
Role of A-Kinase Anchoring Protein 1 in Retinal Ganglion Cells: Neurodegeneration and Neuroprotection
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
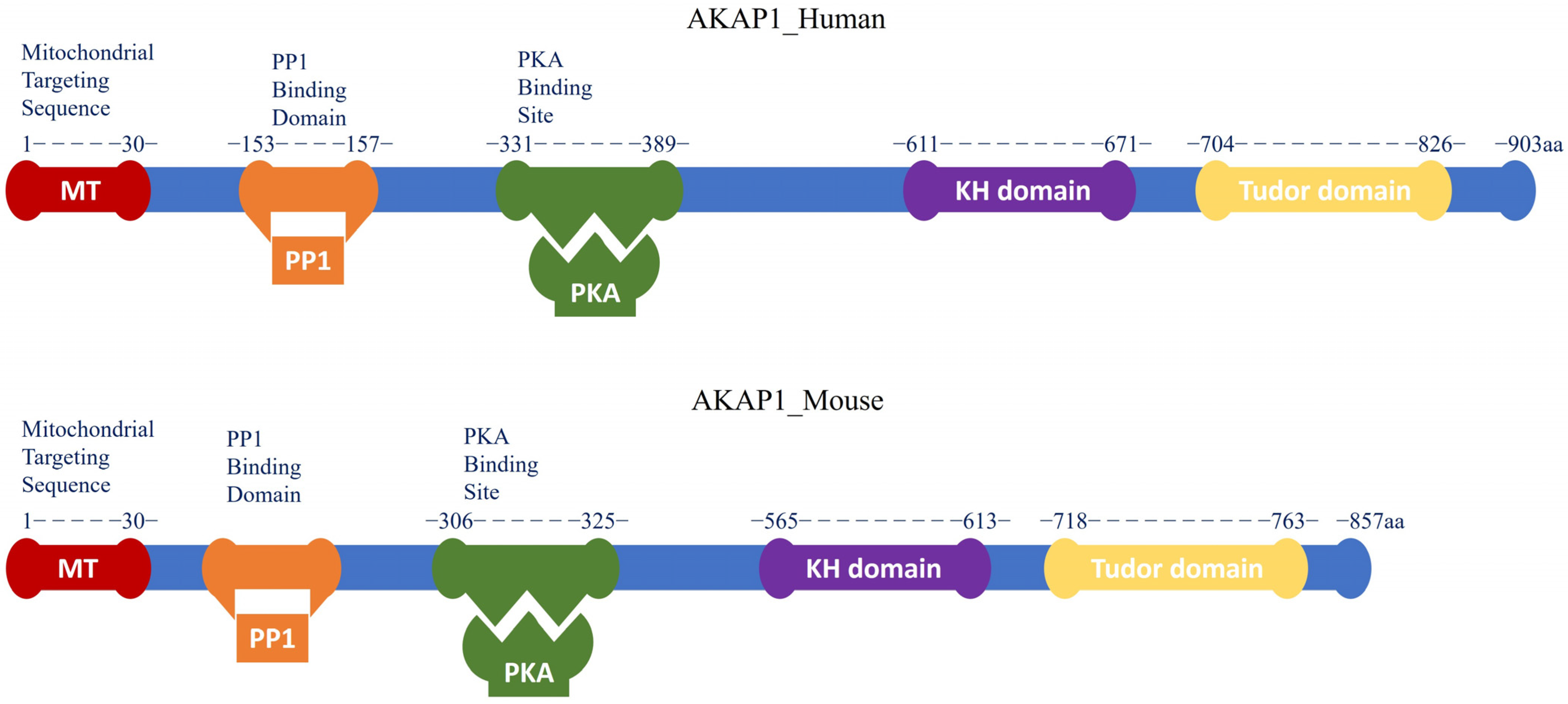
1.1. AKAP1
1.2. cAMP/PKA Signaling and AKAP1 Complex
1.3. AKAP1 and Glaucomatous Retinal Ganglion Cells
2. AKAP1 and Its Role in Mitochondria
2.1. AKAP1 and Mitochondrial Dynamics
2.2. AKAP1 and Mitochondrial Bioenergetics
2.3. AKAP1 and Mitophagy
3. AKAP1 in Neurodegeneration
3.1. AKAP1 in Glaucomatous Neurodegeneration
3.2. AKAP1 in Other Neurodegenerative Diseases
4. AKAP1 in Neuroprotection
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Affaitati, A.; Cardone, L.; De Cristofaro, T.; Carlucci, A.; Ginsberg, M.D.; Varrone, S.; Gottesman, M.E.; Avvedimento, E.V.; Feliciello, A. Essential role of A-kinase anchor protein 121 for cAMP signaling to mitochondria. J. Biol. Chem. 2003, 278, 4286–4294. [Google Scholar] [CrossRef] [Green Version]
- Merrill, R.A.; Dagda, R.K.; Dickey, A.S.; Cribbs, J.T.; Green, S.H.; Usachev, Y.M.; Strack, S. Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS Biol. 2011, 9, e1000612. [Google Scholar] [CrossRef] [Green Version]
- Ju, W.-K.; Perkins, G.A.; Kim, K.-Y.; Bastola, T.; Choi, W.-Y.; Choi, S.-H. Glaucomatous optic neuropathy: Mitochondrial dynamics, dysfunction and protection in retinal ganglion cells. Prog. Retin. Eye Res. 2022, 2022, 101136. [Google Scholar] [CrossRef]
- Carlucci, A.; Lignitto, L.; Feliciello, A. Control of mitochondria dynamics and oxidative metabolism by cAMP, AKAPs and the proteasome. Trends Cell Biol. 2008, 18, 604–613. [Google Scholar] [CrossRef]
- Banky, P.; Huang, L.J.-S.; Taylor, S.S. Dimerization/Docking Domain of the Type Iα Regulatory Subunit of cAMP-dependent Protein Kinase: Requirements for Dimerization and Docking Are Distinct but Overlapping*. J. Biol. Chem. 1998, 273, 35048–35055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feliciello, A.; Rubin, C.S.; Avvedimento, E.V.; Gottesman, M.E. Expression of A Kinase Anchor Protein 121 Is Regulated by Hormones in Thyroid and Testicular Germ Cells*. J. Biol. Chem. 1998, 273, 23361–23366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.-Y.; Moss, S.B.; Rubin, C.S. Characterization of S-AKAP84, a Novel Developmentally Regulated A Kinase Anchor Protein of Male Germ Cells (*). J. Biol. Chem. 1995, 270, 27804–27811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrill, R.A.; Strack, S. Mitochondria: A kinase anchoring protein 1, a signaling platform for mitochondrial form and function. Int. J. Biochem. Cell Biol. 2014, 48, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Cardone, L.; de Cristofaro, T.; Affaitati, A.; Garbi, C.; Ginsberg, M.D.; Saviano, M.; Varrone, S.; Rubin, C.S.; Gottesman, M.E.; Avvedimento, E.V.; et al. A-Kinase Anchor Protein 84/121 are Targeted to Mitochondria and Mitotic Spindles by Overlapping Amino-terminal Motifs. J. Mol. Biol. 2002, 320, 663–675. [Google Scholar] [CrossRef]
- Lemay, J.; Maidou-Peindara, P.; Cancio, R.; Ennifar, E.; Coadou, G.; Maga, G.; Rain, J.-C.; Benarous, R.; Liu, L.X. AKAP149 Binds to HIV-1 Reverse Transcriptase and Is Involved in the Reverse Transcription. J. Mol. Biol. 2008, 383, 783–796. [Google Scholar] [CrossRef]
- Chen, Q.; Lin, R.-Y.; Rubin, C.S. Organelle-specific targeting of protein kinase AII (PKAII): Molecular and in situ characterization of murine A kinase anchor proteins that recruit regulatory subunits of PKAII to the cytoplasmic surface of mitochondria. J. Biol. Chem. 1997, 272, 15247–15257. [Google Scholar] [CrossRef] [Green Version]
- Asirvatham, A.L.; Galligan, S.G.; Schillace, R.V.; Davey, M.P.; Vasta, V.; Beavo, J.A.; Carr, D.W. A-Kinase Anchoring Proteins Interact with Phosphodiesterases in T Lymphocyte Cell Lines1. J. Immunol. 2004, 173, 4806–4814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinderman, F.S.; Kim, C.; von Daake, S.; Ma, Y.; Pham, B.Q.; Spraggon, G.; Xuong, N.-H.; Jennings, P.A.; Taylor, S.S. A Dynamic Mechanism for AKAP Binding to RII Isoforms of cAMP-Dependent Protein Kinase. Mol. Cell 2006, 24, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grozdanov, P.N.; Stocco, D.M. Short RNA Molecules with High Binding Affinity to the KH Motif of A-Kinase Anchoring Protein 1 (AKAP1): Implications for the Regulation of Steroidogenesis. Mol. Endocrinol. 2012, 26, 2104–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, W. A-kinase anchoring protein 1 (AKAP1) and its role in some cardiovascular diseases. J. Mol. Cell. Cardiol. 2020, 138, 99–109. [Google Scholar] [CrossRef]
- Dickey, A.S.; Strack, S. PKA/AKAP1 and PP2A/Bβ2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 15716–15726. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.C.; Benedetto, G.D.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Flippo, K.H.; Gnanasekaran, A.; Perkins, G.A.; Ajmal, A.; Merrill, R.A.; Dickey, A.S.; Taylor, S.S.; McKnight, G.S.; Chauhan, A.K.; Usachev, Y.M.; et al. AKAP1 Protects from Cerebral Ischemic Stroke by Inhibiting Drp1-Dependent Mitochondrial Fission. J. Neurosci. 2018, 38, 8233–8242. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Feng, J.; Ma, D.; Wang, F.; Wang, Y.; Li, C.; Wang, X.; Yin, X.; Zhang, M.; Dagda, R.K.; et al. Neuroprotective mitochondrial remodeling by AKAP121/PKA protects HT22 cell from glutamate-induced oxidative stress. Mol. Nuerobiol. 2019, 56, 5586–5607. [Google Scholar] [CrossRef]
- Liu, Y.; Merrill, R.A.; Strack, S. A-kinase anchoring protein 1: Emerging roles in regulating mitochondrial form and function in health and disease. Cells 2020, 9, 298. [Google Scholar] [CrossRef] [Green Version]
- Bouvet, M.; Blondeau, J.-P.; Lezoualc’h, F. The Epac1 protein: Pharmacological modulators, cardiac signalosome and pathophysiology. Cells 2019, 8, 1543. [Google Scholar] [CrossRef] [Green Version]
- Kamenetsky, M.; Middelhaufe, S.; Bank, E.M.; Levin, L.R.; Buck, J.; Steegborn, C. Molecular Details of cAMP Generation in Mammalian Cells: A Tale of Two Systems. J. Mol. Biol. 2006, 362, 623–639. [Google Scholar] [CrossRef] [Green Version]
- Kopperud, R.; Krakstad, C.; Selheim, F.; Døskeland, S.O. cAMP effector mechanisms. Novel twists for an ‘old’signaling system. FEBS Lett. 2003, 546, 121–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herberg, F.W.; Maleszka, A.; Eide, T.; Vossebein, L.; Tasken, K. Analysis of A-kinase anchoring protein (AKAP) interaction with protein kinase A (PKA) regulatory subunits: PKA isoform specificity in AKAP binding11Edited by J. A. Wells. J. Mol. Biol. 2000, 298, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Thuen, M.; Singstad, T.E.; Pedersen, T.B.; Haraldseth, O.; Berry, M.; Sandvig, A.; Brekken, C. Manganese-enhanced MRI of the optic visual pathway and optic nerve injury in adult rats. J. Magn. Reson. Imaging An. Off. Journal. Int. Soc. Magn. Reson. Med. 2005, 22, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.; Ahmed, Z.; Lorber, B.; Douglas, M.; Logan, A. Regeneration of axons in the visual system. Restor. Neurol. Neurosci. 2008, 26, 147–174. [Google Scholar] [PubMed]
- Nadal-Nicolas, F.M.; Jimenez-Lopez, M.; Salinas-Navarro, M.; Sobrado-Calvo, P.; Alburquerque-Bejar, J.J.; Vidal-Sanz, M.; Agudo-Barriuso, M. Whole number, distribution and co-expression of brn3 transcription factors in retinal ganglion cells of adult albino and pigmented rats. PLoS ONE 2012, 7, e49830. [Google Scholar] [CrossRef] [Green Version]
- You, Y.; Gupta, V.K.; Li, J.C.; Klistorner, A.; Graham, S.L. Optic neuropathies: Characteristic features and mechanisms of retinal ganglion cell loss. Rev. Neurosci. 2013, 24, 301–321. [Google Scholar] [CrossRef]
- Sanes, J.R.; Masland, R.H. The Types of Retinal Ganglion Cells: Current Status and Implications for Neuronal Classification. Annu. Rev. Neurosci. 2015, 38, 221–246. [Google Scholar] [CrossRef]
- Kim, U.S.; Mahroo, O.A.; Mollon, J.D.; Yu-Wai-Man, P. Retinal Ganglion Cells—Diversity of Cell Types and Clinical Relevance. Front. Neurol. 2021, 12, 661938. [Google Scholar] [CrossRef]
- Lazzerini Ospri, L.; Prusky, G.; Hattar, S. Mood, the Circadian System, and Melanopsin Retinal Ganglion Cells. Annu. Rev. Neurosci. 2017, 40, 539–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quigley, H.A.; Addicks, E.M. Chronic experimental glaucoma in primates. II. Effect of extended intraocular pressure elevation on optic nerve head and axonal transport. Investig. Ophthalmol. Vis. Sci. 1980, 19, 137–152. [Google Scholar]
- Martin, K.R.; Quigley, H.A.; Valenta, D.; Kielczewski, J.; Pease, M.E. Optic nerve dynein motor protein distribution changes with intraocular pressure elevation in a rat model of glaucoma. Exp. Eye Res. 2006, 83, 255–262. [Google Scholar] [CrossRef]
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: Mechanisms and consequences. Prog. Retin. Eye Res. 2006, 25, 490–513. [Google Scholar] [CrossRef] [Green Version]
- Gregerson, D.S. Immune privilege in the retina. Ocul. Immunol. Inflamm. 1998, 6, 257–267. [Google Scholar] [CrossRef]
- Streilein, J.W.; Okamoto, S.; Sano, Y.; Taylor, A.W. Neural control of ocular immune privilege. Ann. N. Y. Acad. Sci. 2000, 917, 297–306. [Google Scholar] [CrossRef]
- Tezel, G.; Wax, M.B. Increased production of tumor necrosis factor-α by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J. Neurosci. 2000, 20, 8693–8700. [Google Scholar] [CrossRef] [Green Version]
- Pannicke, T.; Uckermann, O.; Iandiev, I.; Biedermann, B.; Wiedemann, P.; Perlman, I.; Reichenbach, A.; Bringmann, A. Altered membrane physiology in Müller glial cells after transient ischemia of the rat retina. Glia 2005, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.-J.; Guo, Y.; Agey, P.; Wheeler, L.; Hare, W.A. α2 adrenergic modulation of NMDA receptor function as a major mechanism of RGC protection in experimental glaucoma and retinal excitotoxicity. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4515–4522. [Google Scholar] [CrossRef]
- Neufeld, A.H.; Gachie, E.N. The inherent, age-dependent loss of retinal ganglion cells is related to the lifespan of the species. Neurobiol. Aging 2003, 24, 167–172. [Google Scholar] [CrossRef]
- Kim, K.Y.; Perkins, G.A.; Shim, M.S.; Bushong, E.; Alcasid, N.; Ju, S.; Ellisman, M.H.; Weinreb, R.N.; Ju, W.K. DRP1 inhibition rescues retinal ganglion cells and their axons by preserving mitochondrial integrity in a mouse model of glaucoma. Cell Death Dis. 2015, 6, e1839. [Google Scholar] [CrossRef] [Green Version]
- Ju, W.-K.; Kim, K.-Y.; Lindsey, J.D.; Angert, M.; Duong-Polk, K.X.; Scott, R.T.; Kim, J.J.; Kukhmazov, I.; Ellisman, M.H.; Perkins, G.A.; et al. Intraocular pressure elevation induces mitochondrial fission and triggers OPA1 release in glaucomatous optic nerve. Investig. Opthalmol. Vis. Sci. 2008, 49, 4903–4911. [Google Scholar] [CrossRef]
- Edwards, G.; Perkins, G.A.; Kim, K.-Y.; Kong, Y.; Lee, Y.; Choi, S.-H.; Liu, Y.; Skowronska-Krawczyk, D.; Weinreb, R.N.; Zangwill, L.; et al. Loss of AKAP1 triggers Drp1 dephosphorylation-mediated mitochondrial fission and loss in retinal ganglion cells. Cell Death Dis. 2020, 11, 254. [Google Scholar] [CrossRef] [Green Version]
- Czachor, A.; Failla, A.; Lockey, R.; Kolliputi, N. Pivotal role of AKAP121 in mitochondrial physiology. Am. J. Physiol. Cell Physiol. 2016, 310, C625–C628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiattarella, G.G.; Cattaneo, F.; Pironti, G.; Magliulo, F.; Carotenuto, G.; Pirozzi, M.; Polishchuk, R.; Borzacchiello, D.; Paolillo, R.; Oliveti, M.; et al. Akap1 deficiency promotes mitochondrial aberrations and exacerbates cardiac injury following permanent coronary ligation via enhanced mitophagy and apoptosis. PLoS ONE 2016, 11, e0154076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Benedetto, G.; Gerbino, A.; Lefkimmiatis, K. Shaping mitochondrial dynamics: The role of cAMP signalling. Biochem. Biophys. Res. Commun. 2018, 500, 65–74. [Google Scholar] [CrossRef]
- Liesa, M.; Palacín, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flippo, K.H.; Strack, S. Mitochondrial dynamics in neuronal injury, development and plasticity. J. Cell Sci. 2017, 130, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Cereghetti, G.; Stangherlin, A.; De Brito, O.M.; Chang, C.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [Green Version]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppins, S.; Lackner, L.; Nunnari, J.J.A.R.B. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [Green Version]
- Torraco, A.; Peralta, S.; Iommarini, L.; Diaz, F. Mitochondrial Diseases Part I: Mouse models of OXPHOS deficiencies caused by defects in respiratory complex subunits or assembly factors. Mitochondrion 2015, 21, 76–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, S.; De Rasmo, D.; Scacco, S.; Signorile, A.; Technikova-Dobrova, Z.; Palmisano, G.; Sardanelli, A.M.; Papa, F.; Panelli, D.; Scaringi, R.; et al. Mammalian complex I: A regulable and vulnerable pacemaker in mitochondrial respiratory function. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 719–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helling, S.; Vogt, S.; Rhiel, A.; Ramzan, R.; Wen, L.; Marcus, K.; Kadenbach, B. Phosphorylation and Kinetics of Mammalian Cytochrome c Oxidase. Mol. Cell. Proteom. 2008, 7, 1714–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, B.; Bender, E.; Arnold, S.; Hüttemann, M.; Lee, I.; Kadenbach, B. Cytochrome C oxidase and the regulation of oxidative phosphorylation. Chembiochem A Eur. J. Chem. Biol. 2001, 2, 392–403. [Google Scholar] [CrossRef]
- Salvi, M.; Brunati, A.M.; Toninello, A. Tyrosine phosphorylation in mitochondria: A new frontier in mitochondrial signaling. Free Radic. Biol. Med. 2005, 38, 1267–1277. [Google Scholar] [CrossRef]
- Guedouari, H.; Ould Amer, Y.; Pichaud, N.; Hebert-Chatelain, E. Characterization of the interactome of c-Src within the mitochondrial matrix by proximity-dependent biotin identification. Mitochondrion 2021, 57, 257–269. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J.A. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Ould Amer, Y.; Hebert-Chatelain, E. Mitochondrial cAMP-PKA signaling: What do we really know? Biochim. Biophys. Acta Bioenerg. 2018, 1859, 868–877. [Google Scholar] [CrossRef]
- Song, D.H.; Park, J.; Maurer, L.L.; Lu, W.; Philbert, M.A.; Sastry, A.M. Biophysical significance of the inner mitochondrial membrane structure on the electrochemical potential of mitochondria. Phys. Rev. E Stat. Nonlin Soft Matter Phys. 2013, 88, 062723. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopitz, J.; Kisen, G.; Gordon, P.B.; Bohley, P.; Seglen, P. Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J. Cell Biol. 1990, 111, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Kissová, I.; Deffieu, M.k.; Manon, S.; Camougrand, N. Uth1p is involved in the autophagic degradation of mitochondria. J. Biol. Chem. 2004, 279, 39068–39074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban-Martínez, L.; Sierra-Filardi, E.; McGreal, R.S.; Salazar-Roa, M.; Mariño, G.; Seco, E.; Durand, S.; Enot, D.; Graña, O.; Malumbres, M.; et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 2017, 36, 1688–1706. [Google Scholar] [CrossRef] [PubMed]
- Stallons, L.J.; Funk, J.A.; Schnellmann, R.G. Mitochondrial homeostasis in acute organ failure. Curr. Pathobiol. Rep. 2013, 1, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Devi, T.D.; Babu, M.; Mäkinen, P.; Kaikkonen, M.U.; Heinaniemi, M.; Laakso, H.; Ylä-Herttuala, E.; Rieppo, L.; Liimatainen, T.; Naumenko, N.; et al. Aggravated postinfarct heart failure in type 2 diabetes is associated with impaired mitophagy and exaggerated inflammasome activation. Am. J. Pathol. 2017, 187, 2659–2673. [Google Scholar] [CrossRef] [Green Version]
- Kubli, D.A.; Gustafsson, Å.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [Green Version]
- Rüb, C.; Wilkening, A.; Voos, W. Mitochondrial quality control by the Pink1/Parkin system. Cell Tissue Res. 2017, 367, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, Å.B. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Stockum, S.; Marchesan, E.; Ziviani, E. Mitochondrial quality control beyond PINK1/Parkin. Oncotarget 2018, 9, 12550. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An emerging role in aging and age-associated diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Pryde, K.R.; Smith, H.L.; Chau, K.-Y.; Schapira, A.H. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J. Cell Biol. 2016, 213, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Na, J.H.; Sung, K.R.; Baek, S.; Kim, Y.J.; Durbin, M.K.; Lee, H.J.; Kim, H.K.; Sohn, Y.H. Detection of glaucoma progression by assessment of segmented macular thickness data obtained using spectral domain optical coherence tomography. Investig. Opthalmology Vis. Sci. 2012, 53, 3817–3826. [Google Scholar] [CrossRef]
- Tezel, G. Multifactorial Pathogenic Processes of Retinal Ganglion Cell Degeneration in Glaucoma towards Multi-Target Strategies for Broader Treatment Effects. Cells 2021, 10, 1372. [Google Scholar] [CrossRef]
- Ju, W.K.; Kim, K.Y.; Duong-Polk, K.X.; Lindsey, J.D.; Ellisman, M.H.; Weinreb, R.N. Increased optic atrophy type 1 expression protects retinal ganglion cells in a mouse model of glaucoma. Mol. Vis. 2010, 16, 1331–1342. [Google Scholar]
- Lee, D.; Shim, M.S.; Kim, K.Y.; Noh, Y.H.; Kim, H.; Kim, S.Y.; Weinreb, R.N.; Ju, W.K. Coenzyme Q10 inhibits glutamate excitotoxicity and oxidative stress-mediated mitochondrial alteration in a mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 993–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, W.K.; Kim, K.Y.; Noh, Y.H.; Hoshijima, M.; Lukas, T.J.; Ellisman, M.H.; Weinreb, R.N.; Perkins, G.A. Increased mitochondrial fission and volume density by blocking glutamate excitotoxicity protect glaucomatous optic nerve head astrocytes. Glia 2015, 63, 736–753. [Google Scholar] [CrossRef] [PubMed]
- Shim, M.S.; Takihara, Y.; Kim, K.Y.; Iwata, T.; Yue, B.Y.; Inatani, M.; Weinreb, R.N.; Perkins, G.A.; Ju, W.K. Mitochondrial pathogenic mechanism and degradation in optineurin E50K mutation-mediated retinal ganglion cell degeneration. Sci. Rep. 2016, 6, 33830. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef] [Green Version]
- Harun-Or-Rashid, M.; Pappenhagen, N.; Palmer, P.G.; Smith, M.A.; Gevorgyan, V.; Wilson, G.N.; Crish, S.D.; Inman, D.M. Structural and Functional Rescue of Chronic Metabolically Stressed Optic Nerves through Respiration. J. Neurosci. 2018, 38, 5122–5139. [Google Scholar] [CrossRef] [Green Version]
- Osborne, N.N. Pathogenesis of ganglion “cell death” in glaucoma and neuroprotection: Focus on ganglion cell axonal mitochondria. Prog. Brain Res. 2008, 173, 339–352. [Google Scholar] [CrossRef]
- Osborne, N.N.; Nunez-Alvarez, C.; Joglar, B.; Del Olmo-Aguado, S. Glaucoma: Focus on mitochondria in relation to pathogenesis and neuroprotection. Eur. J. Pharmacol. 2016, 787, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-H.; Kim, K.-Y.; Perkins, G.A.; Phan, S.; Edwards, G.; Xia, Y.; Kim, J.; Skowronska-Krawczyk, D.; Weinreb, R.N.; Ellisman, M.H.; et al. AIBP protects retinal ganglion cells against neuroinflammation and mitochondrial dysfunction in glaucomatous neurodegeneration. Redox Biol. 2020, 37, 101703. [Google Scholar] [CrossRef]
- Rice, A.C.; Keeney, P.M.; Algarzae, N.K.; Ladd, A.C.; Thomas, R.R.; Bennett, J.P., Jr. Mitochondrial DNA copy numbers in pyramidal neurons are decreased and mitochondrial biogenesis transcriptome signaling is disrupted in Alzheimer’s disease hippocampi. J. Alzheimers Dis. 2014, 40, 319–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct Membrane Association Drives Mitochondrial Fission by the Parkinson Disease-associated Protein α-Synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [Green Version]
- Johri, A.; Chandra, A.; Beal, M.F. PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free Radic. Biol. Med. 2013, 62, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.; Xu, Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 1998, 18, 3241–3250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagda, R.K.; Banerjee, T.D. Role of protein kinase A in regulating mitochondrial function and neuronal development: Implications to neurodegenerative diseases. Rev. Neurosci. 2015, 26, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Benedetto, G.D.; Iannucci, L.F.; Surdo, N.C.; Zanin, S.; Conca, F.; Grisan, F.; Gerbino, A.; Lefkimmiatis, K. Compartmentalized Signaling in Aging and Neurodegeneration. Cells 2021, 10, 464. [Google Scholar] [CrossRef]
- Banerjee, T.D.; Reihl, K.; Swain, M.; Torres, M.; Dagda, R.K. Mitochondrial PKA Is Neuroprotective in a Cell Culture Model of Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3071–3083. [Google Scholar] [CrossRef]
- Das Banerjee, T.; Dagda, R.Y.; Dagda, M.; Chu, C.T.; Rice, M.; Vazquez-Mayorga, E.; Dagda, R.K. PINK1 regulates mitochondrial trafficking in dendrites of cortical neurons through mitochondrial PKA. J. Neurochem. 2017, 142, 545–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Perry, G.; Smith, M.A.; Wang, X. Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2013, 33 (Suppl. S1), S253–S262. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yin, J.; Ma, X.; Zhao, F.; Siedlak, S.L.; Wang, Z.; Torres, S.; Fujioka, H.; Xu, Y.; Perry, G.; et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 2017, 26, 4118–4131. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liu, M.; Gao, J.; Smith, A.M.; Fujioka, H.; Liang, J.; Perry, G.; Wang, X. Mitochondrial Fusion Suppresses Tau Pathology-Induced Neurodegeneration and Cognitive Decline. J. Alzheimers Dis. 2021, 84, 1057–1069. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef] [PubMed]
- Vitolo, O.V.; Sant’Angelo, A.; Costanzo, V.; Battaglia, F.; Arancio, O.; Shelanski, M. Amyloid beta-peptide inhibition of the PKA/CREB pathway and long-term potentiation: Reversibility by drugs that enhance cAMP signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13217–13221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, Y.; Tiwari, P.; Xie, Z.; Zheng, J.Q. Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J. Neurosci. 2006, 26, 10480–10487. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Jun, S.; Simpkins, J.W. Estrogen amelioration of Abeta-induced defects in mitochondria is mediated by mitochondrial signaling pathway involving ERbeta, AKAP and Drp1. Brain Res. 2015, 1616, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bastola, T.; Perkins, G.A.; Kim, K.-Y.; Choi, S.; Kwon, J.-W.; Shen, Z.; Strack, S.; Ju, W.-K. Role of A-Kinase Anchoring Protein 1 in Retinal Ganglion Cells: Neurodegeneration and Neuroprotection. Cells 2023, 12, 1539. https://doi.org/10.3390/cells12111539
Bastola T, Perkins GA, Kim K-Y, Choi S, Kwon J-W, Shen Z, Strack S, Ju W-K. Role of A-Kinase Anchoring Protein 1 in Retinal Ganglion Cells: Neurodegeneration and Neuroprotection. Cells. 2023; 12(11):1539. https://doi.org/10.3390/cells12111539
Chicago/Turabian StyleBastola, Tonking, Guy A. Perkins, Keun-Young Kim, Seunghwan Choi, Jin-Woo Kwon, Ziyao Shen, Stefan Strack, and Won-Kyu Ju. 2023. "Role of A-Kinase Anchoring Protein 1 in Retinal Ganglion Cells: Neurodegeneration and Neuroprotection" Cells 12, no. 11: 1539. https://doi.org/10.3390/cells12111539