
RANKL Inhibition Reduces Cardiac Hypertrophy in mdx Mice and Possibly in Children with Duchenne Muscular Dystrophy

 , ,
, ,
Abstract
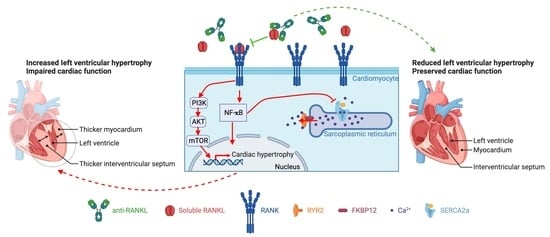
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Echocardiography
2.3. Heart Histology
2.4. Western Blotting
2.5. SERCA Activity
2.6. SERCA2a and PLN Western Blotting
2.7. Children with DMD
2.8. Statistical Analyses
3. Results
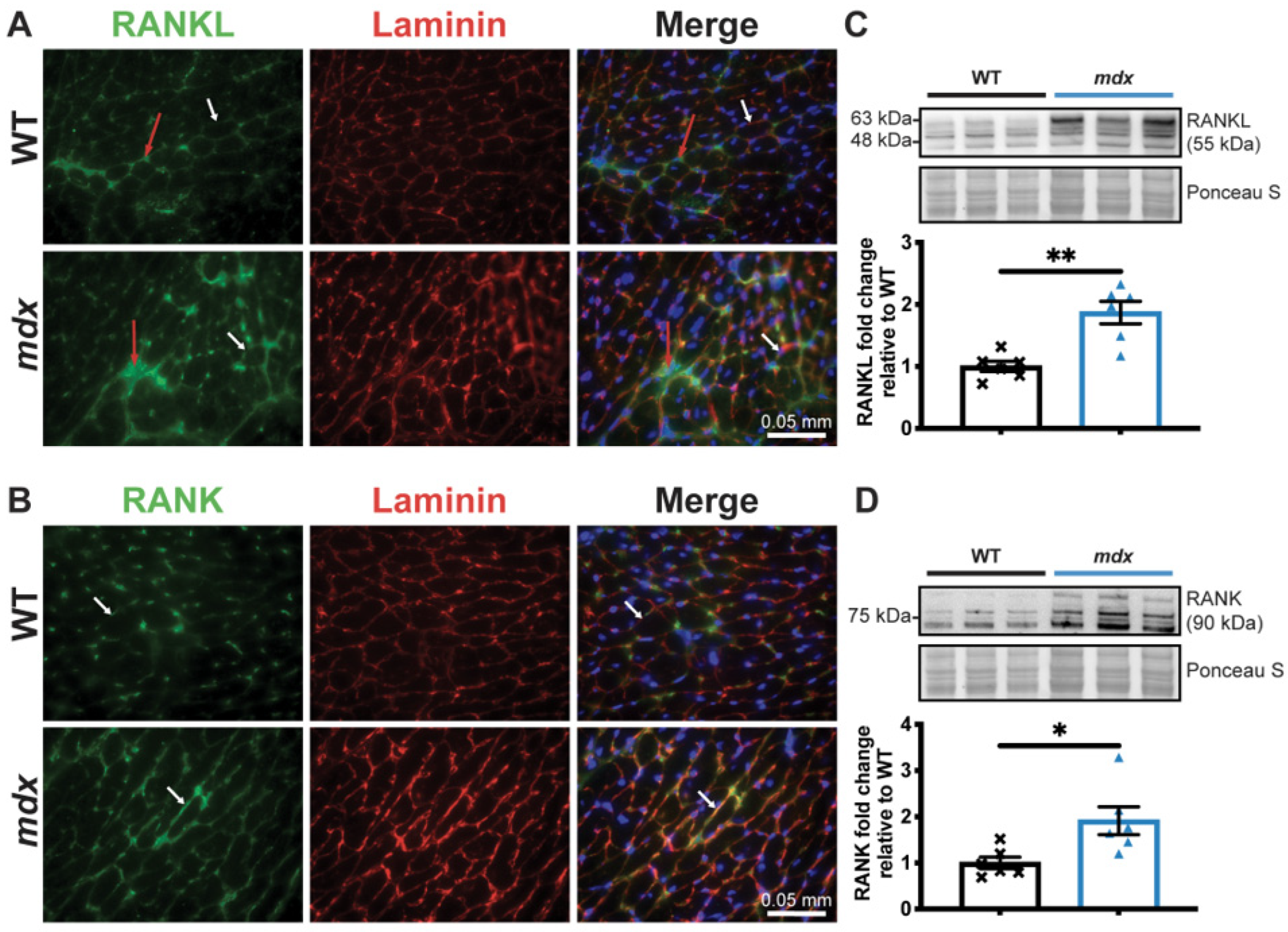
3.1. Dystrophic Mice Develop Left Ventricular Hypertrophy and Exhibit a Decrease in Cardiac Function
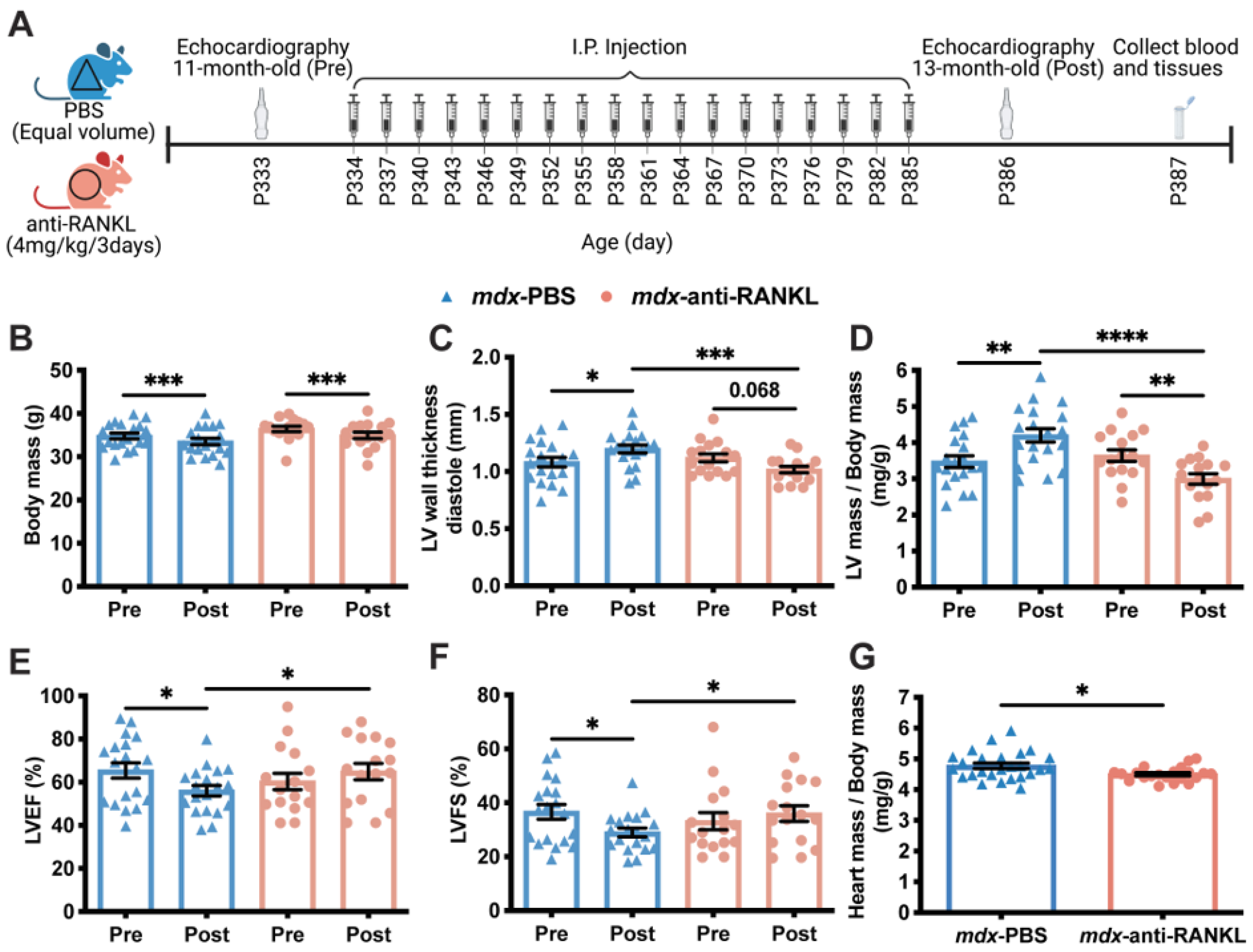
3.2. An Anti-RANKL Treatment Improves Left Ventricular Morphology and Maintains Cardiac Function in Dystrophic Mice
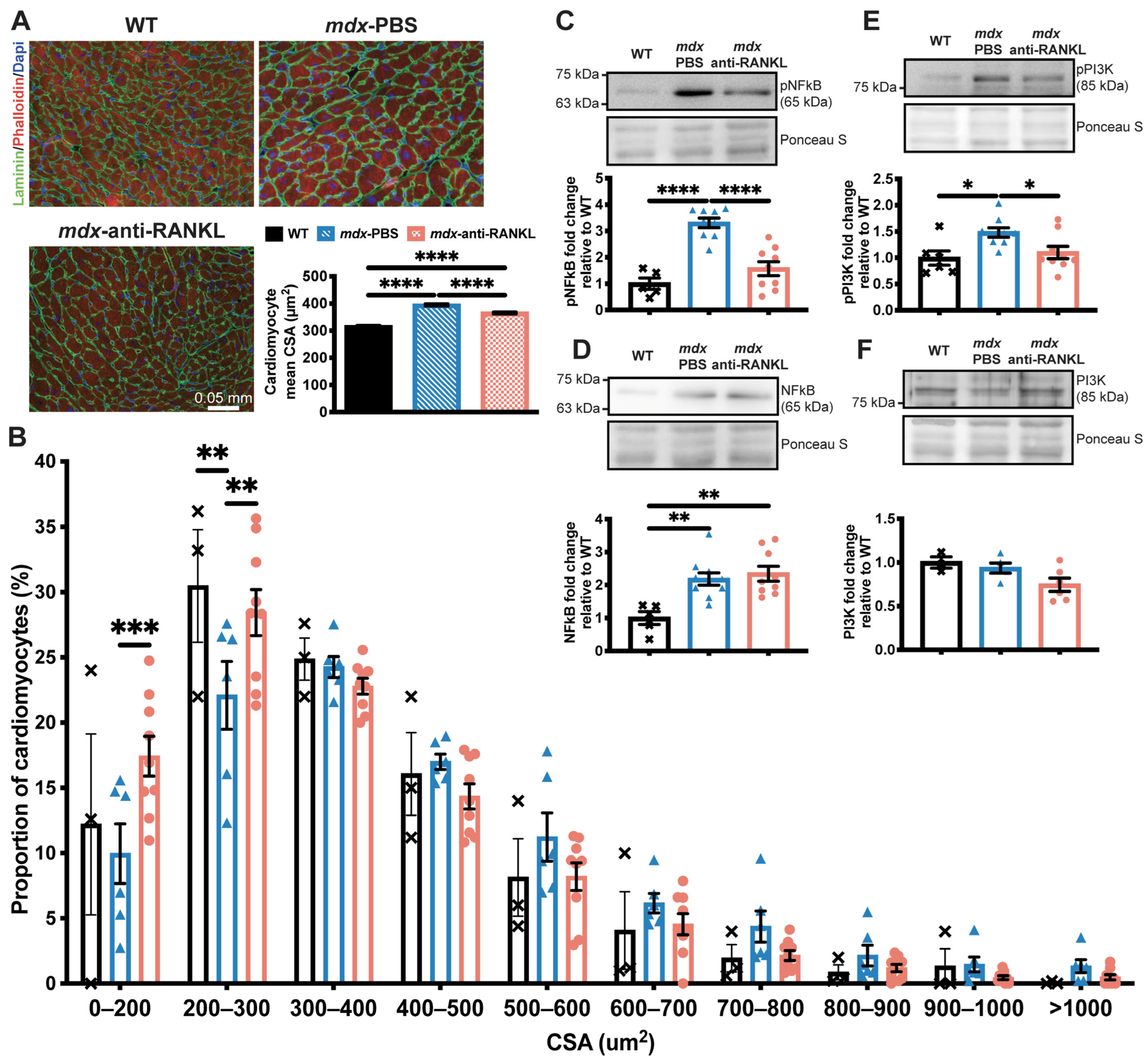
3.3. An Anti-RANKL Treatment Reduces Cardiomyocyte Surface and Inhibits Cardiac Hypertrophy Mediators in Dystrophic Mice
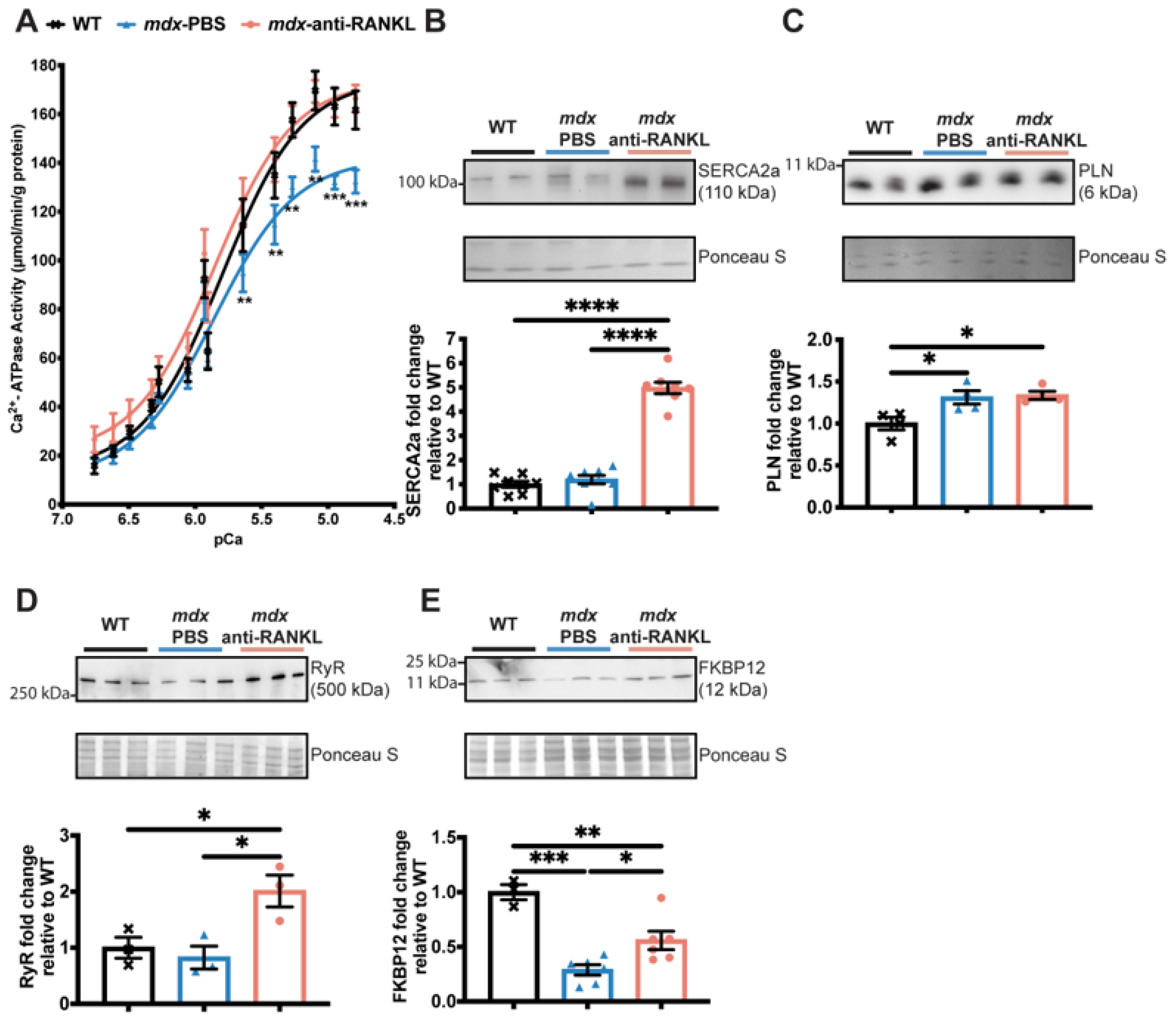
3.4. An Anti-RANKL Treatment Increases SERCA Activity and Modulates Intracellular Calcium Homeostasis Regulators in Dystrophic Hearts
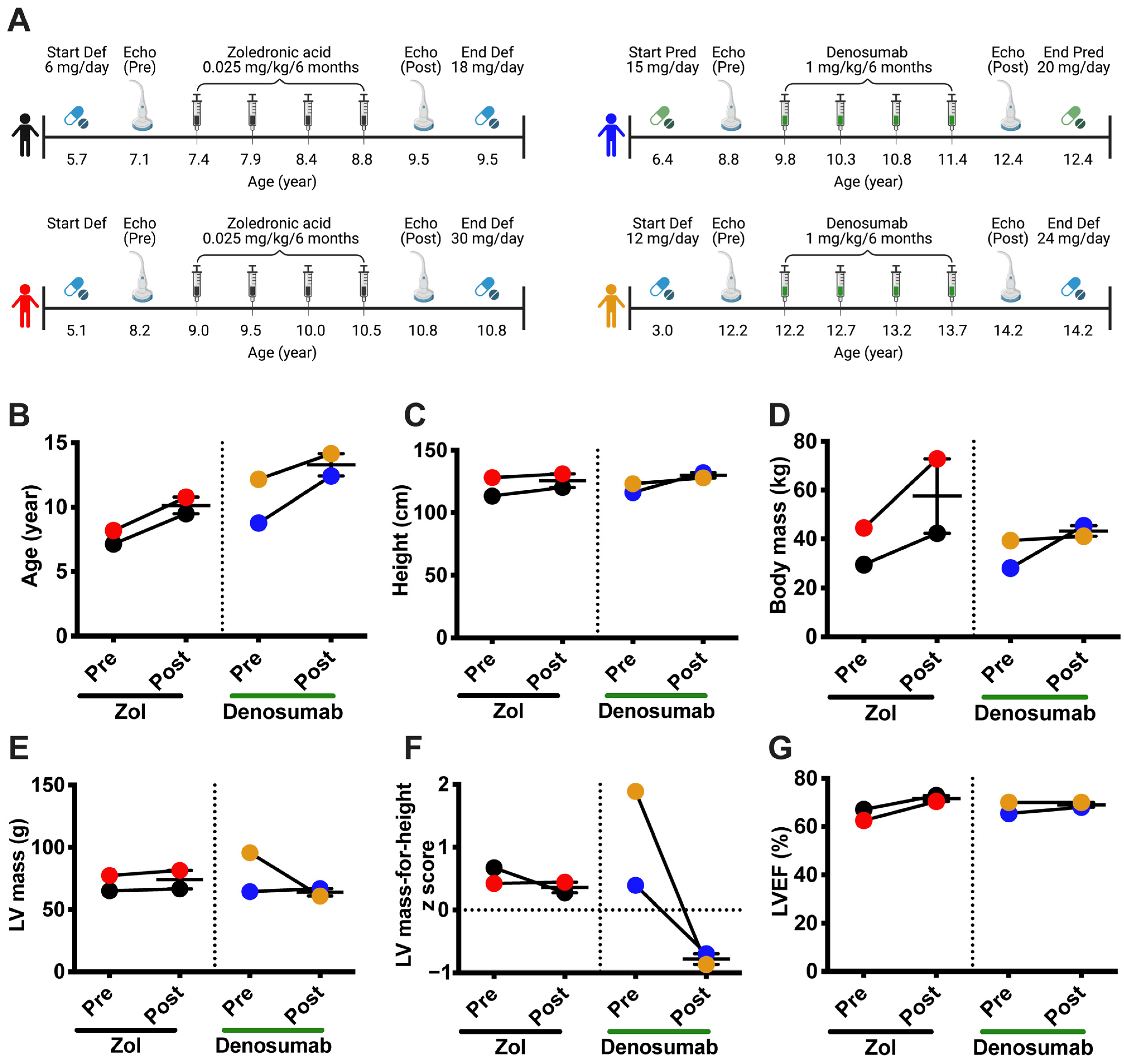
3.5. Denosumab Treatment Reduces Left Ventricular Hypertrophy in Patients with DMD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deconinck, N.; Dan, B. Pathophysiology of Duchenne Muscular Dystrophy: Current Hypotheses. Pediatr. Neurol. 2007, 36, 1–7. [Google Scholar] [CrossRef]
- Judge, D.P.; Kass, D.A.; Thompson, W.R.; Wagner, K.R. Pathophysiology and Therapy of Cardiac Dysfunction in Duchenne Muscular Dystrophy. Am. J. Cardiovasc. Drugs 2011, 11, 287–294. [Google Scholar] [CrossRef]
- Law, M.L.; Cohen, H.; Martin, A.A.; Angulski, A.B.B.; Metzger, J.M. Dysregulation of Calcium Handling in Duchenne Muscular Dystrophy-Associated Dilated Cardiomyopathy: Mechanisms and Experimental Therapeutic Strategies. J. Clin. Med. 2020, 9, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca2+ and Ca2+-Dependent Permeability Transition in Heart Mitochondria in the Early Stages of Duchenne Muscular Dystrophy. Biochim. Biophys. Acta-Bioenerg. 2020, 1861, 148250. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, V.; Poláková, E.; Janíček, R.; Shirokova, N. Mitochondrial Dysfunctions during Progression of Dystrophic Cardiomyopathy. Cell Calcium 2015, 58, 186–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, X.; Ebner, J.; Hilber, K. Voltage-Dependent Sarcolemmal Ion Channel Abnormalities in the Dystrophin-Deficient Heart. Int. J. Mol. Sci. 2018, 19, 3296. [Google Scholar] [CrossRef] [Green Version]
- Danialou, G.; Comtois, A.S.; Dudley, R.; Karpati, G.; Vincent, G.; Des Rosiers, C.; Petrof, B.J. Dystrophin-deficient Cardiomyocytes Are Abnormally Vulnerable to Mechanical Stress-induced Contractile Failure and Injury. FASEB J. 2001, 15, 1655–1657. [Google Scholar] [CrossRef]
- Passamano, L.; Taglia, A.; Palladino, A.; Viggiano, E.; D’Ambrosio, P.; Scutifero, M.; Rosaria Cecio, M.; Torre, V.; DE Luca, F.; Picillo, E.; et al. Improvement of Survival in Duchenne Muscular Dystrophy: Retrospective Analysis of 835 Patients. Acta Myol. Myopathies Cardiomyopathies Off. J. Mediterr. Soc. Myol. 2012, 31, 121–125. [Google Scholar]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. Biology of RANK, RANKL, and Osteoprotegerin. Arthritis Res. Ther. 2007, 9, S1. [Google Scholar] [CrossRef] [Green Version]
- Wolff, J. Concerning the Interrelationship between Form and Function of the Individual Parts of the Organism. By Julius Wolff, 1900. Clin. Orthop. Relat. Res. 1988, 228, 2–11. [Google Scholar]
- Boulanger Piette, A.; Hamoudi, D.; Marcadet, L.; Morin, F.; Argaw, A.; Ward, L.; Frenette, J. Targeting the Muscle-Bone Unit: Filling Two Needs with One Deed in the Treatment of Duchenne Muscular Dystrophy. Curr. Osteoporos. Rep. 2018, 16, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Feng, W.; Wang, F.; Li, W.; Gao, C.; Zhou, B.; Ma, M. Osteoprotegerin/RANK/RANKL Axis in Cardiac Remodeling Due to Immuno-Inflammatory Myocardial Disease. Exp. Mol. Pathol. 2008, 84, 213–217. [Google Scholar] [CrossRef]
- Marcadet, L.; Bouredji, Z.; Argaw, A.; Frenette, J. The Roles of RANK/RANKL/OPG in Cardiac, Skeletal, and Smooth Muscles in Health and Disease. Front. Cell Dev. Biol. 2022, 10, 903657. [Google Scholar] [CrossRef]
- Bonnet, N.; Bourgoin, L.; Biver, E.; Douni, E.; Ferrari, S. RANKL Inhibition Improves Muscle Strength and Insulin Sensitivity and Restores Bone Mass. J. Clin. Investig. 2019, 129, 3214–3223. [Google Scholar] [CrossRef]
- Pin, F.; Jones, A.J.; Huot, J.R.; Narasimhan, A.; Zimmers, T.A.; Bonewald, L.F.; Bonetto, A. RANKL Blockade Reduces Cachexia and Bone Loss Induced by Non-Metastatic Ovarian Cancer in Mice. J. Bone Miner. Res. 2021, 37, 381–396. [Google Scholar] [CrossRef]
- Dufresne, S.S.; Boulanger-Piette, A.; Bossé, S.; Argaw, A.; Hamoudi, D.; Marcadet, L.; Gamu, D.; Fajardo, V.A.; Yagita, H.; Penninger, J.M.; et al. Genetic Deletion of Muscle RANK or Selective Inhibition of RANKL Is Not as Effective as Full-Length OPG-Fc in Mitigating Muscular Dystrophy. Acta Neuropathol. Commun. 2018, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Ueland, T.; Yndestad, A.; Øie, E.; Florholmen, G.; Halvorsen, B.; Frøland, S.S.; Simonsen, S.; Christensen, G.; Gullestad, L.; Aukrust, P. Dysregulated Osteoprotegerin/RANK Ligand/RANK Axis in Clinical and Experimental Heart Failure. Circulation 2005, 111, 2461–2468. [Google Scholar] [CrossRef] [Green Version]
- Ock, S.; Ahn, J.; Lee, S.H.; Park, H.; Son, J.W.; Oh, J.G.; Yang, D.K.; Lee, W.S.; Kim, H.-S.; Rho, J.; et al. Receptor Activator of Nuclear Factor-ΚB Ligand Is a Novel Inducer of Myocardial Inflammation. Cardiovasc. Res. 2012, 94, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, F.; Crowe, L.A.; Roth, A.; Burger, F.; Lenglet, S.; Braunersreuther, V.; Brandt, K.J.; Quercioli, A.; Mach, F.; Vallée, J.-P.; et al. Treatment with Anti-RANKL Antibody Reduces Infarct Size and Attenuates Dysfunction Impacting on Neutrophil-Mediated Injury. J. Mol. Cell. Cardiol. 2016, 94, 82–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufresne, S.S.; Dumont, N.A.; Boulanger-Piette, A.; Fajardo, V.A.; Gamu, D.; Kake-Guena, S.-A.; David, R.O.; Bouchard, P.; Lavergne, É.; Penninger, J.M.; et al. Muscle RANK Is a Key Regulator of Ca 2+ Storage, SERCA Activity, and Function of Fast-Twitch Skeletal Muscles. Am. J. Physiol. Physiol. 2016, 310, C663–C672. [Google Scholar] [CrossRef] [Green Version]
- Fearnley, C.J.; Roderick, H.L.; Bootman, M.D. Calcium Signaling in Cardiac Myocytes. Cold Spring Harb. Perspect. Biol. 2011, 3, a004242. [Google Scholar] [CrossRef] [Green Version]
- Mareedu, S.; Million, E.D.; Duan, D.; Babu, G.J. Abnormal Calcium Handling in Duchenne Muscular Dystrophy: Mechanisms and Potential Therapies. Front. Physiol. 2021, 12, 647010. [Google Scholar] [CrossRef]
- Quinlan, J.G.; Hahn, H.S.; Wong, B.L.; Lorenz, J.N.; Wenisch, A.S.; Levin, L.S. Evolution of the Mdx Mouse Cardiomyopathy: Physiological and Morphological Findings. Neuromuscul. Disord. 2004, 14, 491–496. [Google Scholar] [CrossRef]
- Wehling-Henricks, M.; Jordan, M.C.; Roos, K.P.; Deng, B.; Tidball, J.G. Cardiomyopathy in Dystrophin-Deficient Hearts Is Prevented by Expression of a Neuronal Nitric Oxide Synthase Transgene in the Myocardium. Hum. Mol. Genet. 2005, 14, 1921–1933. [Google Scholar] [CrossRef] [PubMed]
- Fayssoil, A.; Renault, G.; Guerchet, N.; Marchiol-Fournigault, C.; Fougerousse, F.; Richard, I. Cardiac Characterization of Mdx Mice Using High-Resolution Doppler Echocardiography. J. Ultrasound Med. 2013, 32, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Van Erp, C.; Loch, D.; Laws, N.; Trebbin, A.; Hoey, A.J. Timeline of Cardiac Dystrophy in 3-18-Month-Old MDX Mice. Muscle Nerve 2010, 42, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Hamoudi, D.; Marcadet, L.; Piette Boulanger, A.; Yagita, H.; Bouredji, Z.; Argaw, A.; Frenette, J. An Anti-RANKL Treatment Reduces Muscle Inflammation and Dysfunction and Strengthens Bone in Dystrophic Mice. Hum. Mol. Genet. 2019, 28, 3101–3112. [Google Scholar] [CrossRef]
- Wu, J.; Bu, L.; Gong, H.; Jiang, G.; Li, L.; Ma, H.; Zhou, N.; Lin, L.; Chen, Z.; Ye, Y.; et al. Effects of Heart Rate and Anesthetic Timing on High-Resolution Echocardiographic Assessment Under Isoflurane Anesthesia in Mice. J. Ultrasound Med. 2010, 29, 1771–1778. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Kassiri, Z.; Virag, J.A.I.; de Castro Brás, L.E.; Scherrer-Crosbie, M. Guidelines for Measuring Cardiac Physiology in Mice. Am. J. Physiol. Circ. Physiol. 2018, 314, H733–H752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, B.J.; Mackie, A.S.; Mitsnefes, M.; Ali, H.; Mamber, S.; Colan, S.D. A Novel Method of Expressing Left Ventricular Mass Relative to Body Size in Children. Circulation 2008, 117, 2769–2775. [Google Scholar] [CrossRef] [Green Version]
- Spurney, C.F.; Knoblach, S.; Pistilli, E.E.; Nagaraju, K.; Martin, G.R.; Hoffman, E.P. Dystrophin-Deficient Cardiomyopathy in Mouse: Expression of Nox4 and Lox Are Associated with Fibrosis and Altered Functional Parameters in the Heart. Neuromuscul. Disord. 2008, 18, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Putten, M.; van der Pijl, E.M.; Hulsker, M.; Verhaart, I.E.C.; Nadarajah, V.D.; van der Weerd, L.; Aartsma-Rus, A. Low Dystrophin Levels in Heart Can Delay Heart Failure in Mdx Mice. J. Mol. Cell. Cardiol. 2014, 69, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, C.J.; Duhamel, T.A.; Dhalla, N.S. Mechanisms for the Transition from Physiological to Pathological Cardiac Hypertrophy. Can. J. Physiol. Pharmacol. 2020, 98, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Fauconnier, J.; Thireau, J.; Reiken, S.; Cassan, C.; Richard, S.; Matecki, S.; Marks, A.R.; Lacampagne, A. Leaky RyR2 Trigger Ventricular Arrhythmias in Duchenne Muscular Dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 1559–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, T.; Park, S.Y.; Lee, S.H.; Park, J.H.; Suh, S.W. Comparison of Denosumab and Zoledronic Acid in Postmenopausal Women with Osteoporosis: Bone Mineral Density (BMD) and Trabecular Bone Score (TBS). J. Korean Med. Sci. 2022, 37, e68. [Google Scholar] [CrossRef] [PubMed]
- Tissot, C.; Singh, Y.; Sekarski, N. Echocardiographic Evaluation of Ventricular Function—For the Neonatologist and Pediatric Intensivist. Front. Pediatr. 2018, 6, 79. [Google Scholar] [CrossRef] [Green Version]
- Slavic, S.; Andrukhova, O.; Ford, K.; Handschuh, S.; Latic, N.; Reichart, U.; Sasgary, S.; Bergow, C.; Hofbauer, L.C.; Kostenuik, P.J.; et al. Selective Inhibition of Receptor Activator of NF-ΚB Ligand (RANKL) in Hematopoietic Cells Improves Outcome after Experimental Myocardial Infarction. J. Mol. Med. 2018, 96, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; ten Hove, M.; Schneider, J.E.; Stuckey, D.J.; Sebag-Montefiore, L.; Bia, B.L.; Radda, G.K.; Davies, K.E.; Neubauer, S.; Clarke, K. Abnormal Cardiac Morphology, Function and Energy Metabolism in the Dystrophic Mdx Mouse: An MRI and MRS Study. J. Mol. Cell. Cardiol. 2008, 45, 754–760. [Google Scholar] [CrossRef]
- Wada, T.; Nakashima, T.; Hiroshi, N.; Penninger, J.M. RANKL–RANK Signaling in Osteoclastogenesis and Bone Disease. Trends Mol. Med. 2006, 12, 17–25. [Google Scholar] [CrossRef]
- Peterson, J.M.; Wang, D.J.; Shettigar, V.; Roof, S.R.; Canan, B.D.; Bakkar, N.; Shintaku, J.; Gu, J.-M.; Little, S.C.; Ratnam, N.M.; et al. NF-ΚB Inhibition Rescues Cardiac Function by Remodeling Calcium Genes in a Duchenne Muscular Dystrophy Model. Nat. Commun. 2018, 9, 3431. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Chen, M.; Dawood, F.; Zurawska, U.; Li, J.Y.; Parker, T.; Kassiri, Z.; Kirshenbaum, L.A.; Arnold, M.; Khokha, R.; et al. Tumor Necrosis Factor-α Mediates Cardiac Remodeling and Ventricular Dysfunction After Pressure Overload State. Circulation 2007, 115, 1398–1407. [Google Scholar] [CrossRef] [Green Version]
- Kubota, T.; McTiernan, C.F.; Frye, C.S.; Slawson, S.E.; Lemster, B.H.; Koretsky, A.P.; Demetris, A.J.; Feldman, A.M. Dilated Cardiomyopathy in Transgenic Mice with Cardiac-Specific Overexpression of Tumor Necrosis Factor-α. Circ. Res. 1997, 81, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Shioi, T. The Conserved Phosphoinositide 3-Kinase Pathway Determines Heart Size in Mice. EMBO J. 2000, 19, 2537–2548. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.B.; Kim, J.H.; Kim, K.; Youn, B.U.; Ko, A.; Lee, S.Y.; Kim, N. Akt Induces Osteoclast Differentiation through Regulating the GSK3β/NFATc1 Signaling Cascade. J. Immunol. 2012, 188, 163–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.E.; Woo, K.M.; Kim, S.Y.; Kim, H.M.; Kwack, K.; Lee, Z.H.; Kim, H.H. The Phosphatidylinositol 3-Kinase, P38, and Extracellular Signal-Regulated Kinase Pathways Are Involved in Osteoclast Differentiation. Bone 2002, 30, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Houser, S.R. Ca2+ Signaling Domains Responsible For Cardiac Hypertrophy and Arrhythmias. Circ. Res. 2009, 104, 413–415. [Google Scholar] [CrossRef] [Green Version]
- Lipskaia, L.; Chemaly, E.R.; Hadri, L.; Lompre, A.-M.; Hajjar, R.J. Sarcoplasmic Reticulum Ca2+ ATPase as a Therapeutic Target for Heart Failure. Expert Opin. Biol. Ther. 2010, 10, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Mareedu, S.; Pachon, R.; Thilagavathi, J.; Fefelova, N.; Balakrishnan, R.; Niranjan, N.; Xie, L.-H.; Babu, G.J. Sarcolipin Haploinsufficiency Prevents Dystrophic Cardiomyopathy in Mdx Mice. Am. J. Physiol. Circ. Physiol. 2021, 320, H200–H210. [Google Scholar] [CrossRef]
- Wasala, N.B.; Yue, Y.; Lostal, W.; Wasala, L.P.; Niranjan, N.; Hajjar, R.J.; Babu, G.J.; Duan, D. Single SERCA2a Therapy Ameliorated Dilated Cardiomyopathy for 18 Months in a Mouse Model of Duchenne Muscular Dystrophy. Mol. Ther. 2020, 28, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Kolos, J.M.; Voll, A.M.; Bauder, M.; Hausch, F. FKBP Ligands—Where We Are and Where to Go? Front. Pharmacol. 2018, 9, 1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonano, L.A.; Jones, P.P. FK506-Binding Proteins 12 and 12.6 (FKBPs) as Regulators of Cardiac Ryanodine Receptors: Insights from New Functional and Structural Knowledge. Channels 2017, 11, 415–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincenti, M.; Farah, C.; Amedro, P.; Scheuermann, V.; Lacampagne, A.; Cazorla, O. Early Myocardial Dysfunction and Benefits of Cardiac Treatment in Young X-Linked Duchenne Muscular Dystrophy Mice. Cardiovasc. Drugs Ther. 2021, 36, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Lv, F.; Cai, X.; Yang, W.; Gao, L.; Chen, L.; Wu, J.; Ji, L. Denosumab or Romosozumab Therapy and Risk of Cardiovascular Events in Patients with Primary Osteoporosis: Systematic Review and Meta- Analysis. Bone 2020, 130, 115121. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcadet, L.; Juracic, E.S.; Khan, N.; Bouredji, Z.; Yagita, H.; Ward, L.M.; Tupling, A.R.; Argaw, A.; Frenette, J. RANKL Inhibition Reduces Cardiac Hypertrophy in mdx Mice and Possibly in Children with Duchenne Muscular Dystrophy. Cells 2023, 12, 1538. https://doi.org/10.3390/cells12111538
Marcadet L, Juracic ES, Khan N, Bouredji Z, Yagita H, Ward LM, Tupling AR, Argaw A, Frenette J. RANKL Inhibition Reduces Cardiac Hypertrophy in mdx Mice and Possibly in Children with Duchenne Muscular Dystrophy. Cells. 2023; 12(11):1538. https://doi.org/10.3390/cells12111538
Chicago/Turabian StyleMarcadet, Laetitia, Emma Sara Juracic, Nasrin Khan, Zineb Bouredji, Hideo Yagita, Leanne M. Ward, A. Russell Tupling, Anteneh Argaw, and Jérôme Frenette. 2023. "RANKL Inhibition Reduces Cardiac Hypertrophy in mdx Mice and Possibly in Children with Duchenne Muscular Dystrophy" Cells 12, no. 11: 1538. https://doi.org/10.3390/cells12111538
APA StyleMarcadet, L., Juracic, E. S., Khan, N., Bouredji, Z., Yagita, H., Ward, L. M., Tupling, A. R., Argaw, A., & Frenette, J. (2023). RANKL Inhibition Reduces Cardiac Hypertrophy in mdx Mice and Possibly in Children with Duchenne Muscular Dystrophy. Cells, 12(11), 1538. https://doi.org/10.3390/cells12111538