS mutations in metastatic colorectal cancer: Future perspectives for personalized therapy. Gastroenterol. Rep. 2020, 8, 192–205. [Google Scholar] [CrossRef]
- Qiu, L.X.; Mao, C.; Zhang, J.; Zhu, X.D.; Liao, R.Y.; Xue, K.; Li, J.; Chen, Q. Predictive and prognostic value of KRAS mutations in metastatic colorectal cancer patients treated with cetuximab: A meta-analysis of 22 studies. Eur. J. Cancer 2010, 46, 2781–2787. [Google Scholar] [CrossRef] [PubMed]
- Morris, V.K.; Bekaii-Saab, T. Improvements in Clinical Outcomes for BRAF V600E -Mutant Metastatic Colorectal Cancer. Clin. Cancer Res. 2020, 26, 4435–4441. [Google Scholar] [CrossRef]
- Allegra, C.J.; Rumble, R.B.; Hamilton, S.R.; Mangu, P.B.; Roach, N.; Hantel, A.; Schilsky, R.L. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J. Clin. Oncol. 2016, 34, 179–185. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Arain, M.A.; Chen, Y.J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Farkas, L.; et al. Colon Cancer, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2021, 19, 329–359. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodríguez, C.; Brozos, E.; et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef]
- García-Foncillas, J.; Tabernero, J.; Élez, E.; Aranda, E.; Benavides, M.; Camps, C.; Jantus-Lewintre, E.; López, R.; Muinelo-Romay, L.; Montagut, C.; et al. Prospective multicenter real-world RAS mutation comparison between OncoBEAM-based liquid biopsy and tissue analysis in metastatic colorectal cancer. Br. J. Cancer 2018, 119, 1464–1470. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Durkee, K.H.; Moore, K.J.; Goodman, S.N.; Shuber, A.P.; Kinzler, K.W.; Vogelstein, B. Analysis of mutations in DNA isolated from plasma and stool of colorectal cancer patients. Gastroenterology 2008, 135, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- El Messaoudi, S.; Mouliere, F.; Du Manoir, S.; Bascoul-Mollevi, C.; Gillet, B.; Nouaille, M.; Fiess, C.; Crapez, E.; Bibeau, F.; Theillet, C.; et al. Circulating DNA as a Strong Multimarker Prognostic Tool for Metastatic Colorectal Cancer Patient Management Care. Clin. Cancer Res. 2016, 22, 3067–3077. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef]
- Spindler, K.L.G.; Pallisgaard, N.; Vogelius, I.; Jakobsen, A. Quantitative cell-free DNA, KRAS, and BRAF mutations in plasma from patients with metastatic colorectal cancer during treatment with cetuximab and irinotecan. Clin. Cancer Res. 2012, 18, 1177–1185. [Google Scholar] [CrossRef]
- Spindler, K.L.G.; Appelt, A.L.; Pallisgaard, N.; Andersen, R.F.; Brandslund, I.; Jakobsen, A. Cell-free DNA in healthy individuals, noncancerous disease and strong prognostic value in colorectal cancer. Int. J. cancer 2014, 135, 2984–2991. [Google Scholar] [CrossRef]
- Cremolini, C.; Antoniotti, C.; Lonardi, S.; Aprile, G.; Bergamo, F.; Masi, G.; Grande, R.; Tonini, G.; Mescoli, C.; Cardellino, G.G.; et al. Activity and Safety of Cetuximab Plus Modified FOLFOXIRI Followed by Maintenance With Cetuximab or Bevacizumab for RAS and BRAF Wild-type Metastatic Colorectal Cancer: A Randomized Phase 2 Clinical Trial. JAMA Oncol. 2018, 4, 529–536. [Google Scholar] [CrossRef]
- Kim, T.W.; Peeters, M.; Thomas, A.; Gibbs, P.; Hool, K.; Zhang, J.; Ang, A.L.; Bach, B.A.; Price, T. Impact of Emergent Circulating Tumor DNA RAS Mutation in Panitumumab-Treated Chemoresistant Metastatic Colorectal Cancer. Clin. Cancer Res. 2018, 24, 5602–5609. [Google Scholar] [CrossRef] [PubMed]
- Maurel, J.; Alonso, V.; Escudero, P.; Fernández-Martos, C.; Salud, A.; Méndez, M.; Gallego, J.; Rodriguez, J.R.; Martín-Richard, M.; Fernández-Plana, J.; et al. Clinical Impact of Circulating Tumor RAS and BRAF Mutation Dynamics in Patients with Metastatic Colorectal Cancer Treated with First-Line Chemotherapy Plus Anti-Epidermal Growth Factor Receptor Therapy. JCO Precis. Oncol. 2019, 3, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Siena, S.; Sartore-Bianchi, A.; Garcia-Carbonero, R.; Karthaus, M.; Smith, D.; Tabernero, J.; Van Cutsem, E.; Guan, X.; Boedigheimer, M.; Ang, A.; et al. Dynamic molecular analysis and clinical correlates of tumor evolution within a phase II trial of panitumumab-based therapy in metastatic colorectal cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Toledo, R.A.; Cubillo, A.; Vega, E.; Garralda, E.; Alvarez, R.; de la Varga, L.U.; Pascual, J.R.; Sánchez, G.; Sarno, F.; Prieto, S.H.; et al. Clinical validation of prospective liquid biopsy monitoring in patients with wild-type RAS metastatic colorectal cancer treated with FOLFIRI-cetuximab. Oncotarget 2017, 8, 35289–35300. [Google Scholar] [CrossRef]
- Van Emburgh, B.O.; Arena, S.; Siravegna, G.; Lazzari, L.; Crisafulli, G.; Corti, G.; Mussolin, B.; Baldi, F.; Buscarino, M.; Bartolini, A.; et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat. Commun. 2016, 7, 13665. [Google Scholar] [CrossRef]
- Lastraioli, E.; Lavacchi, D.; Palmieri, V.E.; Castiglione, F.; Messerini, L.; Di Costanzo, F.; Antonuzzo, L. Evaluation of RAS mutational status through BEAMing assay to monitor disease progression of metastatic colorectal cancer: A case report. Anticancer. Drugs 2020, 31, 979–982. [Google Scholar] [CrossRef]
- Lastraioli, E.; Antonuzzo, L.; Fantechi, B.; Di Cerbo, L.; Di Costanzo, A.; Lavacchi, D.; Armenio, M.; Arcangeli, A.; Castiglione, F.; Messerini, L.; et al. KRAS and NRAS mutation detection in circulating DNA from patients with metastatic colorectal cancer using BEAMing assay: Concordance with standard biopsy and clinical evaluation. Oncol. Lett. 2021, 21, 15. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Köhne, C.H.; Láng, I.; Folprecht, G.; Nowacki, M.P.; Cascinu, S.; Shchepotin, I.; Maurel, J.; Cunningham, D.; Tejpar, S.; et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: Updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J. Clin. Oncol. 2011, 29, 2011–2019. [Google Scholar] [CrossRef]
- Cercek, A.; Braghiroli, M.I.; Chou, J.F.; Hechtman, J.F.; Kemeny, N.; Saltz, L.; Capanu, M.; Yaeger, R. Clinical Features and Outcomes of Patients with Colorectal Cancers Harboring NRAS Mutations. Clin. Cancer Res. 2017, 23, 4753–4760. [Google Scholar] [CrossRef]
- Bielski, C.M.; Taylor, B.S. Mutant Allele Imbalance in Cancer. Annu. Rev. Cancer Biol. 2020, 5, 221–234. [Google Scholar] [CrossRef]
- Burgess, M.R.; Hwang, E.; Mroue, R.; Bielski, C.M.; Wandler, A.M.; Huang, B.J.; Firestone, A.J.; Young, A.; Lacap, J.A.; Crocker, L.; et al. KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer. Cell 2017, 168, 817–829.e15. [Google Scholar] [CrossRef] [PubMed]
- Ambrogio, C.; Köhler, J.; Zhou, Z.W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q.; et al. KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell 2018, 172, 857–868.e15. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Elez, E.; Argiles, G.; Matos, I.; Sanz-Garcia, E.; Ortiz, C.; Macarulla, T.; Capdevila, J.; Alsina, M.; Sauri, T.; et al. Analysis of mutant allele fractions in driver genes in colorectal cancer—Biological and clinical insights. Mol. Oncol. 2017, 11, 1263. [Google Scholar] [CrossRef] [PubMed]
- Basnet, S.; Zhang, Z.Y.; Liao, W.Q.; Li, S.H.; Li, P.S.; Ge, H.Y. The Prognostic Value of Circulating Cell-Free DNA in Colorectal Cancer: A Meta-Analysis. J. Cancer 2016, 7, 1105–1113. [Google Scholar] [CrossRef]
- Germano, G.; Mauri, G.; Siravegna, G.; Dive, C.; Pierce, J.; Di Nicolantonio, F.; D’Incalci, M.; Bardelli, A.; Siena, S.; Sartore-Bianchi, A. Parallel Evaluation of Circulating Tumor DNA and Circulating Tumor Cells in Metastatic Colorectal Cancer. Clin. Color. Cancer 2018, 17, 80–83. [Google Scholar] [CrossRef]
- Schmiegel, W.; Scott, R.J.; Dooley, S.; Lewis, W.; Meldrum, C.J.; Pockney, P.; Draganic, B.; Smith, S.; Hewitt, C.; Philimore, H.; et al. Blood-based detection of RAS mutations to guide anti-EGFR therapy in colorectal cancer patients: Concordance of results from circulating tumor DNA and tissue-based RAS testing. Mol. Oncol. 2017, 11, 208–219. [Google Scholar] [CrossRef]
- Valladares-Ayerbes, M.; Garcia-Alfonso, P.; Muñoz Luengo, J.; Pimentel Caceres, P.P.; Castillo Trujillo, O.A.; Vidal-Tocino, R.; Llanos, M.; Llorente Ayala, B.; Limon Miron, M.L.; Salud, A.; et al. Evolution of RAS Mutations in Cell-Free DNA of Patients with Tissue RAS Wild-Type Metastatic Colorectal Cancer Receiving First-Line Treatment: The PERSEIDA Study. Cancers 2022, 14, 6075. [Google Scholar] [CrossRef]
- Garciá-Foncillas, J.; Alba, E.; Aranda, E.; Diáz-Rubio, E.; López-López, R.; Tabernero, J.; Vivancos, A. Incorporating BEAMing technology as a liquid biopsy into clinical practice for the management of colorectal cancer patients: An expert taskforce review. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 2943–2949. [Google Scholar] [CrossRef]
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef]
- Toledano-Fonseca, M.; Cano, M.T.; Inga, E.; Rodríguez-Alonso, R.; Gómez-España, M.A.; Guil-Luna, S.; Mena-Osuna, R.; de la Haba-Rodríguez, J.R.; Rodríguez-Ariza, A.; Aranda, E. Circulating Cell-Free DNA-Based Liquid Biopsy Markers for the Non-Invasive Prognosis and Monitoring of Metastatic Pancreatic Cancer. Cancers 2020, 12, 1754. [Google Scholar] [CrossRef]
- Kruger, S.; Heinemann, V.; Ross, C.; Diehl, F.; Nagel, D.; Ormanns, S.; Liebmann, S.; Prinz-Bravin, I.; Westphalen, C.B.; Haas, M.; et al. Repeated mutKRAS ctDNA measurements represent a novel and promising tool for early response prediction and therapy monitoring in advanced pancreatic cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 2348–2355. [Google Scholar] [CrossRef] [PubMed]
- Taus, Á.; Camacho, L.; Rocha, P.; Hardy-Werbin, M.; Pijuan, L.; Piquer, G.; López, E.; Dalmases, A.; Longarón, R.; Clavé, S.; et al. Dynamics of EGFR Mutation Load in Plasma for Prediction of Treatment Response and Disease Progression in Patients With EGFR-Mutant Lung Adenocarcinoma. Clin. Lung Cancer 2018, 19, 387–394.e2. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.A.; Croessmann, S.; Park, B.H. The breast is yet to come: Current and future utility of circulating tumour DNA in breast cancer. Br. J. Cancer 2021, 125, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Bortolini Silveira, A.; Bidard, F.C.; Tanguy, M.L.; Girard, E.; Trédan, O.; Dubot, C.; Jacot, W.; Goncalves, A.; Debled, M.; Levy, C.; et al. Multimodal liquid biopsy for early monitoring and outcome prediction of chemotherapy in metastatic breast cancer. NPJ Breast Cancer 2021, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Olsson, E.; Winter, C.; George, A.; Chen, Y.; Howlin, J.; Tang, M.E.; Dahlgren, M.; Schulz, R.; Grabau, D.; Westen, D.; et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol. Med. 2015, 7, 1034–1047. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tot (n = 62) * | |
|---|---|
| Demographics | |
| Male sex | 35 (56.5%) |
| Age at inclusion, median (IQR) | 67 (61–74) |
| Histology | |
| Adenocarcinoma | 55 (88.7%) |
| Mucinous adenocarcinoma | 7 (11.3%) |
| Grading | |
| G2 | 26 (41.9%) |
| G3 | 11 (17.7%) |
| G4 | 1 (1.6%) |
| Missing | 24 (38.7%) |
| Site of primary lesion | |
| Colon | 46 (74.2%) |
| Rectal | 10 (16.1%) |
| Transverse colon | 2 (3.2%) |
| Missing | 4 (6.5%) |
| Staging | |
| IV (new diagnosis) | 21 (33.9%) |
| IV (relapse) | 14 (22.6%) |
| Missing | 27 (43.6%) |
| Number of metastases | |
| 1 | 28 (45.2%) |
| 2 | 24 (38.7%) |
| 3+ | 10 (16.1%) |
| Site of metastasis | |
| Liver | 37 (59.7%) |
| Lung | 18 (29.0%) |
| Loco-regional | 12 (19.4%) |
| Lymph nodes | 12 (19.4%) |
| Peritoneum | 12 (19.4%) |
| Pleura | 5 (8.1%) |
| Adrenal gland | 3 (4.8%) |
| Bone | 2 (3.2%) |
| Kidney | 2 (3.2%) |
| Pancreas | 1 (1.6%) |
| Endometrium | 1 (1.6%) |
| Bladder | 1 (1.6%) |
| Brain | 1 (1.6%) |
| Surgery on primary site | 48 (77.4%) |
| Chemotherapy | |
| Yes | 47 (75.8%) |
| No | 1 (1.6%) |
| Missing | 14 (22.6%) |
| Chemotherapy agents | |
| Only synthetic agents | 17 (27.4%) |
| Only targeted biologics | 5 (8.1%) |
| Combination of synthetic and biologics | 25 (40.3%) |
| (a) | |||||
| Tissue KRAS | |||||
| WT | Mutated | Value (95% CI) | |||
| Plasma KRAS | |||||
| WT | 9 (14.5%) | 4 (6.5%) | Cohen’s K: 0.43 (0.17–0.68) | ||
| Mutated | 9 (14.5%) | 34 (54.8%) | Sensitivity: 89.5% (75.2–97.1%) | ||
| Specificity: 50.0% (26.0–74.0%) | |||||
| Not Informative | 3 (4.8%) | 3 (4.8%) | PPV: 79.1% (70.2–85.9%) | ||
| NPV: 69.2% (44.4–86.4%) | |||||
| (b) | |||||
| Tissue KRAS | |||||
| Cd12 | Cd13 | Cd146 | WT | Concordance | |
| Plasma KRAS | |||||
| Cd12 | 28 (45.2%) | 1 (1.6%) | 0 | 9 (14.5%) | Kappa: 0.54 (95% CI: 0.33–0.75); % agreement: 75% |
| Cd13 | 0 | 4 (6.5%) | 0 | 0 | |
| Cd146 | 0 | 0 | 1 (1.6%) | 0 | |
| WT | 3 (4.8%) | 1 (1.6%) | 0 | 9 (14.5%) | |
| Not Informative | 3 (4.8%) | 0 | 0 | 3 (4.8%) | |
| (a) | |||
| Tissue NRAS | |||
| WT | Mutated | Value (95% CI) | |
| Plasma NRAS | |||
| WT | 55 (88.7%) | 0 | Cohen’s K: 0.27 (−0.15–0.68) |
| Mutated | 5 (8.1%) | 1 (1.6%) | Sensitivity: 100.0% (2.5–100%) |
| Specificity: 91.7% (81.6–97.2%) | |||
| Not Informative | 1 (1.6%) | 0 | PPV: 16.7% (8.0–31.6%) |
| NPV: 100% | |||
| (b) | |||
| Tissue NRAS | |||
| Cd12 | WT | Concordance | |
| Plasma NRAS | |||
| Cd12 | 1 (1.6%) | 2 (3.2%) | Kappa: 0.32 (95% CI: −0.15–0.80); % agreement: 93.4% |
| Cd61 | 0 | 3 (4.8%) | |
| WT | 0 | 55 (88.7%) | |
| Not Informative | 0 | 1 (1.6%) | |
| MAF KRAS (Median, IQR) | p-Value § | MAF NRAS (Median, IQR) | p-Value § | |||
|---|---|---|---|---|---|---|
| Overall | n = 55 | 0.16 (IQR 0.01–4.79; range 0–28.15) | n = 61 | 0.007 (IQ1 0.003–0.010; range 0.001–0.516) | ||
| Demographics | ||||||
| Male sex | n = 23 | 0.22 (0.01–5.46) | 0.511 | n = 34 | 0.006 (0.003–0.009) | 0.425 |
| Female sex | n = 32 | 0.06 (0.01–2.05) | n = 27 | 0.007 (0.002–0.014) | ||
| Histology | ||||||
| ADK | n = 48 | 0.17 (0.02–3.90) | 0.990 | n = 54 | 0.006 (0.002–0.008) | 0.004 * |
| Colloid ADK | n = 7 | 0.05 (0.01–13.0) | n = 7 | 0.027 (0.009–0.310) | ||
| Grading | ||||||
| G2 | n = 242 | 0.49 (0.02–7.37) | 0.025 * | n = 25 | 0.005 (0.003–0.010) | 0.828 |
| G3 | n = 8 | 0.01 (0.01–0.14) | n = 11 | 0.007 (0.002–0.011) | ||
| G4 | n = 1 | 0.00 | n = 1 | 0.006 | ||
| Missing | n = 22 | 0.25 (0.03–3.10) | n = 24 | 0.007 (0.004–0.009) | ||
| Site of primary lesion | ||||||
| Colon | n = 43 | 0.22 (0.01–5.46) | 0.660 | n = 45 | 0.007 (0.003–0.010) | 0.189 |
| Rectal | n = 8 | 0.06 (0.04–0.60) | n = 10 | 0.007 (0.003–0.010) | ||
| Transverse colon | n = 2 | 0.04 (0.01–0.06) | n = 2 | 0.002 | ||
| Missing | n = 2 | 1.62 (1.62–3.10) | n = 4 | 0.007 (0.005–0.007) | ||
| Staging | ||||||
| IV (new diagnosis) | n = 20 | 0.71 (0.04–9.01) | 0.330 | n = 20 | 0.008 (0.006–0.105) | 0.575 |
| IV (relapse) | n = 12 | 0.48 (0.01–5.07) | n = 14 | 0.011 (0.004–0.030) | ||
| Missing | n = 23 | 0.06 (0.01–0.33) | n = 27 | 0.004 (0.002–0.007) | ||
| Number of metastases | ||||||
| 1 | n = 26 | 0.23 (0.01–1.00) | 0.776 | n = 28 | 0.005 (0.002–0.008) | 0.243 |
| 2 | n = 21 | 0.06 (0.01–7.66) | n = 23 | 0.007 (0.003–0.010) | ||
| 3+ | n = 8 | 0.15 (0.04–5.07) | n = 10 | 0.007 (0.004–0.014) | ||
| Site of metastasis | ||||||
| Liver | No: n = 22; Yes: n = 33 | No: 0.05 (0.01–0.44) Yes: 0.33 (0.02–6.76) | 0.049 * | No: n = 24; Yes: n = 37 | No: 0.005 (0.003–0.007) Yes: 0.007 (0.003–0.13) | 0.061 |
| Lung | No: n = 41; Yes: n = 14 | No: 0.05 (0.07–0.77) Yes: 0.96 (0.04–5.61) | 0.113 | No: n = 43; Yes: n = 18 | No: 0.005 (0.002–0.009) Yes: 0.008 (0.006–0.017) | 0.025 * |
| Loco-regional | No: n = 45; Yes: n = 10 | No: 0.16 (0.01–5.26) Yes: 0.24 (0.02–1.00) | 0.785 | No: n = 49; Yes: n = 12 | No: 0.007 (0.003–0.011) Yes: 0.007 (0.003–0.007) | 0.315 |
| Lymph nodes | No: n = 43; Yes: n = 12 | No: 0.16 (0.01–1.00) Yes: 0.23 (0.01–6.11) | 0.514 | No: n = 50; Yes: n = 11 | No: 0.006 (0.002–0.009) Yes: 0.009 (0.004–0.014) | 0.100 |
| Peritoneum | No: n = 43; Yes: n = 12 | No: 0.32 (0.01–5.26) Yes: 0.05 (0.01–0.15) | 0.139 | No: n = 50; Yes: n = 11 | No: 0.007 (0.003–0.009) Yes: 0.007 (0.002–0.056) | 0.799 |
| Bone | No: n = 53 Yes: n = 2 | No: 0.16 (0.01–4.69) Yes: 0.09 (0.01–0.17) | - | No: n = 59 Yes: n = 2 | No: 0.007 (0.003–0.010) Yes: 0.04 (0.004–0.004) | - |
| Surgery on primary site | ||||||
| Yes | n = 42 | 0.06 (0.01–0.92) | 0.010 * | n = 47 | 0.008 (0.006–0.010) | 0.126 |
| No | n = 13 | 5.46 (0.07–9.86) | n = 14 | 0.006 (0.002–0.010) | ||
| Chemotherapy | ||||||
| No | n = 1 | 0.01 | - | n = 1 | 0.007 | - |
| Yes (any) | n = 42 | 0.17 (0.01–5.26) | n = 47 | 0.007 (0.003–0.010) | ||
| Only synthetic agents | n = 14 | 0.36 (0.01–8.16) | 0.359 ** | n = 17 | 0.007 (0.005–0.007) | 0.343 ** |
| Only targeted biologics | n = 4 | 0.02 (0.01–0.12) | n = 5 | 0.013 (0.011–0.469) | ||
| Combination of synthetic and biologics | n = 24 | 0.17 (0.01–3.13) | n = 25 | 0.005 (0.002–0.10) | ||
| Missing | n = 12 | 0.25 (0.04–3.90) | n = 13 | 0.006 (0.002–0.009) | ||
| Response | ||||||
| Complete response | n = 3 | 0.01 (0.01–0.02) | 0.160 | n = 3 | 0.011 (0.004–0.469) | 0.552 |
| Partial response | n = 4 | 0.03 (0.00–3.58) | n = 4 | 0.006 (0.004–0.007) | ||
| Stable disease | n = 6 | 0.09 (0.01–0.23) | n = 8 | 0.006 (0.003–0.012) | ||
| Progressive disease | n = 17 | 0.44 (0.03–1.00) | n = 20 | 0.007 (0.003–0.010) | ||
| TE | n = 7 | 0.33 (0.16–3.10) | ||||
| Missing | n = 18 | 0.11 (0.01–5.46) | n = 18 | 0.005 (0.002–0.008) | ||
| Survival | ||||||
| Survived | n = 22 | 0.11 (0.01–0.39) | 0.459 | n = 25 | 0.004 (0.002–0.007) | 0.182 |
| Deceased | n = 9 | 0.22 (0.01–8.16) | n = 11 | 0.007 (0.003–0.009) | ||
| Missing | n = 24 | 0.53 (0.02–6.18) | n = 25 | 0.008 (0.004–0.017) |
| Baseline | 4 Weeks | 8 Weeks | 12 Weeks | 48 Weeks | Therapy | Best Response | |
|---|---|---|---|---|---|---|---|
| Oncobio001 | M (0.042) | Low DNA | M (0.340) | M (0.418 + 0.156 Cd61) | END | FOLFIRI + BEVACIZUMAB | PD |
| Oncobio002 | M (0.173) | WT (0.010) | M (0.127) | FOLFIRI + BEVACIZUMAB | SD | ||
| Oncobio003 | M (0.012) | M (0.254) | Low DNA | FOLFIRI | CR | ||
| Oncobio004 | M (7.103) | Low DNA | M (0.604) | XELOX | PR | ||
| Oncobio005 | M (0.060) | M (0.530) | M (0.082) | CAPOX + BEVACIZUMAB | PR | ||
| Oncobio006 | Low plasma vol | Low DNA | XELOX | SD | |||
| Oncobio007 | WT (0.005) | WT (0.006) | M (0.102) | FOLFOX + VECTIBIX | PR | ||
| Oncobio008 | WT (0.010) | M (0.105) | Low plasma vol | 0.011 | FOLFIRI + VECTIBIX | SD | |
| Oncobio009 | WT (0.005) | WT (0.010) | M (0.503) | FOLFIRI + VECTIBIX | SD | ||
| Oncobio010 | M (0.012) | M (0.040) | END | CAPOX + BEVACIZUMAB | PD | ||
| Oncobio011 | M (0.233) | M (0.038) | M (0.451) | CAPECITABINE + PANITUMUMAB | SD | ||
| Oncobio012 | M (0.771) | END | CAPOX | PD | |||
| Oncobio013 | M (8.159) | M (0.382) | END | FOLFOXIRI | PD | ||
| Oncobio014 | Low plasma vol | Low plasma vol | M (0.218) | M (1.995) | CAPOX + BEVACIZUMAB | SD | |
| Oncobio015 | WT (0.010) | END | CAPOX + BEVACIZUMAB | PD | |||
| Oncobio016 | WT (0.005) | M (0.050) | WT (0.011) | FOLFIRI + BEVACIZUMAB | |||
| Oncobio017 | M (0.334) | M (0.461 + 0.065 Cd117) | FOLFOX | ||||
| Oncobio018 | Low plasma vol | M (0.251) | M (0.290) | END | FOLFIRI + BEVACIZUMAB | PD | |
| Oncobio019 | M (0.317) | M (0.351) | M (0.242) | CAPECITABINE + BEVACIZUMAB | |||
| Oncobio020 | M (0.026) | M (0.045) | M (0.137) | END | CAPOX + BEVACIZUMAB | PD | |
| Oncobio021 | M (0.038 + 0.056 NRAS Cd12) | M (0.054) | Low plasma vol | END | CAPOX + BEVACIZUMAB | PD | |
| Oncobio022 | M (0.437) | M (0.112) | Low DNA | CAPOX + BEVACIZUMAB | SD | ||
| Oncobio023 | M (0.393) | M (0.110) | M (0.017) | FOLFOX | PR | ||
| Oncobio024 | WT (0.003) | M (0.018) | WT (0.008) | OXALIPLATIN | PR | ||
| Oncobio025 | M (0.162) | M (0.063) | DEGRAMONT + BEVA | ||||
| Oncobio026 | Low plasma vol | Low plasma vol | Low plasma vol | CAPECITABINE + BEVACIZUMAB | |||
| Oncobio027 | M (1.004) | Low plasma vol | M (1.044) | END | FOLFOX + BEVACIZUMAB | PD | |
| Oncobio028 | Low plasma vol | M (0.015) | M (0.038) | FOLFOX | |||
| Oncobio029 | M (0.218) | WT (0.005) | END | PEMBROLIZUMAB | PD | ||
| Oncobio030 | M (3.102) | M (0.358 + 0.047 NRAS Cd12) | |||||
| Oncobio031 | M (0.021) | M (0.012) | WT (0.008) | FOLFIRI + BEVACIZUMAB | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lastraioli, E.; Bettiol, A.; Iorio, J.; Limatola, E.; Checcacci, D.; Parisi, E.; Bianchi, C.; Arcangeli, A.; Iannopollo, M.; Di Costanzo, F.; et al. Evaluation of RAS Mutational Status in Liquid Biopsy to Monitor Disease Progression in Metastatic Colorectal Cancer Patients. Cells 2023, 12, 1458. https://doi.org/10.3390/cells12111458
Lastraioli E, Bettiol A, Iorio J, Limatola E, Checcacci D, Parisi E, Bianchi C, Arcangeli A, Iannopollo M, Di Costanzo F, et al. Evaluation of RAS Mutational Status in Liquid Biopsy to Monitor Disease Progression in Metastatic Colorectal Cancer Patients. Cells. 2023; 12(11):1458. https://doi.org/10.3390/cells12111458
Chicago/Turabian StyleLastraioli, Elena, Alessandra Bettiol, Jessica Iorio, Elvira Limatola, Daniele Checcacci, Erica Parisi, Cristina Bianchi, Annarosa Arcangeli, Mauro Iannopollo, Francesco Di Costanzo, and et al. 2023. "Evaluation of RAS Mutational Status in Liquid Biopsy to Monitor Disease Progression in Metastatic Colorectal Cancer Patients" Cells 12, no. 11: 1458. https://doi.org/10.3390/cells12111458