

Aberrant DNA Methylation, Expression, and Occurrence of Transcript Variants of the ABC Transporter ABCA7 in Breast Cancer

 , , , , , , ,
, , , , , , ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.1.1. Breast Tissue Samples
2.1.2. Cell Lines
2.1.3. Treatment of MCF-7, BT-474, SK-BR3, and MDA-MB-231 Cells with Dox or Tax
2.2. DNA and RNA Extraction
2.3. DNA Methylation Analysis
2.4. Gene Expression and Analysis of Intron-Containing Transcripts
2.5. Mass Spectrometry-Based Shotgun Proteomics
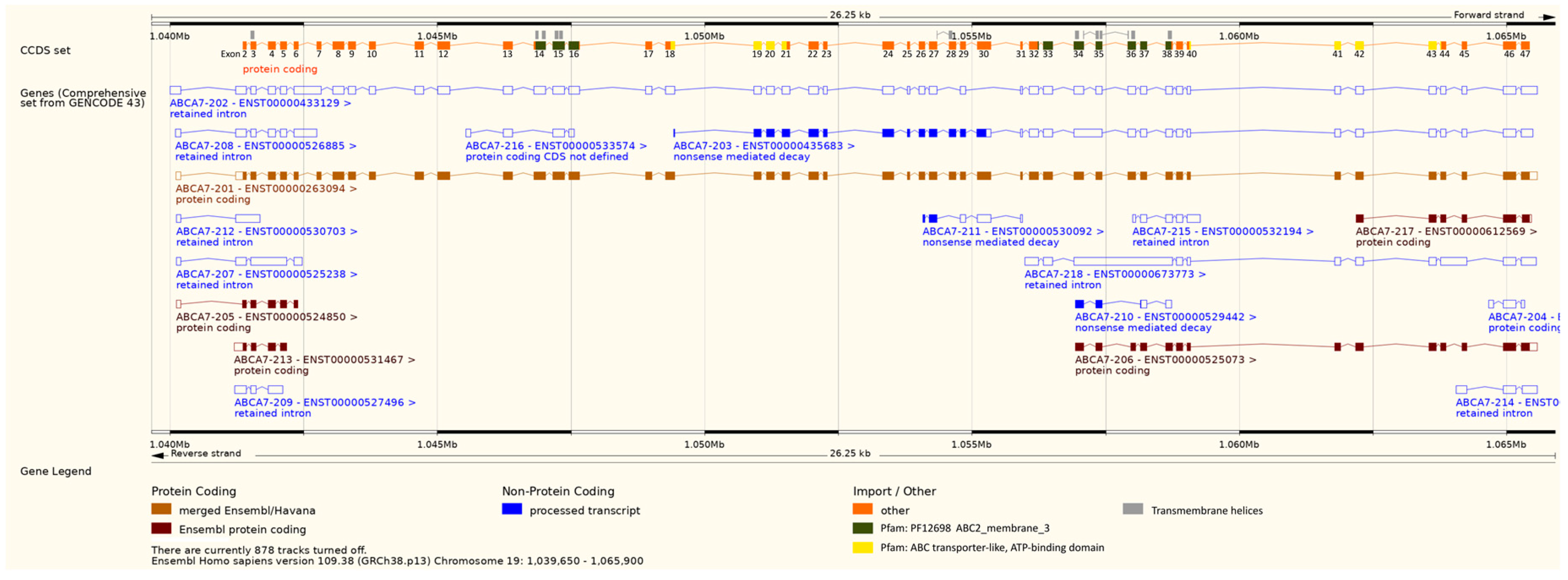
2.6. Bioinformatics
2.7. Statistical Analysis
3. Results
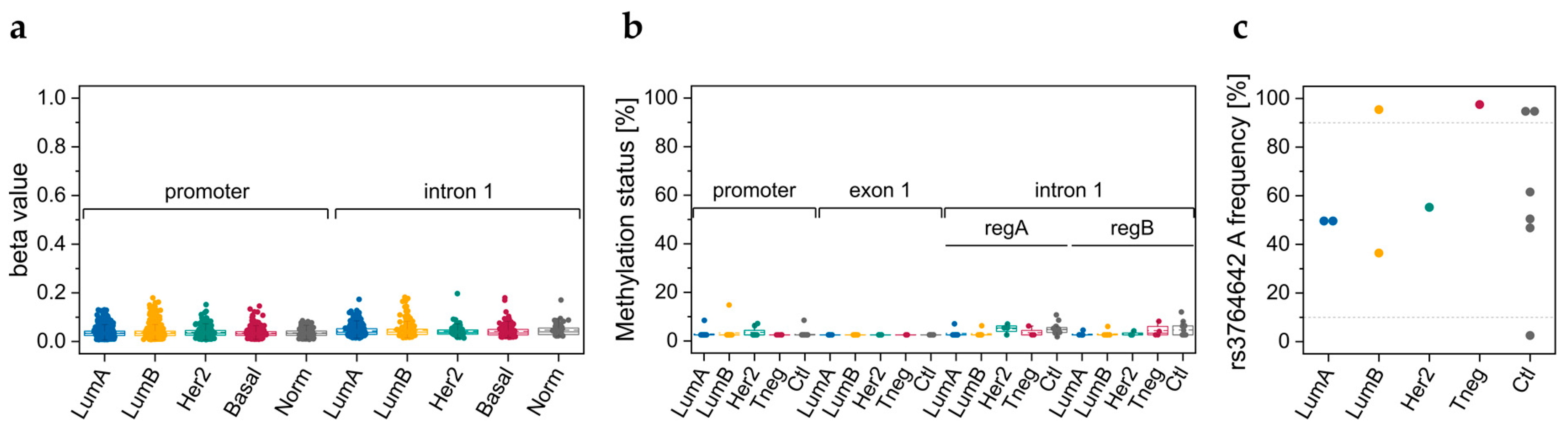
3.1. DNA Methylation Data Retrieved from TCGA
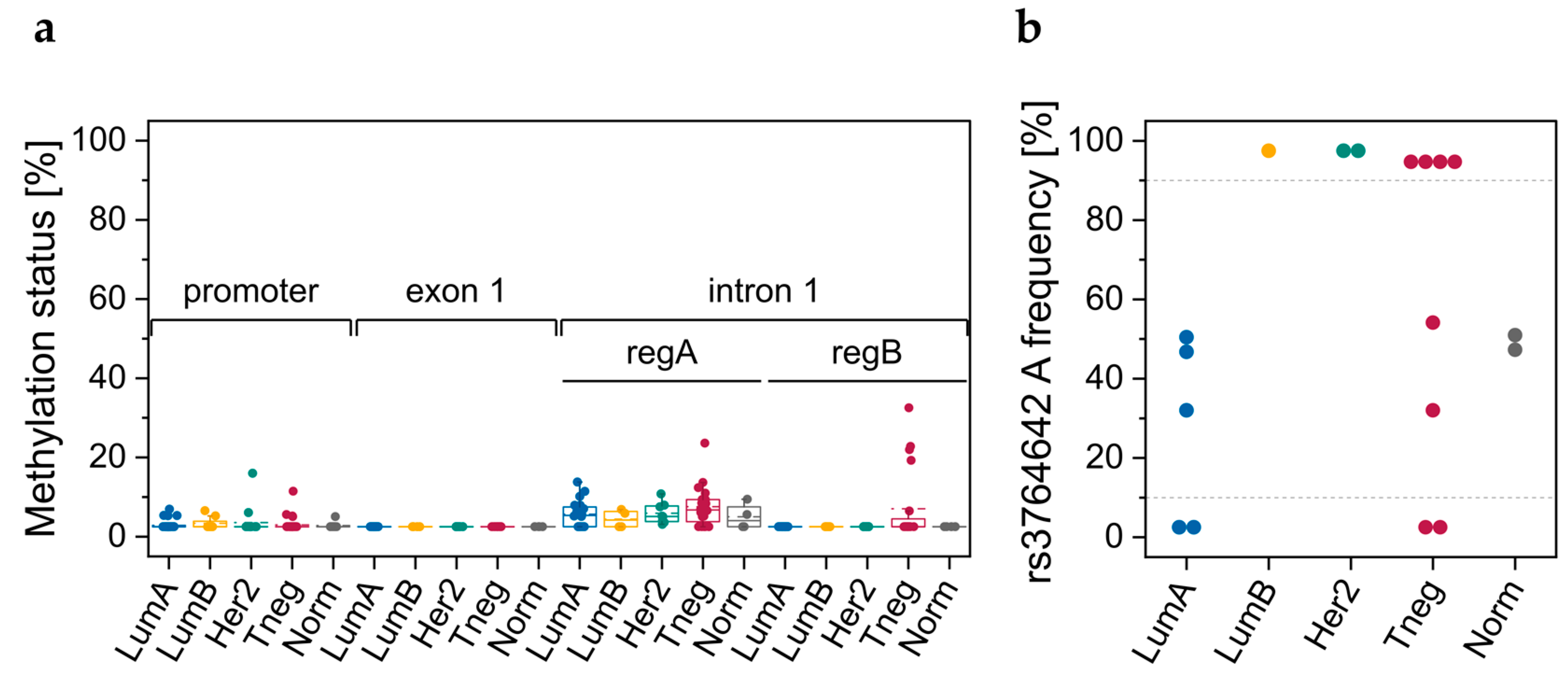
3.2. Breast Tissue Samples
3.2.1. DNA Methylation Status of CpGs in ABCA7 Promoter–Exon 1 and Intron 1
3.2.2. Genotypes of SNPs rs531508435, rs549725064, and rs3764642
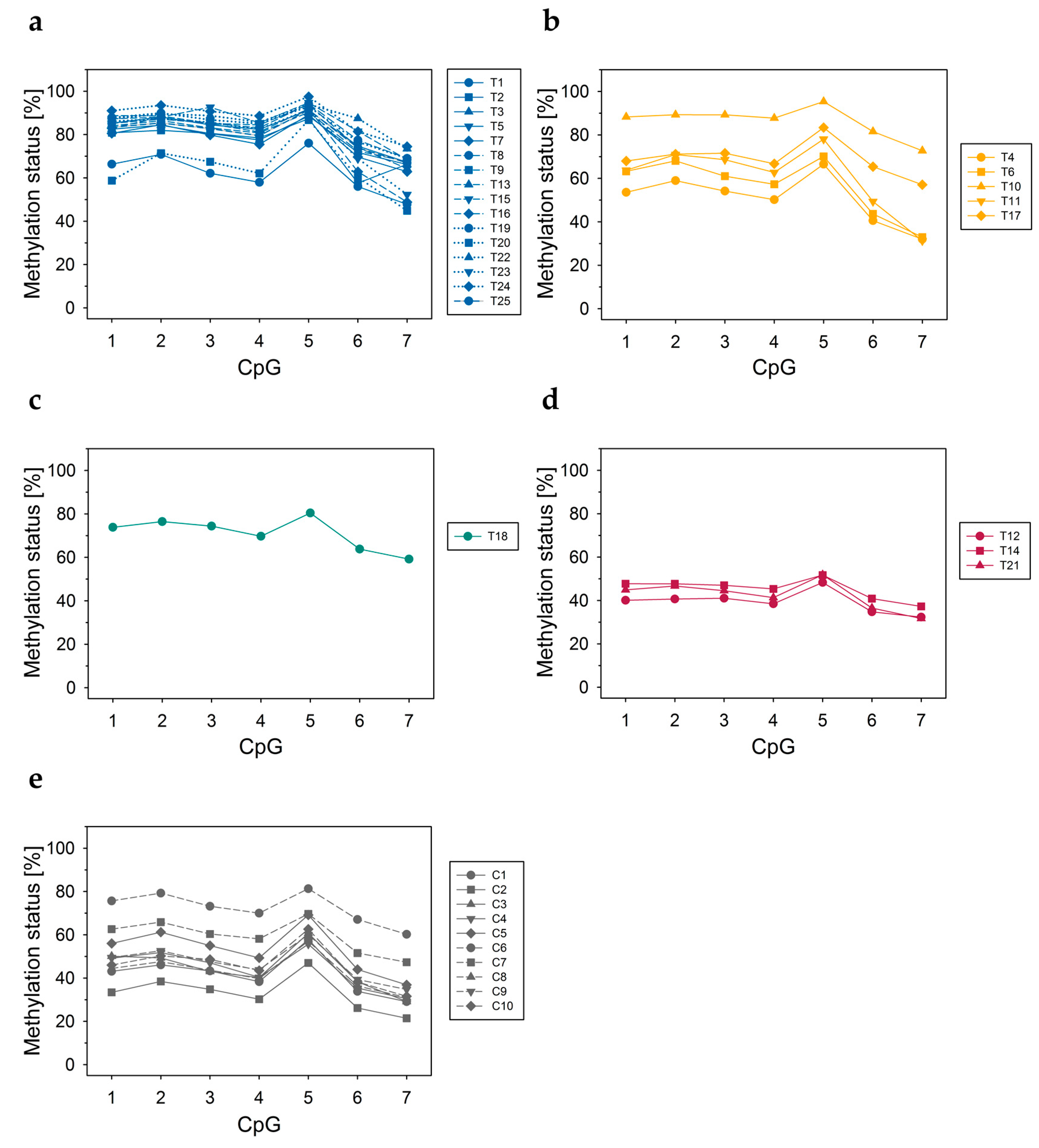
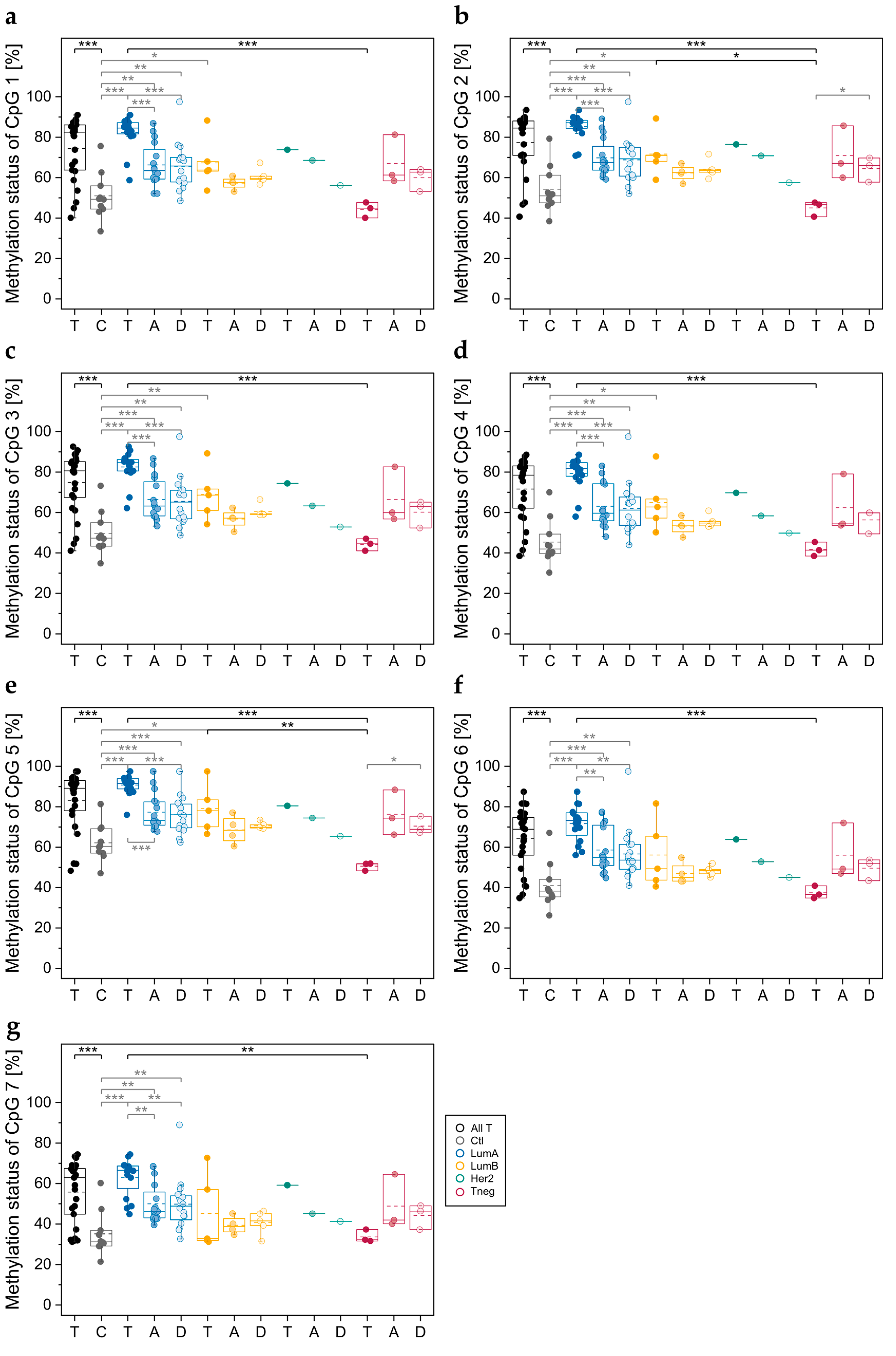
3.2.3. DNA Methylation Status of CpGs at ABCA7 Exon 5–Intron 5 Boundary
3.2.4. Association of the DNA Methylation Status at the Exon 5–Intron 5 Boundary in Breast Tissues with Cell Proliferation and Age
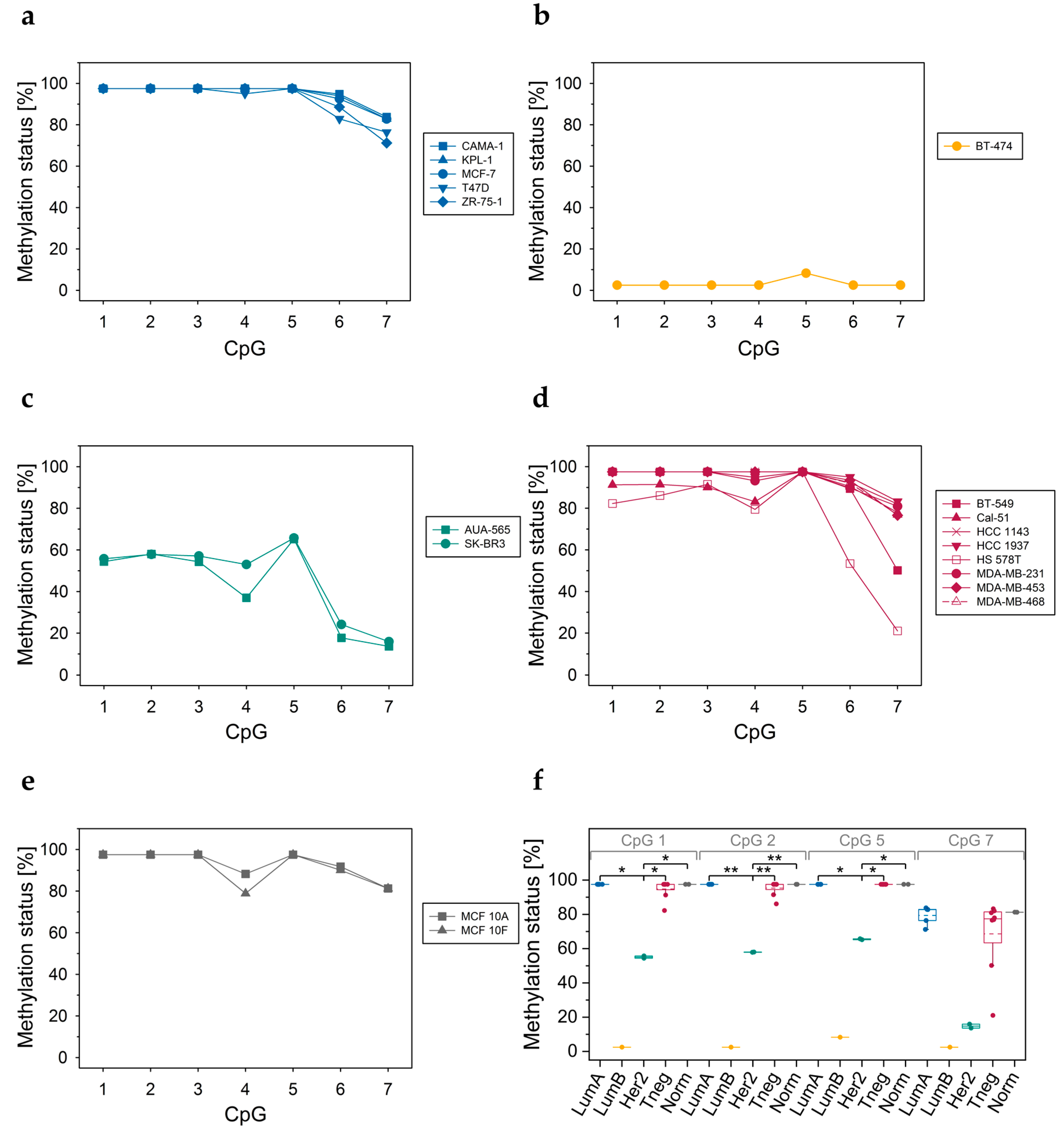
3.3. Commercial Breast Cell Lines
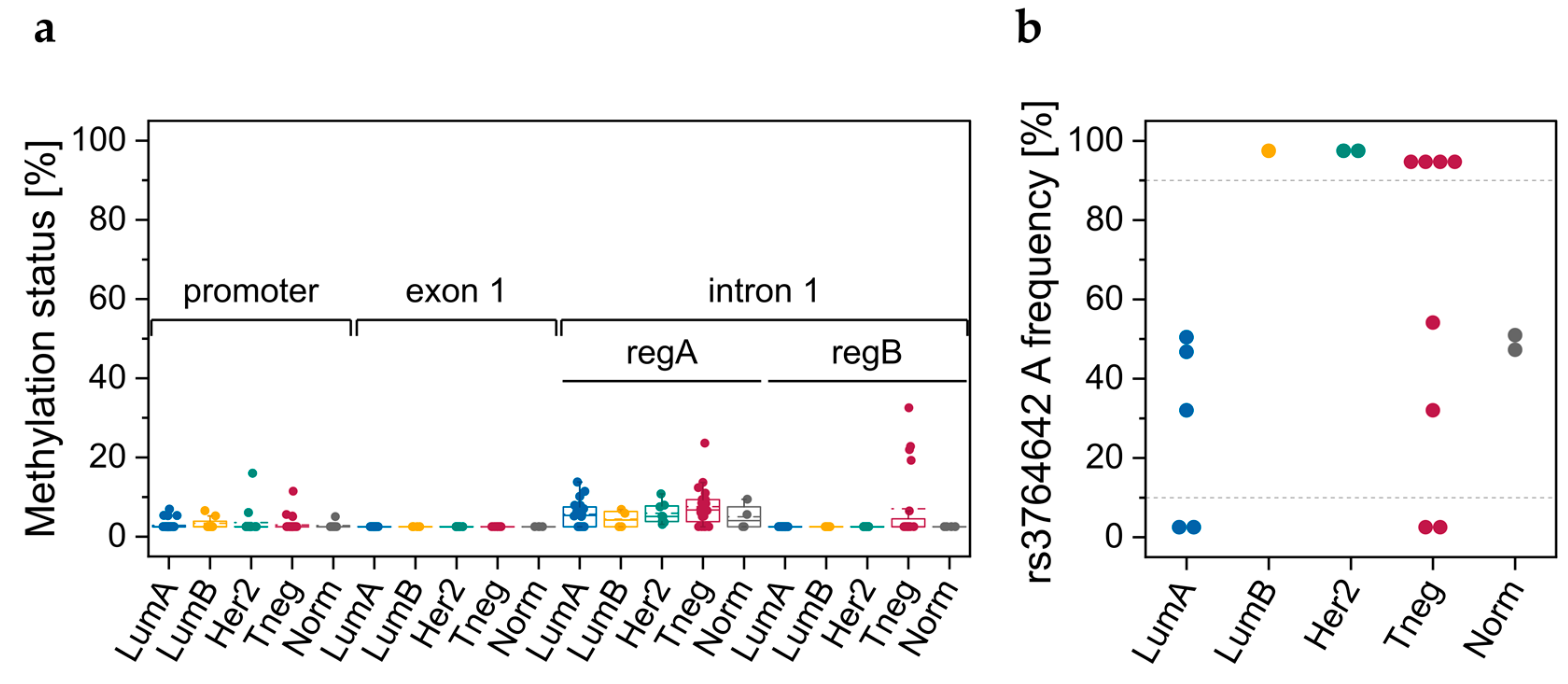
3.3.1. DNA Methylation Status of CpGs in Promoter–Exon 1 and Intron 1
3.3.2. Genotypes of SNPs rs531508435, rs549725064, and rs3764642
3.3.3. DNA Methylation Status of CpGs at Exon 5–Intron 5 Boundary
3.3.4. DNA Methylation Status of CpGs in ABCA7 Exon 12
3.3.5. ABAC7 mRNA Levels
3.3.6. Intron-Containing Transcripts
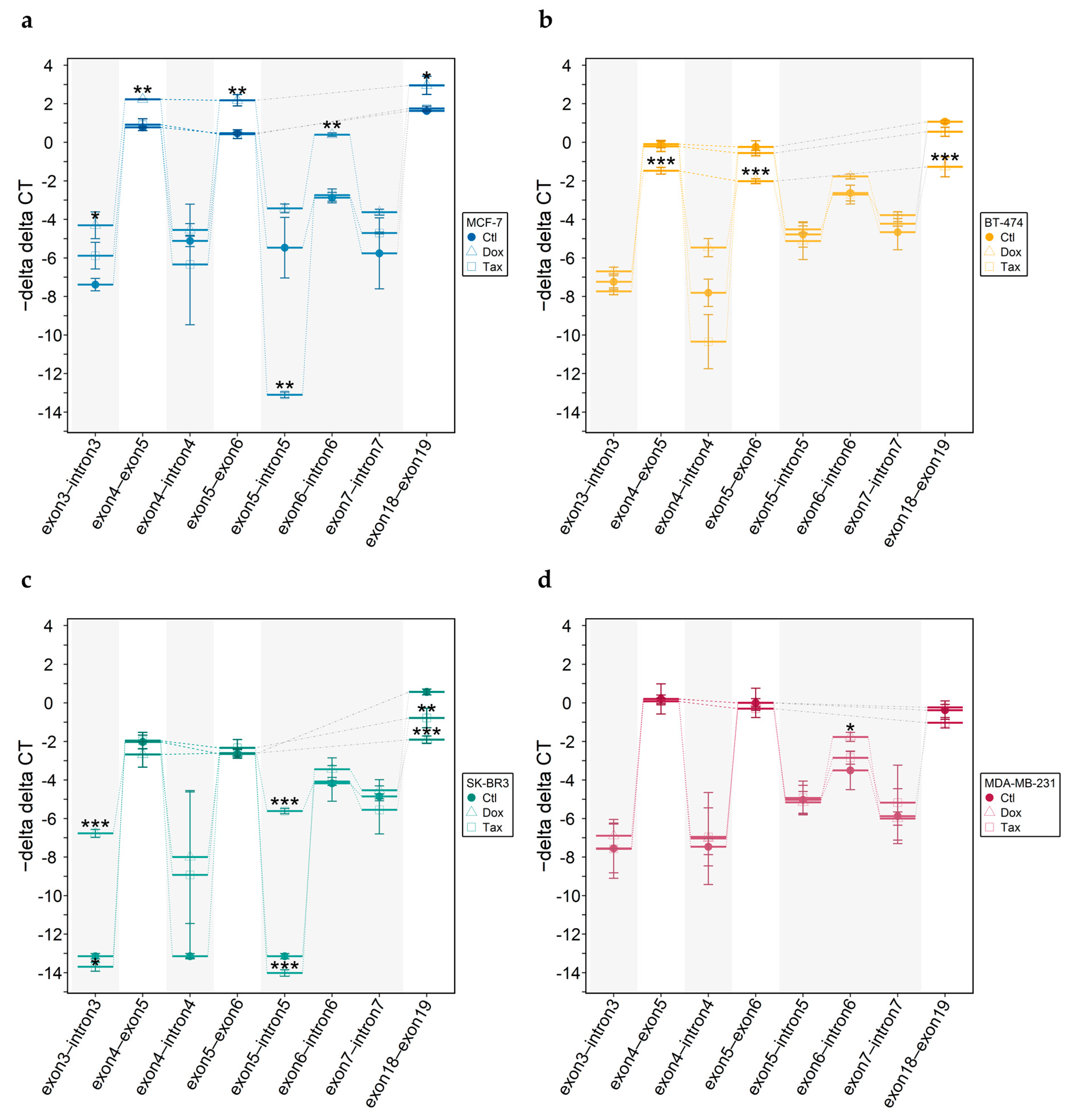
3.4. Breast Cancer Cell Lines Treated with Dox or Tax
3.4.1. DNA Methylation Status of CpGs at the Exon 5–Intron 5 Boundary
3.4.2. ABCA7 mRNA Levels
3.4.3. Intron-Containing Transcripts
3.4.4. Shotgun Proteomics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC transporters: The power to change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Linton, K.J. Structure and Function of ABC Transporters. Physiology 2007, 22, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Locher, K.P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 2016, 23, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, W.E.; Orsó, E.; Diederich, W.; Klucken, J.; Drobnik, W.; Schmitz, G. Identification of a Novel Human Sterol-Sensitive ATP-Binding Cassette Transporter (ABCA7). Biochem. Biophys. Res. Commun. 2000, 273, 532–538. [Google Scholar] [CrossRef]
- Kim, W.S.; Guillemin, G.J.; Glaros, E.N.; Lim, C.K.; Garner, B. Quantitation of ATP-binding cassette subfamily-A transporter gene expression in primary human brain cells. NeuroReport 2006, 17, 891–896. [Google Scholar] [CrossRef]
- Kim, W.S.; Li, H.; Ruberu, K.; Chan, S.; Elliott, D.A.; Low, J.K.; Cheng, D.; Karl, T.; Garner, B. Deletion of ABCA7 Increases Cerebral Amyloid-β Accumulation in the J20 Mouse Model of Alzheimer’s Disease. J. Neurosci. 2013, 33, 4387–4394. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C. Is ABCA1 a lipid transfer protein? J. Lipid Res. 2018, 59, 749–763. [Google Scholar] [CrossRef]
- Abe-Dohmae, S.; Ikeda, Y.; Matsuo, M.; Hayashi, M.; Okuhira, K.-I.; Ueda, K.; Yokoyama, S. Human ABCA7 Supports Apolipoprotein-mediated Release of Cellular Cholesterol and Phospholipid to Generate High Density Lipoprotein. J. Biol. Chem. 2004, 279, 604–611. [Google Scholar] [CrossRef]
- Iwamoto, N.; Abe-Dohmae, S.; Sato, R.; Yokoyama, S. ABCA7 expression is regulated by cellular cholesterol through the SREBP2 pathway and associated with phagocytosis. J. Lipid Res. 2006, 47, 1915–1927. [Google Scholar] [CrossRef]
- Linsel-Nitschke, P.; Jehle, A.W.; Shan, J.; Cao, G.; Bacic, D.; Lan, D.; Wang, N.; Tall, A.R. Potential role of ABCA7 in cellular lipid efflux to apoA-I. J. Lipid Res. 2005, 46, 86–92. [Google Scholar] [CrossRef]
- Quazi, F.; Molday, R.S. Differential Phospholipid Substrates and Directional Transport by ATP-binding Cassette Proteins ABCA1, ABCA7, and ABCA4 and Disease-causing Mutants. J. Biol. Chem. 2013, 288, 34414–34426. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Kim, W.S.; Kwok, J.B.; Hill, A.F.; Cappai, R.; Rye, K.-A.; Garner, B. ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J. Neurochem. 2008, 106, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Picataggi, A.; Rodrigues, A.; Cromley, D.A.; Wang, H.; Wiener, J.P.; Garliyev, V.; Billheimer, J.T.; Grabiner, B.C.; Hurt, J.A.; Chen, A.C.; et al. Specificity of ABCA7-mediated cell lipid efflux. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2022, 1867, 159157. [Google Scholar] [CrossRef]
- Jehle, A.W.; Gardai, S.J.; Li, S.; Linsel-Nitschke, P.; Morimoto, K.; Janssen, W.J.; Vandivier, R.W.; Wang, N.; Greenberg, S.; Dale, B.M.; et al. ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J. Cell Biol. 2006, 174, 547–556. [Google Scholar] [CrossRef]
- Tanaka, N.; Abe-Dohmae, S.; Iwamoto, N.; Fitzgerald, M.L.; Yokoyama, S. HMG-CoA reductase inhibitors enhance phagocytosis by upregulating ATP-binding cassette transporter A7. Atherosclerosis 2011, 217, 407–414. [Google Scholar] [CrossRef] [PubMed]
- De Roeck, A.; Van Broeckhoven, C.; Sleegers, K. The role of ABCA7 in Alzheimer’s disease: Evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 2019, 138, 201–220. [Google Scholar] [CrossRef]
- Satoh, K.; Abe-Dohmae, S.; Yokoyama, S.; George-Hyslop, P.S.; Fraser, P.E. ATP-binding Cassette Transporter A7 (ABCA7) Loss of Function Alters Alzheimer Amyloid Processing. J. Biol. Chem. 2015, 290, 24152–24165. [Google Scholar] [CrossRef]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef]
- Humphries, C.; Kohli, M.A.; Whitehead, P.; Mash, D.C.; Pericak-Vance, M.A.; Gilbert, J. Alzheimer disease (AD) specific transcription, DNA methylation and splicing in twenty AD associated loci. Mol. Cell. Neurosci. 2015, 67, 37–45. [Google Scholar] [CrossRef]
- Yu, L.; Chibnik, L.B.; Srivastava, G.P.; Pochet, N.; Yang, J.; Xu, J.; Kozubek, J.; Obholzer, N.; Leurgans, S.E.; Schneider, J.A.; et al. Association of Brain DNA Methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 With Pathological Diagnosis of Alzheimer Disease. JAMA Neurol. 2015, 72, 15–24. [Google Scholar] [CrossRef]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.-S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.-C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.-C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- De Roeck, A.; Duchateau, L.; Van Dongen, J.; Cacace, R.; Bjerke, M.; Van den Bossche, T.; Cras, P.; Vandenberghe, R.; De Deyn, P.P.; Engelborghs, S.; et al. An intronic VNTR affects splicing of ABCA7 and increases risk of Alzheimer’s disease. Acta Neuropathol. 2018, 135, 827–837. [Google Scholar] [CrossRef]
- Mohelnikova-Duchonova, B.; Brynychova, V.; Oliverius, M.; Honsova, E.; Kala, Z.; Muckova, K.; Soucek, P. Differences in Transcript Levels of ABC Transporters Between Pancreatic Adenocarcinoma and Nonneoplastic Tissues. Pancreas 2013, 42, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Elsnerova, K.; Bartakova, A.; Tihlarik, J.; Bouda, J.; Rob, L.; Skapa, P.; Hruda, M.; Gut, I.; Mohelnikova-Duchonova, B.; Soucek, P.; et al. Gene Expression Profiling Reveals Novel Candidate Markers of Ovarian Carcinoma Intraperitoneal Metastasis. J. Cancer 2017, 8, 3598–3606. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Q.; Zhou, J.; Zhang, S. ATP-binding cassette transporter A7 accelerates epithelial-to-mesenchymal transition in ovarian cancer cells by upregulating the transforming growth factor-β signaling pathway. Oncol. Lett. 2018, 16, 5868–5874. [Google Scholar] [CrossRef]
- Xie, W.; Shui, C.; Fang, X.; Peng, Y.; Qin, L. miR-197-3p reduces epithelial–mesenchymal transition by targeting ABCA7 in ovarian cancer cells. 3 Biotech 2020, 10, 375. [Google Scholar] [CrossRef]
- Hlavata, I.; Mohelnikova-Duchonova, B.; Vaclavikova, R.; Liska, V.; Pitule, P.; Novak, P.; Bruha, J.; Vycital, O.; Holubec, L.; Treska, V.; et al. The role of ABC transporters in progression and clinical outcome of colorectal cancer. Mutagenesis 2012, 27, 187–196. [Google Scholar] [CrossRef]
- Hlaváč, V.; Brynychová, V.; Václavíková, R.; Ehrlichová, M.; Vrána, D.; Pecha, V.; Koževnikovová, R.; Trnková, M.; Gatěk, J.; Kopperová, D.; et al. The expression profile of ATP-binding cassette transporter genes in breast carcinoma. Pharmacogenomics 2013, 14, 515–529. [Google Scholar] [CrossRef]
- Llaverias, G.; Danilo, C.; Mercier, I.; Daumer, K.; Capozza, F.; Williams, T.M.; Sotgia, F.; Lisanti, M.P.; Frank, P.G. Role of Cholesterol in the Development and Progression of Breast Cancer. Am. J. Pathol. 2011, 178, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Danilo, C.; Frank, P.G. Cholesterol and breast cancer development. Curr. Opin. Pharmacol. 2012, 12, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Baek, A.E.; Nelson, E.R. The Contribution of Cholesterol and Its Metabolites to the Pathophysiology of Breast Cancer. Horm. Cancer 2016, 7, 219–228. [Google Scholar] [CrossRef]
- Nelson, E.R. The Significance of Cholesterol and its Metabolite, 27-Hydroxycholesterol in Breast Cancer. Mol. Cell. Endocrinol. 2018, 466, 73–80. [Google Scholar] [CrossRef]
- Garcia-Estevez, L.; Moreno-Bueno, G. Updating the role of obesity and cholesterol in breast cancer. Breast Cancer Res. 2019, 21, 35. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.-T.; Phuong, T.N.T.; Tien, N.L.B.; Tran, D.-K.; Nguyen, T.-T.; Van Thanh, V.; Quang, T.L.; Minh, L.B.; Pham, V.H.; Ngoc, V.T.N.; et al. The Effects of Adipocytes on the Regulation of Breast Cancer in the Tumor Microenvironment: An Update. Cells 2019, 8, 857. [Google Scholar] [CrossRef] [PubMed]
- Bernard, J.J.; Wellberg, E.A. The Tumor Promotional Role of Adipocytes in the Breast Cancer Microenvironment and Macroenvironment. Am. J. Pathol. 2021, 191, 1342–1352. [Google Scholar] [CrossRef]
- Rybinska, I.; Agresti, R.; Trapani, A.; Tagliabue, E.; Triulzi, T. Adipocytes in Breast Cancer, the Thick and the Thin. Cells 2020, 9, 560. [Google Scholar] [CrossRef]
- Rybinska, I.; Mangano, N.; Tagliabue, E.; Triulzi, T. Cancer-Associated Adipocytes in Breast Cancer: Causes and Consequences. Int. J. Mol. Sci. 2021, 22, 3775. [Google Scholar] [CrossRef]
- Spitzwieser, M.; Entfellner, E.; Werner, B.; Pulverer, W.; Pfeiler, G.; Hacker, S.; Cichna-Markl, M. Hypermethylation of CDKN2A exon 2 in tumor, tumor-adjacent and tumor-distant tissues from breast cancer patients. BMC Cancer 2017, 17, 260. [Google Scholar] [CrossRef]
- Pacher, M.; Seewald, M.J.; Mikula, M.; Oehler, S.; Mogg, M.; Vinatzer, U.; Eger, A.; Schweifer, N.; Varecka, R.; Sommergruber, W.; et al. Impact of constitutive IGF1/IGF2 stimulation on the transcriptional program of human breast cancer cells. Carcinogenesis 2007, 28, 49–59. [Google Scholar] [CrossRef] [PubMed]
- NCBI. Available online: https://www.ncbi.nlm.nih.gov (accessed on 15 January 2022).
- Auton, A.; Abecasis, G.; Altshuler, D.M.; Durbin, R.M.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [PubMed]
- Zappe, K.; Pointner, A.; Switzeny, O.J.; Magnet, U.; Tomeva, E.; Heller, J.; Mare, G.; Wagner, K.-H.; Knasmueller, S.; Haslberger, A.G. Counteraction of Oxidative Stress by Vitamin E Affects Epigenetic Regulation by Increasing Global Methylation and Gene Expression of MLH1 and DNMT1 Dose Dependently in Caco-2 Cells. Oxidative Med. Cell. Longev. 2018, 2018, 3734250. [Google Scholar] [CrossRef] [PubMed]
- Kılıç, Y.; Çelebiler, A.; Sakızlı, M. Selecting housekeeping genes as references for the normalization of quantitative PCR data in breast cancer. Clin. Transl. Oncol. 2014, 16, 184–190. [Google Scholar] [CrossRef]
- Tilli, T.M.; Castro, C.d.S.; Tuszynski, J.A.; Carels, N. A strategy to identify housekeeping genes suitable for analysis in breast cancer diseases. BMC Genom. 2016, 17, 639. [Google Scholar] [CrossRef]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef]
- Meier-Menches, S.M.; Neuditschko, B.; Zappe, K.; Schaier, M.; Gerner, M.C.; Schmetterer, K.G.; Del Favero, G.; Bonsignore, R.; Cichna-Markl, M.; Koellensperger, G.; et al. An Organometallic Gold(I) Bis-N-Heterocyclic Carbene Complex with Multimodal Activity in Ovarian Cancer Cells. Chem.Eur. J. 2020, 26, 15528–15537. [Google Scholar] [CrossRef]
- Zougman, A.; Selby, P.J.; Banks, R.E. Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics 2014, 14, 1006. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Paz, I.; Kosti, I.; Ares, M., Jr.; Cline, M.; Mandel-Gutfreund, Y. RBPmap: A web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014, 42, W361–W367. [Google Scholar] [CrossRef] [PubMed]
- Alboukadel, K. Rstatix: Pipe-Friendly Framework for Basic Statistical Tests. R Package Version 0.7.0. Available online: https://CRAN.R-project.org/package=rstatix (accessed on 15 January 2022).
- Office of Pesticide Programs, U.S. EPA. Assigning Values to Non-Detected/Non-Quantified Pesticide Residues in Human Health Food Exposure Assessments; U.S. EPA: Washington, DC, USA, 2000.
- Bretz, F.; Hothorn, T.; Westfall, P. Multiple Comparisons Using R; CRC Press: Boca Raton, FL, USA, 2016; pp. 1–183. [Google Scholar]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Luu, P.-L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; A Jones, P. DNA methylation and breast carcinogenesis. Oncogene 2002, 21, 5462–5482. [Google Scholar] [CrossRef] [PubMed]
- Mikeska, T.; Craig, J.M. DNA Methylation Biomarkers: Cancer and Beyond. Genes 2014, 5, 821–864. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Ohm, J.E. Epigenetic gene silencing in cancer—A mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 2006, 6, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Murtha, M.; Esteller, M. Extraordinary Cancer Epigenomics: Thinking Outside the Classical Coding and Promoter Box. Trends Cancer 2016, 2, 572–584. [Google Scholar] [CrossRef]
- Aran, D.; Sabato, S.; Hellman, A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013, 14, R21. [Google Scholar] [CrossRef]
- Rauscher, G.H.; Kresovich, J.K.; Poulin, M.; Yan, L.; Macias, V.; Mahmoud, A.M.; Al-Alem, U.; Kajdacsy-Balla, A.; Wiley, E.L.; Tonetti, D.; et al. Exploring DNA methylation changes in promoter, intragenic, and intergenic regions as early and late events in breast cancer formation. BMC Cancer 2015, 15, 816. [Google Scholar] [CrossRef]
- Bradnam, K.R.; Korf, I. Longer First Introns Are a General Property of Eukaryotic Gene Structure. PLoS ONE 2008, 3, e3093. [Google Scholar] [CrossRef]
- Maor, G.L.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015, 31, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Bates, D.O. Hallmarks of alternative splicing in cancer. Oncogene 2014, 33, 5311–5318. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zeng, Z.; Wei, H.; Wang, Z. Alternative splicing in cancers: From aberrant regulation to new therapeutics. Semin. Cell Dev. Biol. 2018, 75, 13–22. [Google Scholar] [CrossRef]
- Yang, Q.; Zhao, J.; Zhang, W.; Chen, D.; Wang, Y. Aberrant alternative splicing in breast cancer. J. Mol. Cell Biol. 2019, 11, 920–929. [Google Scholar] [CrossRef]
- Dvinge, H.; Bradley, R.K. Widespread intron retention diversifies most cancer transcriptomes. Genome Med. 2015, 7, 45. [Google Scholar] [CrossRef]
- Kim, D.; Shivakumar, M.; Han, S.; Sinclair, M.S.; Lee, Y.-J.; Zheng, Y.; Olopade, O.I.; Kim, D.; Lee, Y. Population-dependent Intron Retention and DNA Methylation in Breast Cancer. Mol. Cancer Res. 2018, 16, 461–469. [Google Scholar] [CrossRef]
- Monteuuis, G.; Wong, J.J.L.; Bailey, C.G.; Schmitz, U.; Rasko, J.E.J. The changing paradigm of intron retention: Regulation, ramifications and recipes. Nucleic Acids Res. 2019, 47, 11497–11513. [Google Scholar] [CrossRef]
- Kumari, A.; Sedehizadeh, S.; Brook, J.D.; Kozlowski, P.; Wojciechowska, M. Differential fates of introns in gene expression due to global alternative splicing. Hum. Genet. 2022, 141, 31–47. [Google Scholar] [CrossRef]
- Pal, S.; Gupta, R.; Davuluri, R.V. Alternative transcription and alternative splicing in cancer. Pharmacol. Ther. 2012, 136, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, U.; Barbosa-Morais, N.L.; Pan, Q.; Nachman, E.N.; Alipanahi, B.; Gonatopoulos-Pournatzis, T.; Frey, B.; Irimia, M.; Blencowe, B.J. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 2014, 24, 1774–1786. [Google Scholar] [CrossRef] [PubMed]
- Holm, K.; Hegardt, C.; Staaf, J.; Vallon-Christersson, J.; Jönsson, G.; Olsson, H.; Borg, A.; Ringnér, M. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 2010, 12, R36. [Google Scholar] [CrossRef] [PubMed]
- Bediaga, N.G.; Acha-Sagredo, A.; Guerra, I.; Viguri, A.; Albaina, C.; Diaz, I.R.; Rezola, R.; Alberdi, M.J.; Dopazo, J.; Montaner, D.; et al. DNA methylation epigenotypes in breast cancer molecular subtypes. Breast Cancer Res. 2010, 12, R77. [Google Scholar] [CrossRef]
- Widschwendter, M.; Siegmund, K.D.; Müller, H.M.; Fiegl, H.; Marth, C.; Müller-Holzner, E.; Jones, P.A.; Laird, P.W. Association of Breast Cancer DNA Methylation Profiles with Hormone Receptor Status and Response to Tamoxifen. Cancer Res. 2004, 64, 3807–3813. [Google Scholar] [CrossRef]
- Fiegl, H.; Millinger, S.; Goebel, G.; Müller-Holzner, E.; Marth, C.; Laird, P.W.; Widschwendter, M. Breast Cancer DNA Methylation Profiles in Cancer Cells and Tumor Stroma: Association with HER-2/neu Status in Primary Breast Cancer. Cancer Res. 2006, 66, 29–33. [Google Scholar] [CrossRef]
- Feng, W.; Shen, L.; Wen, S.; Rosen, D.G.; Jelinek, J.; Hu, X.; Huan, S.; Huang, M.; Liu, J.; Sahin, A.A.; et al. Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res. 2007, 9, R57. [Google Scholar] [CrossRef]
- Branham, M.T.; Marzese, D.M.; Laurito, S.R.; Gago, F.E.; Orozco, J.I.; Tello, O.M.; Vargas-Roig, L.M.; Roqué, M. Methylation profile of triple-negative breast carcinomas. Oncogenesis 2012, 1, e17. [Google Scholar] [CrossRef]
- Chai, H.; Brown, R.E. Field effect in cancer-an update. Ann Clin. Lab. Sci. 2009, 39, 331–337. [Google Scholar]
- Gadaleta, E.; Thorn, G.J.; Ross-Adams, H.; Jones, L.J.; Chelala, C. Field cancerization in breast cancer. J. Pathol. 2022, 257, 561–574. [Google Scholar] [CrossRef]
- Pashayan, N.; Antoniou, A.C.; Ivanus, U.; Esserman, L.J.; Easton, D.F.; French, D.; Sroczynski, G.; Hall, P.; Cuzick, J.; Evans, D.G.; et al. Personalized early detection and prevention of breast cancer: ENVISION consensus statement. Nat. Rev. Clin. Oncol. 2020, 17, 687–705. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Gao, Y.; Jones, A.; Ruebner, M.; Beckmann, M.W.; Wachter, D.L.; Fasching, P.A.; Widschwendter, M. DNA methylation outliers in normal breast tissue identify field defects that are enriched in cancer. Nat. Commun. 2016, 7, 10478. [Google Scholar] [CrossRef] [PubMed]
- Curtius, K.; Wright, N.A.; Graham, T.A. An evolutionary perspective on field cancerization. Nat. Rev. Cancer 2018, 18, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Rao, R.; Beebe, K.; Neckers, L.; Han, I.; Nahta, R.; Liu, J.O. Selective Inhibition of HER2-Positive Breast Cancer Cells by the HIV Protease Inhibitor Nelfinavir. J. Natl. Cancer Inst. 2012, 104, 1576–1590. [Google Scholar] [CrossRef] [PubMed]
- Arriola, E.; Marchio, C.; Tan, D.S.; Drury, S.C.; Lambros, M.B.; Natrajan, R.; Rodriguez-Pinilla, S.M.; Mackay, A.; Tamber, N.; Fenwick, K.; et al. Genomic analysis of the HER2/TOP2A amplicon in breast cancer and breast cancer cell lines. Lab. Investig. 2008, 88, 491–503. [Google Scholar] [CrossRef]
- Vranic, S.; Gatalica, Z.; Wang, Z.-Y. Update on the molecular profile of the MDA-MB-453 cell line as a model for apocrine breast carcinoma studies. Oncol. Lett. 2011, 2, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Abe-Dohmae, S.; Munehira, Y.; Aoki, R.; Kawamoto, S.; Furuya, A.; Shitara, K.; Amachi, T.; Kioka, N.; Matsuo, M.; et al. Posttranscriptional regulation of human ABCA7 and its function for the apoA-I-dependent lipid release. Biochem. Biophys. Res. Commun. 2003, 311, 313–318. [Google Scholar] [CrossRef]
- Vasquez, J.B.; Simpson, J.F.; Harpole, R.; Estus, S. Alzheimer’s disease genetics and ABCA7 splicing. J. Alzheimer’s Dis. 2017, 59, 633–641. [Google Scholar] [CrossRef]
- Anastasiadi, D.; Esteve-Codina, A.; Piferrer, F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin 2018, 11, 37. [Google Scholar] [CrossRef]
- Gimeno-Valiente, F.; López-Rodas, G.; Castillo, J.; Franco, L. Alternative Splicing, Epigenetic Modifications and Cancer: A Dangerous Triangle, or a Hopeful One? Cancers 2022, 14, 560. [Google Scholar] [CrossRef]
- Gelfman, S.; Cohen, N.; Yearim, A.; Ast, G. DNA-methylation effect on cotranscriptional splicing is dependent on GC architecture of the exon–intron structure. Genome Res. 2013, 23, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Hollander, D.; Naftelberg, S.; Lev-Maor, G.; Kornblihtt, A.R.; Ast, G. How Are Short Exons Flanked by Long Introns Defined and Committed to Splicing? Trends Genet. 2016, 32, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Hou, J.; Liu, S.; Zhang, Y.; Li, W.; Zhang, Y.; Ding, Y. Estrogen Receptor α Mediates Doxorubicin Sensitivity in Breast Cancer Cells by Regulating E-Cadherin. Front. Cell Dev. Biol. 2021, 9, 583572. [Google Scholar] [CrossRef]
- Zasadil, L.M.; Andersen, K.A.; Yeum, D.; Rocque, G.B.; Wilke, L.G.; Tevaarwerk, A.J.; Raines, R.T.; Burkard, M.E.; Weaver, B.A. Cytotoxicity of Paclitaxel in Breast Cancer Is due to Chromosome Missegregation on Multipolar Spindles. Sci. Transl. Med. 2014, 6, 229ra43. [Google Scholar] [CrossRef] [PubMed]
- Yokochi, T.; Robertson, K.D. Doxorubicin Inhibits DNMT1, Resulting in Conditional Apoptosis. Mol. Pharmacol. 2004, 66, 1415–1420. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Liu, X.; Zeng, Y.; Liu, J.; Wu, F. DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: Mechanism and clinical application. Clin. Epigenetics 2021, 13, 166. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Shi, Y.G. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development 2012, 139, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Baubec, T.; Ivánek, R.; Lienert, F.; Schübeler, D. Methylation-Dependent and -Independent Genomic Targeting Principles of the MBD Protein Family. Cell 2013, 153, 480–492. [Google Scholar] [CrossRef]
- Müller, H.M.; Fiegl, H.; Goebel, G.; Hubalek, M.M.; Widschwendter, A.; Müller-Holzner, E.; Marth, C.; Widschwendter, M. MeCP2 and MBD2 expression in human neoplastic and non-neoplastic breast tissue and its association with oestrogen receptor status. Br. J. Cancer 2003, 89, 1934–1939. [Google Scholar] [CrossRef]
- Saltzman, A.L.; Pan, Q.; Blencowe, B.J. Regulation of alternative splicing by the core spliceosomal machinery. Genes Dev. 2011, 25, 373–384. [Google Scholar] [CrossRef]
- El Marabti, E.; Younis, I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef] [PubMed]
- Anufrieva, K.S.; Shender, V.O.; Arapidi, G.P.; Pavlyukov, M.S.; Shakhparonov, M.I.; Shnaider, P.V.; Butenko, I.O.; Lagarkova, M.A.; Govorun, V.M. Therapy-induced stress response is associated with downregulation of pre-mRNA splicing in cancer cells. Genome Med. 2018, 10, 49. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
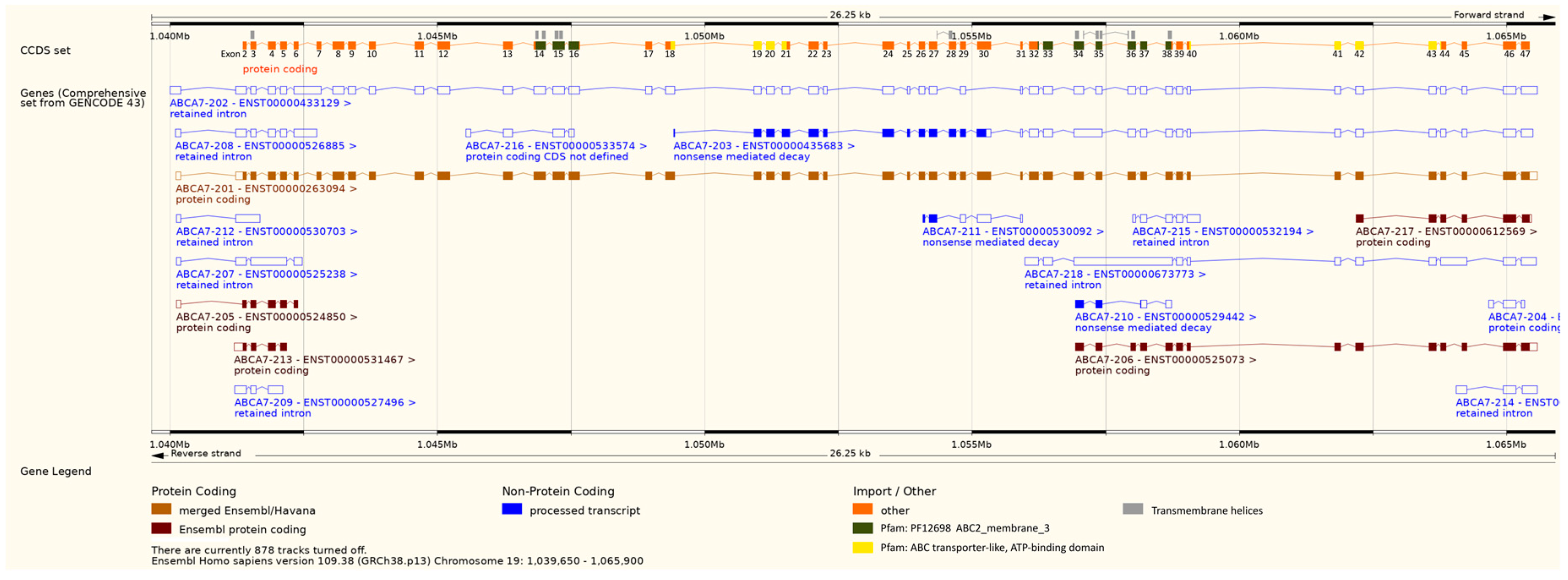{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age [y] | Menopause | Histological | Histological | MIB-1 [%] | Receptor Status | Molecular | ||
|---|---|---|---|---|---|---|---|---|---|
| Status | Type | Grading | ER | PR | Her2 | Subtype | |||
| 1 | 75 | Post | IDC | 2 | 10 | + | + | − | Luminal A |
| 2 | 65 | Post | IDC | 2 | 10 | + | + | − | Luminal A |
| 3 | 54 | Peri | IDC | 3 | 30 | + | − | − | Luminal A |
| 4 | 39 | Pre | IDC | 2 | 40 | + | + | + | Luminal B |
| 5 | 66 | Post | IDC | 2 | 60 | + | + | − | Luminal A |
| 6 | 50 | Pre | IDC | 3 | 50 | + | + | + | Luminal B |
| 7 | 73 | Post | IDC | 3 | 20 | + | + | − | Luminal A |
| 8 | 76 | Post | IDC | 2 | 20 | + | + | − | Luminal A |
| 9 | 63 | Post | IDC | 3 | 30 | + | + | − | Luminal A |
| 10 | 48 | Post | IDC | 3 | 20 | + | + | + | Luminal B |
| 11 | 58 | Post | IDC | 1 | 20 | + | + | + | Luminal B |
| 12 | 61 | Post | IDC | 3 | 70 | − | − | − | Triple-negative |
| 13 | 52 | Pre | ILC | 2 | 50 | + | + | − | Luminal A |
| 14 | 42 | Pre | IDC | 3 | 80 | +/− | − | − | Triple-negative 1 |
| 15 | 67 | Post | IDC | 3 | 40 | + | + | − | Luminal A |
| 16 | 61 | Post | ILC | 2 | 30 | + | + | − | Luminal A |
| 17 | 41 | Pre | Mucinous | 2 | 50 | + | + | + | Luminal B |
| 18 | 59 | Post | IDC | 3 | 50 | − | − | + | Her2-positive |
| 19 | 82 | Post | IDC | 3 | 30 | + | − | − | Luminal A |
| 20 | 86 | Post | IDC | 3 | 40 | + | + | − | Luminal A |
| 21 | 43 | Pre | IDC | 3 | 30 | − | − | − | Triple-negative |
| 22 | 50 | Pre | IDC | 3 | 60 | + | + | − | Luminal A |
| 23 | 55 | Post | IDC | 3 | 10 | + | + | − | Luminal A |
| 24 | 62 | Post | IDC | 3 | 70 | + | − | − | Luminal A |
| 25 | 32 | Pre | IDC | 2 | 30 | + | + | − | Luminal A |
| Primer Set | Primer Sequence (5′→3′) | Amplicon Length [bp] | Number of CpGs Analyzed | Primer Conc. [nM] | Ta [°C] |
|---|---|---|---|---|---|
| Prom | F: GTTTYGAYGGAGTAGGTTAGTGA | 156 | 12 | 200 | 58.5 |
| R: [Btn]CTTCCCTCCTCCCAACAACA | |||||
| S: GAGTAGGTTAGTGAG | |||||
| A | F: GGTGGATAGGTTTAAGGAAAATAGA | 138 | 5 | 400 | 57.0 |
| R: [Btn]CCAACTCTACCCACACCTTATA | |||||
| S: GGTTTAAGGAAAATAGA | |||||
| B | F: AGATAGTTTTGGTAGTTAATTAGATG | 78 | 4 | 400 | 57.0 |
| R: [Btn]CTACCCCAAAAATAAAATAAACCAATA | |||||
| S: AGTTTTGGTAGTTAATTAGATG | |||||
| C | F: TTGGTTGGTTTAGGGAAGTTG | 131 | 7 | 200 | 58.3 |
| R: [Btn]CCCACCCTATACCCATTTC | |||||
| S: GGTTTAGGGAAGTTGAT | |||||
| D | F: TTAATTAAGTAGTTTTTATTGGAATTAT | 130 | 9 | 400 | 51.0 |
| R: [Btn]AAAACATCCCCAATACAATATC | |||||
| S: TTTATTGGAATTATTTATGT | |||||
| E | F: TAGTGTTTGTTTTTGGATAAGTTGGA | 164 | 8 | 200 | 55.4 |
| R: [Btn]AAACCCAAATCTAAAATTAAATACTCTATA | |||||
| S: GGTATTTTTAGAGGTAG |
| Gene | Primer Set | Primer Sequence (5′→3′) | Location | Amplicon Length [bp] |
|---|---|---|---|---|
| ABCA7 | exon 1–exon 2 | F: GGAGCCCCTGGAAGAGC | boundary | 127 |
| R: CAGAGAGGTCAGGACAACG | exon 2 | |||
| exon 2–exon 3 | F: ACCGTTGTCCTGACCTCTC | exon 2 | 118 | |
| R: GCTGGACCGGCTGTCTC | boundary | |||
| exon 3–exon 4 | F: CACCATGAATGCCACTTCC | boundary | 150 | |
| R: GGAGTCGTTGAAGTTGCTCA | exon 4 | |||
| exon 4–exon 5 set 1 | F: CAACTTCAACGACTCCCTGGT | boundary | 82 | |
| R: CAGCGTCCTGTGGGCACT | exon 5 | |||
| exon 4–exon 5 set 2 | F: GCTCCAGGGTCTCATCTGTA | exon 4 | 103 | |
| R: GGAGACCAGGGAGTCGTTG | boundary | |||
| exon 5–exon 6 | F: TAGCCGATGCCCGCACTG | exon 5 | 109 | |
| R: TGAGGCTGGGCCGTGCT | boundary | |||
| exon 6–exon 7 | F: ACCAACCAAGCAGTCTCCAC | exon 6 | 88 | |
| R: CAACCCCAGGGATTCCGTG | boundary | |||
| exon 18–exon 19 | F: ACCACCACCCTGTCCATC | boundary | 137 | |
| R: AACAGCACGTTGTACTGAGG | exon 19 | |||
| PUM1 | housekeeping gene | F: TACCGCTCCGCCCGTGT | boundary | 130 |
| (exon 1–exon 2) | R: CAGGCATGTTGGGATTAGCTG | exon 2 |
| Gene | Primer Set | Primer Sequence (5′→3′) | Location | Amplicon Length [bp] |
|---|---|---|---|---|
| ABCA7 | exon 3–intron 3 | F: CAGCTCCTGGTCGAATTGC | exon 3 | 192 |
| R: CTGGTGAAGGCTCCCTGA | intron 3 | |||
| exon 4–intron 4 | F: GCTCCAGGGTCTCATCTGTA | exon 4 | 119 | |
| R: CGCACCCACTGCCTCTG | intron 4 | |||
| exon 5–intron 5 | F: TAGCCGATGCCCGCACTG | exon 5 | 176 | |
| R: CACCCTGTGCCCATTTCAC | intron 5 | |||
| exon 6–intron 6 | F: ACCAACCAAGCAGTCTCCAC | exon 6 | 218 | |
| R: CAGACGGACAAGCAGCATCA | intron 6 | |||
| exon 7–intron 7 | F: CCTTGCACAGCTTGTTGGAG | exon 7 | 168 | |
| R: CTCGCTGTCTAATCCTCCTG | intron 7 |
| Gene | Primer Set | Primer Sequence (5′→3′) | Location | Amplicon Length 1 [bp] |
|---|---|---|---|---|
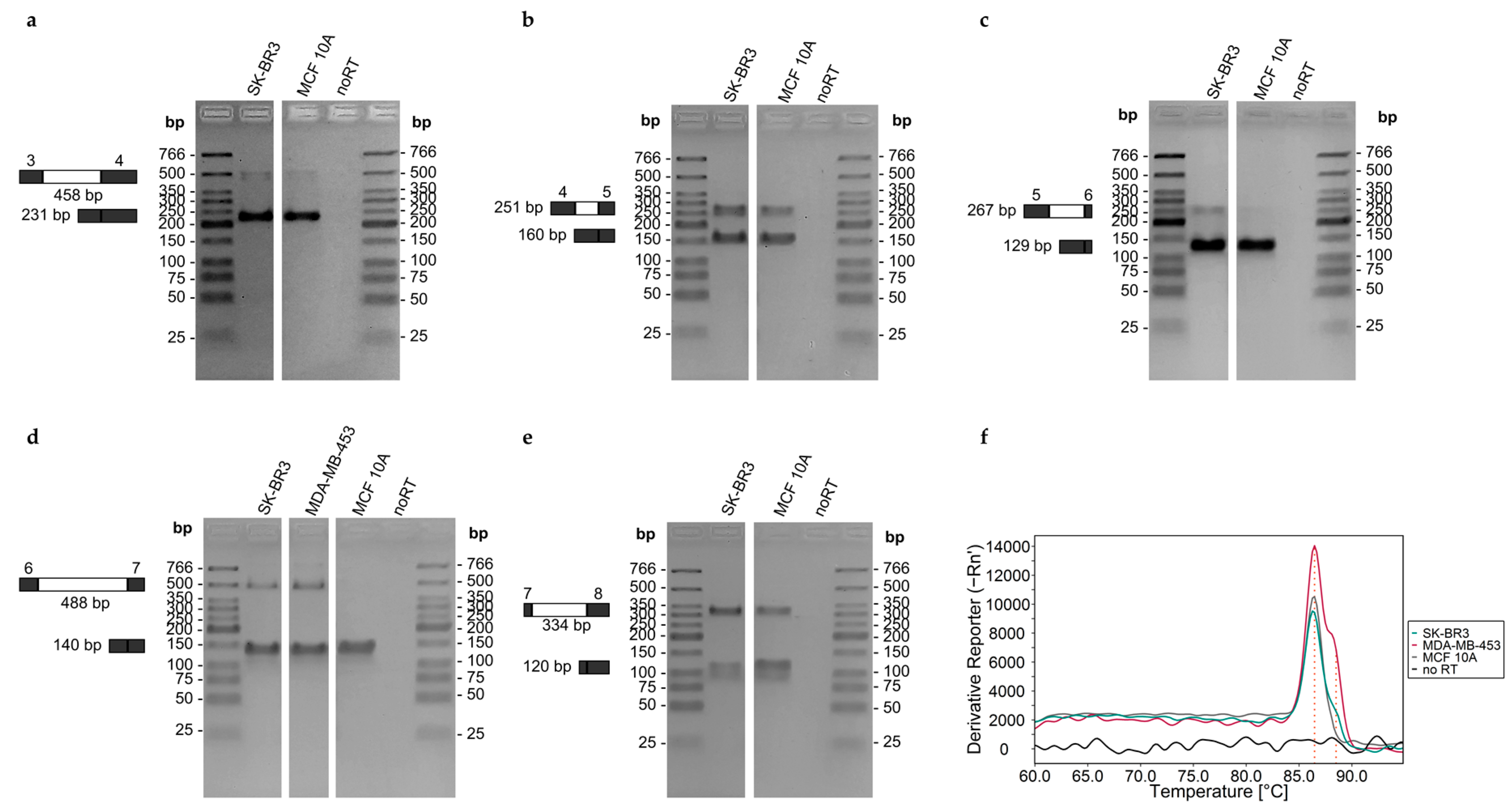
| ABCA7 | exon 3–intron 3–exon 4 | F: CAGCTCCTGGTCGAATTGC | exon 3 | 458/231 |
| R: GGAGTCGTTGAAGTTGCTCA | exon 4 | |||
| exon 4–intron 4–exon 5 | F: GCTCCAGGGTCTCATCTGTA | exon 4 | 251/160 | |
| R: CAGCGTCCTGTGGGCACT | exon 5 | |||
| exon 5–intron 5–exon 6 | F: TAGCCGATGCCCGCACTG | exon 5 | 267/129 | |
| R: GTGGAGACTGCTTGGTTGGT | exon 6 | |||
| exon 6–intron 6–exon 7 | F: ACCAACCAAGCAGTCTCCAC | exon 6 | 488/140 | |
| R: CCTCAGCGGCCTCCAACA | exon 7 | |||
| exon 7–intron 7–exon 8 | F: TGTTGGAGGCCGCTGAGG | exon 7 | 334/120 | |
| R: CCTCTGACAGCAACTCCAG | exon 8 |
| Cell Line | Treatment | Fraction | Protein | Fold Change (Log 2) | p Value 1 |
|---|---|---|---|---|---|
| MCF-7 | Dox | CYT | TET3 | −4.89 | <0.001 |
| NE | DNMT1 | −2.19 | <0.01 | ||
| DNMT3A | −1.12 | <0.01 | |||
| DNMT3B | −2.36 | <0.05 | |||
| TET2 | 3.53 | <0.01 | |||
| TET3 | −1.54 | <0.01 | |||
| MeCP2 | 1.20 | <0.05 | |||
| MBD1 | 4.34 | <0.05 | |||
| MBD3 | −3.15 | <0.001 | |||
| SETDB1 | −2.53 | <0.05 | |||
| Tax | NE | MBD3 | −0.63 | <0.01 | |
| BT-474 | Dox | NE | TET2 | −2.80 | <0.001 |
| MBD3 | −2.35 | <0.01 | |||
| Tax | NE | DNMT1 | −1.32 | <0.001 | |
| SK-BR3 | Dox | CYT | TET3 | 6.48 | <0.001 |
| NE | DNMT1 | −3.48 | <0.01 | ||
| MBD2 | −2.05 | <0.01 | |||
| MBD3 | −2.16 | <0.001 | |||
| MDA-MB-231 | Dox | NE | MeCP2 | 2.61 | <0.05 |
| MBD4 | 3.65 | <0.05 | |||
| Tax | NE | MBD3 | −1.92 | <0.01 |
| Cell Line | Treatment | Fraction | Protein | Fold Change (Log 2) | p Value 1 | Predicted Binding Site | ||
|---|---|---|---|---|---|---|---|---|
| Intron 3 | Intron 5 | Intron 6 | ||||||
| MCF-7 | Dox | NE | CELF1 | −2.36 | <0.001 | + | − | + |
| ESRP1 | −2.09 | <0.001 | + | + | − | |||
| EWSR1 | 1.44 | <0.01 | + | + | − | |||
| FUBP1 | −4.67 | <0.001 | + | − | − | |||
| FXR2 | 2.30 | <0.01 | − | − | + | |||
| HNRNPA0 | 1.41 | <0.01 | − | − | + | |||
| HNRNPA2B1 | 1.10 | <0.001 | + | − | + | |||
| HNRNPF | −0.57 | <0.01 | + | + | + | |||
| HNRNPH2 | 0.82 | <0.05 | + | + | + | |||
| HNRNPU | 1.49 | <0.001 | − | − | + | |||
| ILF2 | 1.64 | <0.001 | + | + | + | |||
| KHSRP | −2.88 | <0.01 | + | − | − | |||
| PCBP1 | −1.14 | <0.01 | + | − | + | |||
| PCBP2 | −0.93 | <0.05 | + | − | + | |||
| PUF60 | −4.10 | <0.05 | − | − | + | |||
| RBFOX1/ RBFOX2 | −1.29 | <0.05 | + | − | − | |||
| RBM8A | 0.71 | <0.05 | − | − | + | |||
| RBM24/ RBM38 | −3.49 | <0.01 | + | + | + | |||
| SNRNP70 | −0.45 | <0.01 | − | − | − | |||
| SRSF5 | 0.62 | <0.01 | + | − | + | |||
| TAF15 | 1.08 | <0.05 | + | − | + | |||
| TARDBP | −1.18 | <0.01 | + | − | + | |||
| ZC3H14 | −1.77 | <0.001 | + | − | − | |||
| Tax | NE | ESRP1 | −0.88 | <0.001 | + | + | − | |
| EWSR1 | 1.04 | <0.01 | + | + | − | |||
| HNRNPF | −0.90 | <0.001 | + | + | + | |||
| HNRNPK | 0.64 | <0.01 | − | + | − | |||
| ILF2 | 1.07 | <0.01 | + | + | + | |||
| RBM23 | 1.84 | <0.05 | − | + | + | |||
| RBM42 | 1.01 | <0.01 | − | + | − | |||
| SK-BR3 | Dox | CYT | HNRNPH2 | 1.29 | <0.01 | + | + | + |
| NE | ESRP1 | 2.05 | <0.05 | + | + | − | ||
| ESRP2 | −4.71 | <0.001 | − | + | − | |||
| FUS | 4.55 | <0.001 | − | + | + | |||
| HNRNPM | 1.25 | <0.05 | − | + | − | |||
| PUM1 | −2.74 | <0.01 | + | − | − | |||
| RBFOX1/ RBFOX2 | −2.24 | <0.05 | + | − | − | |||
| RBM24/ RBM38 | −2.65 | <0.001 | + | + | + | |||
| SRSF2 | 1.06 | <0.05 | + | + | + | |||
| ZC3H14 | −4.81 | <0.001 | + | − | − | |||
| MDA-MB-231 | Dox | NE | CELF1 | −1.80 | <0.05 | + | − | + |
| FUS | 0.70 | <0.05 | − | + | + | |||
| FXR2 | 1.06 | <0.001 | − | − | + | |||
| HNRNPA0 | 0.48 | <0.01 | − | − | + | |||
| RBM6 | 0.51 | <0.01 | − | − | + | |||
| RBM23 | 1.77 | <0.01 | − | + | + | |||
| RBM25 | 0.89 | <0.05 | − | − | + | |||
| SRSF2 | 1.52 | <0.05 | + | + | + | |||
| SRSF5 | 0.61 | <0.01 | + | − | + | |||
| TAF15 | 1.15 | <0.05 | + | − | + | |||
| ZNF326 | 0.93 | <0.05 | − | − | + | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zappe, K.; Kopic, A.; Scheichel, A.; Schier, A.-K.; Schmidt, L.E.; Borutzki, Y.; Miedl, H.; Schreiber, M.; Mendrina, T.; Pirker, C.; et al. Aberrant DNA Methylation, Expression, and Occurrence of Transcript Variants of the ABC Transporter ABCA7 in Breast Cancer. Cells 2023, 12, 1462. https://doi.org/10.3390/cells12111462
Zappe K, Kopic A, Scheichel A, Schier A-K, Schmidt LE, Borutzki Y, Miedl H, Schreiber M, Mendrina T, Pirker C, et al. Aberrant DNA Methylation, Expression, and Occurrence of Transcript Variants of the ABC Transporter ABCA7 in Breast Cancer. Cells. 2023; 12(11):1462. https://doi.org/10.3390/cells12111462
Chicago/Turabian StyleZappe, Katja, Antonio Kopic, Alexandra Scheichel, Ann-Katrin Schier, Lukas Emanuel Schmidt, Yasmin Borutzki, Heidi Miedl, Martin Schreiber, Theresa Mendrina, Christine Pirker, and et al. 2023. "Aberrant DNA Methylation, Expression, and Occurrence of Transcript Variants of the ABC Transporter ABCA7 in Breast Cancer" Cells 12, no. 11: 1462. https://doi.org/10.3390/cells12111462
APA StyleZappe, K., Kopic, A., Scheichel, A., Schier, A.-K., Schmidt, L. E., Borutzki, Y., Miedl, H., Schreiber, M., Mendrina, T., Pirker, C., Pfeiler, G., Hacker, S., Haslik, W., Pils, D., Bileck, A., Gerner, C., Meier-Menches, S., Heffeter, P., & Cichna-Markl, M. (2023). Aberrant DNA Methylation, Expression, and Occurrence of Transcript Variants of the ABC Transporter ABCA7 in Breast Cancer. Cells, 12(11), 1462. https://doi.org/10.3390/cells12111462