
The Ying and Yang of Hydrogen Sulfide as a Paracrine/Autocrine Agent in Neurodegeneration: Focus on Amyotrophic Lateral Sclerosis

 , ,
, ,  ,
, 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Hydrogen Sulfide, the Vasculature, and the Tripartite Synapse
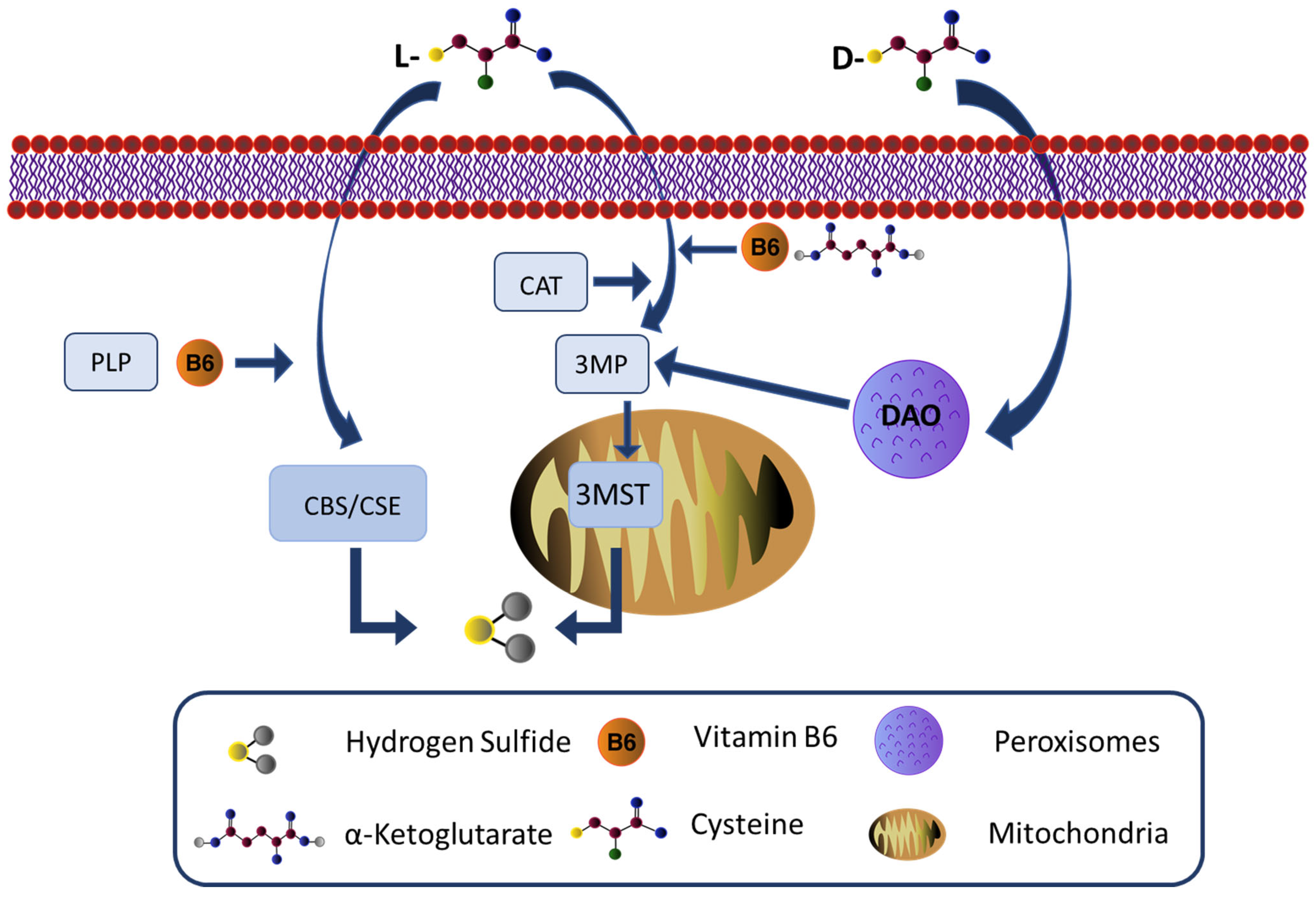
3. Hydrogen Sulfide and Neurons
4. Hydrogen Sulfide and Glial Cells
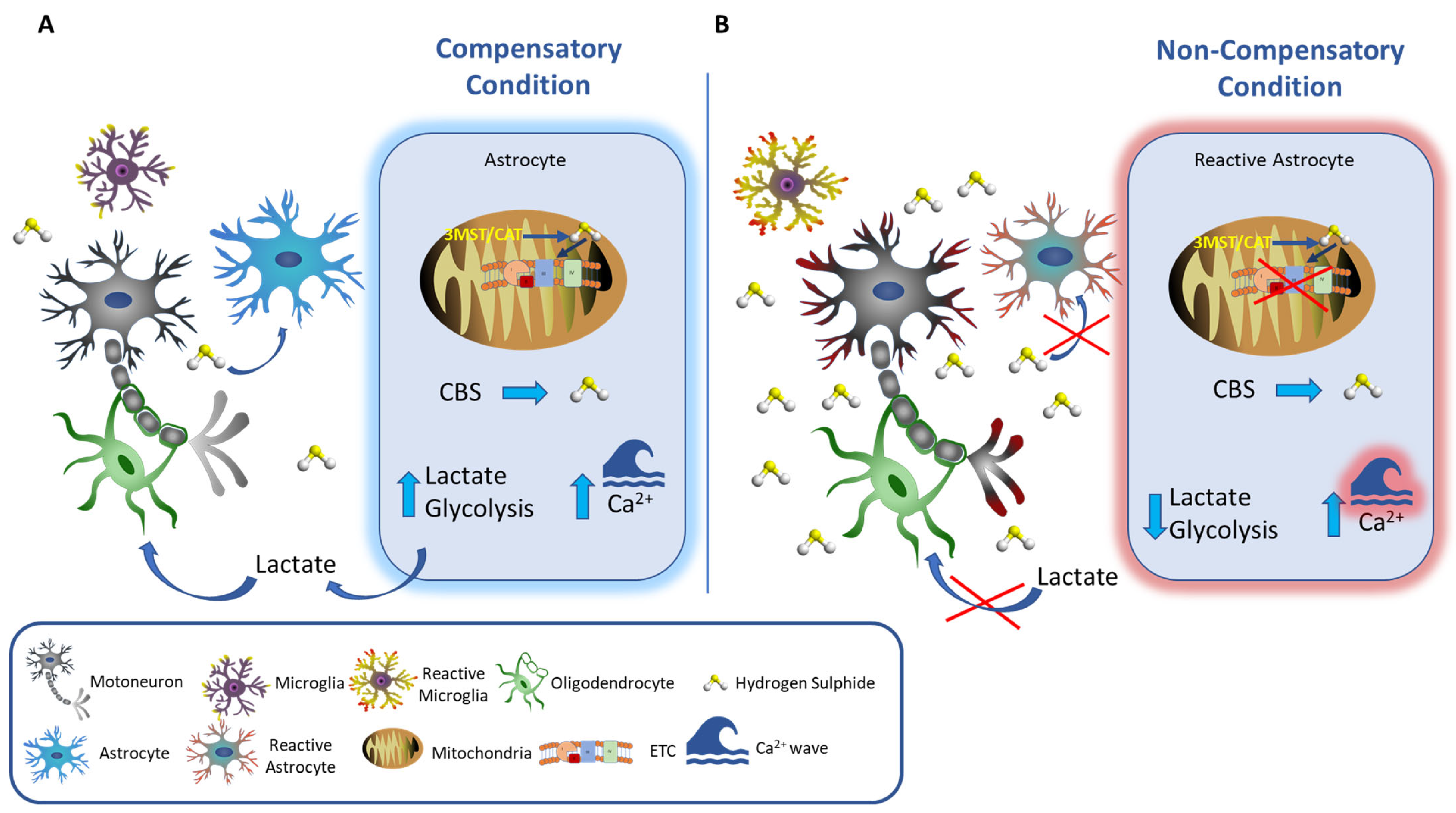
4.1. Astrocytes
4.2. Microglia
4.3. Oligodendrocytes
5. Hydrogen Sulfide and the Resolution of Inflammation
6. Hydrogen Sulfide and Amyotrophic Lateral Sclerosis
6.1. Homocysteine
6.2. Glutathione (GSH)
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Warenycia, M.W.; Goodwin, L.R.; Benishin, C.G.; Reiffenstein, R.J.; Francom, D.M.; Taylor, J.D.; Dieken, F.P. Acute Hydrogen Sulfide Poisoning. Biochem. Pharmacol. 1989, 38, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Hydrogen Sulfide, an Enhancer of Vascular Nitric Oxide Signaling: Mechanisms and Implications. Am. J. Physiol. Cell Physiol. 2017, 312, C3–C15. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen Sulfide-Linked Sulfhydration of NF-ΚB Mediates Its Antiapoptotic Actions. Mol. Cell 2012, 45, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, S.; Ogasawara, Y. Sulfur Atom in Its Bound State Is a Unique Element Involved in Physiological Functions in Mammals. Molecules 2016, 21, 1753. [Google Scholar] [CrossRef] [Green Version]
- Abe, K.; Kimura, H. The Possible Role of Hydrogen Sulfide as an Endogenous Neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Liu, D.-X.; Wang, F.-W.; Zhang, Q.; Du, Z.-X.; Zhan, J.-M.; Yuan, Q.-H.; Ling, E.-A.; Hao, A.-J. L-Cysteine Promotes the Proliferation and Differentiation of Neural Stem Cells via the CBS/H2S Pathway. Neuroscience 2013, 237, 106–117. [Google Scholar] [CrossRef]
- Liu, D.; Wang, Z.; Zhan, J.; Zhang, Q.; Wang, J.; Zhang, Q.; Xian, X.; Luan, Q.; Hao, A. Hydrogen Sulfide Promotes Proliferation and Neuronal Differentiation of Neural Stem Cells and Protects Hypoxia-Induced Decrease in Hippocampal Neurogenesis. Pharmacol. Biochem. Behav. 2014, 116, 55–63. [Google Scholar] [CrossRef]
- Fukami, K.; Kawabata, A. Hydrogen Sulfide and Neuronal Differentiation: Focus on Ca2+ Channels. Nitric Oxide 2015, 46, 50–54. [Google Scholar] [CrossRef]
- Kimura, H.; Shibuya, N.; Kimura, Y. Hydrogen Sulfide Is a Signaling Molecule and a Cytoprotectant. Antioxid. Redox Signal. 2012, 17, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Sbodio, J.I.; Xu, R.; Vandiver, M.S.; Cha, J.Y.; Snowman, A.M.; Snyder, S.H. Cystathionine γ-Lyase Deficiency Mediates Neurodegeneration in Huntington’s Disease. Nature 2014, 509, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Banerjee, R. PLP-Dependent H2S Biogenesis. Biochim. Biophys. Acta BBA 2011, 1814, 1518–1527. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Chan, S.-J.; Ng, Y.-K.; Wong, P.T.-H. Brain 3-Mercaptopyruvate Sulfurtransferase (3MST): Cellular Localization and Downregulation after Acute Stroke. PLoS ONE 2013, 8, e67322. [Google Scholar] [CrossRef] [Green Version]
- Furne, J.; Saeed, A.; Levitt, M.D. Whole Tissue Hydrogen Sulfide Concentrations Are Orders of Magnitude Lower than Presently Accepted Values. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1479–R1485. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate Sulfurtransferase Produces Hydrogen Sulfide and Bound Sulfane Sulfur in the Brain. Antioxid. Redox Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef]
- Shibuya, N.; Koike, S.; Tanaka, M.; Ishigami-Yuasa, M.; Kimura, Y.; Ogasawara, Y.; Fukui, K.; Nagahara, N.; Kimura, H. A Novel Pathway for the Production of Hydrogen Sulfide from D-Cysteine in Mammalian Cells. Nat. Commun. 2013, 4, 1366. [Google Scholar] [CrossRef] [Green Version]
- Gould, S.J.; Keller, G.A.; Subramani, S. Identification of Peroxisomal Targeting Signals Located at the Carboxy Terminus of Four Peroxisomal Proteins. J. Cell Biol. 1988, 107, 897–905. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.C. Glutathione Synthesis. Biochim. Biophys. Acta BBA 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, P.; Kim, J.-K. Oxidation of Sulphide by Cytochrome aa3. Biochim. Biophys. Acta BBA 1981, 637, 312–320. [Google Scholar] [CrossRef]
- Goubern, M.; Andriamihaja, M.; Nübel, T.; Blachier, F.; Bouillaud, F. Sulfide, the First Inorganic Substrate for Human Cells. FASEB J. 2007, 21, 1699–1706. [Google Scholar] [CrossRef]
- Huerta de la Cruz, S.; Medina-Terol, G.J.; Tapia-Martínez, J.A.; Silva-Velasco, D.L.; Beltran-Ornelas, J.H.; Sánchez-López, A.; Sancho, M.; Centurión, D. Hydrogen Sulfide as a Neuromodulator of the Vascular Tone. Eur. J. Pharmacol. 2023, 940, 175455. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite Synapses: Astrocytes Process and Control Synaptic Information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–Endothelial Interactions at the Blood–Brain Barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Zonta, M.; Angulo, M.C.; Gobbo, S.; Rosengarten, B.; Hossmann, K.-A.; Pozzan, T.; Carmignoto, G. Neuron-to-Astrocyte Signaling Is Central to the Dynamic Control of Brain Microcirculation. Nat. Neurosci. 2003, 6, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen Sulfide Is an Endogenous Stimulator of Angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977. [Google Scholar] [CrossRef] [Green Version]
- Katsouda, A.; Bibli, S.-I.; Pyriochou, A.; Szabo, C.; Papapetropoulos, A. Regulation and Role of Endogenously Produced Hydrogen Sulfide in Angiogenesis. Pharmacol. Res. 2016, 113, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Gao, Y.; Huang, X.; Wang, X. GYY4137, a Hydrogen Sulfide (H2S) Donor, Shows Potent Anti-Hepatocellular Carcinoma Activity through Blocking the STAT3 Pathway. Int. J. Oncol. 2014, 44, 1259–1267. [Google Scholar] [CrossRef] [Green Version]
- Leskova, A.; Pardue, S.; Glawe, J.D.; Kevil, C.G.; Shen, X. Role of Thiosulfate in Hydrogen Sulfide-Dependent Redox Signaling in Endothelial Cells. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H256–H264. [Google Scholar] [CrossRef] [Green Version]
- Lambrechts, D.; Storkebaum, E.; Morimoto, M.; Del-Favero, J.; Desmet, F.; Marklund, S.L.; Wyns, S.; Thijs, V.; Andersson, J.; van Marion, I.; et al. VEGF Is a Modifier of Amyotrophic Lateral Sclerosis in Mice and Humans and Protects Motoneurons against Ischemic Death. Nat. Genet. 2003, 34, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Disbrow, E.; Stokes, K.Y.; Ledbetter, C.; Patterson, J.; Kelley, R.; Pardue, S.; Reekes, T.; Larmeu, L.; Batra, V.; Yuan, S.; et al. Plasma Hydrogen Sulfide: A Biomarker of Alzheimer’s Disease and Related Dementias. Alzheimer’s Dement. 2021, 17, 1391–1402. [Google Scholar] [CrossRef]
- Reekes, T.H.; Ledbetter, C.R.; Alexander, J.S.; Stokes, K.Y.; Pardue, S.; Bhuiyan, M.A.N.; Patterson, J.C.; Lofton, K.T.; Kevil, C.G.; Disbrow, E.A. Elevated Plasma Sulfides Are Associated with Cognitive Dysfunction and Brain Atrophy in Human Alzheimer’s Disease and Related Dementias. Redox Biol. 2023, 62, 102633. [Google Scholar] [CrossRef]
- Giovinazzo, D.; Bursac, B.; Sbodio, J.I.; Nalluru, S.; Vignane, T.; Snowman, A.M.; Albacarys, L.M.; Sedlak, T.W.; Torregrossa, R.; Whiteman, M.; et al. Hydrogen Sulfide Is Neuroprotective in Alzheimer’s Disease by Sulfhydrating GSK3β and Inhibiting Tau Hyperphosphorylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017225118. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, C.; Manuel, M.L.; Yuan, S.; Kevil, C.G.; McCarter, K.D.; Lu, W.; Sun, H. Role of Hydrogen Sulfide in Early Blood-Brain Barrier Disruption Following Transient Focal Cerebral Ischemia. PLoS ONE 2015, 10, e0117982. [Google Scholar] [CrossRef]
- Qu, K.; Chen, C.P.L.H.; Halliwell, B.; Moore, P.K.; Wong, P.T.-H. Hydrogen Sulfide Is a Mediator of Cerebral Ischemic Damage. Stroke 2006, 37, 889–893. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.T.H.; Qu, K.; Chimon, G.N.; Seah, A.B.H.; Chang, H.M.; Wong, M.C.; Ng, Y.K.; Rumpel, H.; Halliwell, B.; Chen, C.P.L.H. High Plasma Cyst(e)Ine Level May Indicate Poor Clinical Outcome in Patients with Acute Stroke: Possible Involvement of Hydrogen Sulfide. J. Neuropathol. Exp. Neurol. 2006, 65, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Longone, P.; Davoli, A.; Greco, V.; Spalloni, A.; Guatteo, E.; Neri, C.; Ricciardo Rizzo, G.; Cordella, A.; Romigi, A.; Cortese, C.; et al. Hydrogen Sulphide “a Double-Faced Janus” in Amyotrophic Lateral Sclerosis (ALS). Ther. Targets Neurol. Dis. 2015. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, R.; Wu, L.; Yang, G. Hydrogen Sulfide Signaling in Regulation of Cell Behaviors. Nitric Oxide 2020, 103, 9–19. [Google Scholar] [CrossRef]
- Guidotti, T.L. Hydrogen Sulphide. Occup. Med. 1996, 46, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Kilburn, K.H.; Thrasher, J.D.; Gray, M.R. Low-Level Hydrogen Sulfide and Central Nervous System Dysfunction. Toxicol. Ind. Health 2010, 26, 387–405. [Google Scholar] [CrossRef]
- Elwood, M. The Scientific Basis for Occupational Exposure Limits for Hydrogen Sulphide—A Critical Commentary. Int. J. Environ. Res. Public. Health 2021, 18, 2866. [Google Scholar] [CrossRef]
- Goodwin, L.R.; Francom, D.; Dieken, F.P.; Taylor, J.D.; Warenycia, M.W.; Reiffenstein, R.J.; Dowling, G. Determination of Sulfide in Brain Tissue by Gas Dialysis/Ion Chromatography: Postmortem Studies and Two Case Reports. J. Anal. Toxicol. 1989, 13, 105–109. [Google Scholar] [CrossRef]
- Savage, J.C.; Gould, D.H. Determination of Sulfide in Brain Tissue and Rumen Fluid by Ion-Interaction Reversed-Phase High-Performance Liquid Chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1990, 526, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Dallas, M.L.; Al-Owais, M.M.; Hettiarachchi, N.T.; Vandiver, M.S.; Jarosz-Griffiths, H.H.; Scragg, J.L.; Boyle, J.P.; Steele, D.; Peers, C. Hydrogen Sulfide Regulates Hippocampal Neuron Excitability via S-Sulfhydration of Kv2.1. Sci. Rep. 2021, 11, 8194. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-S.; Pessah, I.N.; Santana, C.M.; Purnell, B.S.; Li, R.; Buchanan, G.F.; Rumbeiha, W.K. Investigations into Hydrogen Sulfide-Induced Suppression of Neuronal Activity In Vivo and Calcium Dysregulation In Vitro. Toxicol. Sci. 2023, 192, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Marutani, E.; Morita, M.; Hirai, S.; Kai, S.; Grange, R.M.H.; Miyazaki, Y.; Nagashima, F.; Traeger, L.; Magliocca, A.; Ida, T.; et al. Sulfide Catabolism Ameliorates Hypoxic Brain Injury. Nat. Commun. 2021, 12, 3108. [Google Scholar] [CrossRef]
- Sheng, Z.-H.; Cai, Q. Mitochondrial Transport in Neurons: Impact on Synaptic Homeostasis and Neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [Green Version]
- Vitvitsky, V.; Kumar, R.; Libiad, M.; Maebius, A.; Landry, A.P.; Banerjee, R. The Mitochondrial NADH Pool Is Involved in Hydrogen Sulfide Signaling and Stimulation of Aerobic Glycolysis. J. Biol. Chem. 2021, 296, 100736. [Google Scholar] [CrossRef]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The Bioenergetic and Antioxidant Status of Neurons Is Controlled by Continuous Degradation of a Key Glycolytic Enzyme by APC/C–Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Steinhäuser, C. Ion Channels in Glial Cells. Brain Res. Rev. 2000, 32, 380–412. [Google Scholar] [CrossRef]
- Liddelow, S.; Barres, B. SnapShot: Astrocytes in Health and Disease. Cell 2015, 162, 1170. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [Green Version]
- Magistretti, P.J.; Allaman, I. Lactate in the Brain: From Metabolic End-Product to Signalling Molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Supplie, L.M.; Düking, T.; Campbell, G.; Diaz, F.; Moraes, C.T.; Götz, M.; Hamprecht, B.; Boretius, S.; Mahad, D.; Nave, K.-A. Respiration-Deficient Astrocytes Survive As Glycolytic Cells In Vivo. J. Neurosci. 2017, 37, 4231–4242. [Google Scholar] [CrossRef] [Green Version]
- Bittar, P.G.; Charnay, Y.; Pellerin, L.; Bouras, C.; Magistretti, P.J. Selective Distribution of Lactate Dehydrogenase Isoenzymes in Neurons and Astrocytes of Human Brain. J. Cereb. Blood Flow Metab. 1996, 16, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Mongeon, R.; Venkatachalam, V.; Yellen, G. Cytosolic NADH-NAD+ Redox Visualized in Brain Slices by Two-Photon Fluorescence Lifetime Biosensor Imaging. Antioxid. Redox Signal. 2016, 25, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Haim, L.B.; Rowitch, D.H. Functional Diversity of Astrocytes in Neural Circuit Regulation. Nat. Rev. Neurosci. 2017, 18, 31–41. [Google Scholar] [CrossRef]
- Doyle, J.P.; Dougherty, J.D.; Heiman, M.; Schmidt, E.F.; Stevens, T.R.; Ma, G.; Bupp, S.; Shrestha, P.; Shah, R.D.; Doughty, M.L.; et al. Application of a Translational Profiling Approach for the Comparative Analysis of CNS Cell Types. Cell 2008, 135, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Fouillet, A.; Mawson, J.; Suliman, O.; Sharrack, B.; Romero, I.A.; Woodroofe, M.N. CCL2 Binding Is CCR2 Independent in Primary Adult Human Astrocytes. Brain Res. 2012, 1437, 115–126. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive Astrocyte Nomenclature, Definitions, and Future Directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Lee, M.; Schwab, C.; Yu, S.; McGeer, E.; McGeer, P.L. Astrocytes Produce the Antiinflammatory and Neuroprotective Agent Hydrogen Sulfide. Neurobiol. Aging 2009, 30, 1523–1534. [Google Scholar] [CrossRef]
- Nagai, Y.; Tsugane, M.; Oka, J.; Kimura, H. Hydrogen Sulfide Induces Calcium Waves in Astrocytes. FASEB J. 2004, 18, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Nii, T.; Eguchi, R.; Yamaguchi, S.; Otsuguro, K. Hydrogen Sulfide Induces Ca2+ Release from the Endoplasmic Reticulum and Suppresses ATP-Induced Ca2+ Signaling in Rat Spinal Cord Astrocytes. Eur. J. Pharmacol. 2021, 891, 173684. [Google Scholar] [CrossRef]
- Bauer, C.C.; Boyle, J.P.; Porter, K.E.; Peers, C. Modulation of Ca2+ Signalling in Human Vascular Endothelial Cells by Hydrogen Sulfide. Atherosclerosis 2010, 209, 374–380. [Google Scholar] [CrossRef] [PubMed]
- de Pascual, R.; Baraibar, A.M.; Méndez-López, I.; Pérez-Ciria, M.; Polo-Vaquero, I.; Gandía, L.; Ohia, S.E.; García, A.G.; de Diego, A.M.G. Hydrogen Sulphide Facilitates Exocytosis by Regulating the Handling of Intracellular Calcium by Chromaffin Cells. Pflugers Arch. 2018, 470, 1255–1270. [Google Scholar] [CrossRef] [PubMed]
- Yong, Q.C.; Choo, C.H.; Tan, B.H.; Low, C.-M.; Bian, J.-S. Effect of Hydrogen Sulfide on Intracellular Calcium Homeostasis in Neuronal Cells. Neurochem. Int. 2010, 56, 508–515. [Google Scholar] [CrossRef]
- Ujike, A.; Otsuguro, K.; Miyamoto, R.; Yamaguchi, S.; Ito, S. Bidirectional Effects of Hydrogen Sulfide via ATP-Sensitive K+ Channels and Transient Receptor Potential A1 Channels in RIN14B Cells. Eur. J. Pharmacol. 2015, 764, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Krueger, D.; Foerster, M.; Mueller, K.; Zeller, F.; Slotta-huspenina, J.; Donovan, J.; Grundy, D.; Schemann, M. Signaling Mechanisms Involved in the Intestinal Pro-Secretory Actions of Hydrogen Sulfide. Neurogastroenterol. Motil. 2010, 22, 1224–1331, e319–e320. [Google Scholar] [CrossRef]
- Trevisani, M.; Patacchini, R.; Nicoletti, P.; Gatti, R.; Gazzieri, D.; Lissi, N.; Zagli, G.; Creminon, C.; Geppetti, P.; Harrison, S. Hydrogen Sulfide Causes Vanilloid Receptor 1-Mediated Neurogenic Inflammation in the Airways. Br. J. Pharmacol. 2005, 145, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, R.; Otsuguro, K.; Ito, S. Time- and Concentration-Dependent Activation of TRPA1 by Hydrogen Sulfide in Rat DRG Neurons. Neurosci. Lett. 2011, 499, 137–142. [Google Scholar] [CrossRef]
- Ogawa, H.; Takahashi, K.; Miura, S.; Imagawa, T.; Saito, S.; Tominaga, M.; Ohta, T. H2S Functions as a Nociceptive Messenger through Transient Receptor Potential Ankyrin 1 (TRPA1) Activation. Neuroscience 2012, 218, 335–343. [Google Scholar] [CrossRef]
- Tsugane, M.; Nagai, Y.; Kimura, Y.; Oka, J.-I.; Kimura, H. Differentiated Astrocytes Acquire Sensitivity to Hydrogen Sulfide That Is Diminished by the Transformation into Reactive Astrocytes. Antioxid. Redox Signal. 2007, 9, 257–269. [Google Scholar] [CrossRef]
- Wang, P.; Wang, F.; Ni, L.; Wu, P.; Chen, J. Targeting Redox-Altered Plasticity to Reactivate Synaptic Function: A Novel Therapeutic Strategy for Cognitive Disorder. Acta Pharm. Sin. B 2021, 11, 599–608. [Google Scholar] [CrossRef]
- Tarui, T.; Fukami, K.; Nagasawa, K.; Yoshida, S.; Sekiguchi, F.; Kawabata, A. Involvement of Src Kinase in T-Type Calcium Channel-Dependent Neuronal Differentiation of NG108-15 Cells by Hydrogen Sulfide. J. Neurochem. 2010, 114, 512–519. [Google Scholar] [CrossRef]
- Cheung, N.S.; Peng, Z.F.; Chen, M.J.; Moore, P.K.; Whiteman, M. Hydrogen Sulfide Induced Neuronal Death Occurs via Glutamate Receptor and Is Associated with Calpain Activation and Lysosomal Rupture in Mouse Primary Cortical Neurons. Neuropharmacology 2007, 53, 505–514. [Google Scholar] [CrossRef]
- Chen, M.J.; Peng, Z.F.; Manikandan, J.; Melendez, A.J.; Tan, G.S.; Chung, C.M.; Li, Q.-T.; Tan, T.M.; Deng, L.W.; Whiteman, M.; et al. Gene Profiling Reveals Hydrogen Sulphide Recruits Death Signaling via the N-Methyl-D-Aspartate Receptor Identifying Commonalities with Excitotoxicity. J. Cell. Physiol. 2011, 226, 1308–1322. [Google Scholar] [CrossRef]
- Wildburger, N.C.; Lin-Ye, A.; Baird, M.A.; Lei, D.; Bao, J. Neuroprotective Effects of Blockers for T-Type Calcium Channels. Mol. Neurodegener. 2009, 4, 44. [Google Scholar] [CrossRef] [Green Version]
- Bordey, A.; Sontheimer, H. Ion Channel Expression by Astrocytes in Situ: Comparison of Different CNS Regions. Comp. Study 2000, 30, 27–38. [Google Scholar] [CrossRef]
- Chao, D.; He, X.; Yang, Y.; Balboni, G.; Salvadori, S.; Kim, D.H.; Xia, Y. Hydrogen Sulfide Induced Disruption of Na+ Homeostasis in the Cortex. Toxicol. Sci. 2012, 128, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Li, Y.; Zhao, Z.; Li, P.; Xie, Y. Hydrogen Sulfide Overproduction Is Involved in Acute Ischemic Cerebral Injury Under Hyperhomocysteinemia. Front. Neurosci. 2020, 14, 582851. [Google Scholar] [CrossRef]
- Luo, Y.; Wu, P.-F.; Zhou, J.; Xiao, W.; He, J.-G.; Guan, X.-L.; Zhang, J.-T.; Hu, Z.-L.; Wang, F.; Chen, J.-G. Aggravation of Seizure-like Events by Hydrogen Sulfide: Involvement of Multiple Targets That Control Neuronal Excitability. CNS Neurosci. Ther. 2014, 20, 411–419. [Google Scholar] [CrossRef]
- Deitmer, J.W.; Rose, C.R. PH Regulation and Proton Signalling by Glial Cells. Prog. Neurobiol. 1996, 48, 73–103. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Choo, C.H.; Hu, L.-F.; Tan, B.H.; Hu, G.; Bian, J.-S. Hydrogen Sulfide Regulates Intracellular PH in Rat Primary Cultured Glia Cells. Neurosci. Res. 2010, 66, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Kiyama, H.; Ueno, M. Dual Functions of Microglia in the Formation and Refinement of Neural Circuits during Development. Int. J. Dev. Neurosci. 2019, 77, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. V Local Self-Renewal Can Sustain CNS Microglia Maintenance and Function throughout Adult Life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; El Khoury, J. Analysis of the Microglial Sensome. Methods Mol. Biol. 2019, 2034, 305–323. [Google Scholar]
- Satoh, J.; Kino, Y.; Asahina, N.; Takitani, M.; Miyoshi, J.; Ishida, T.; Saito, Y. TMEM119 Marks a Subset of Microglia in the Human Brain. Neuropathology 2016, 36, 39–49. [Google Scholar] [CrossRef]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.; Means, T.K.; El Khoury, J. The Microglial Sensome Revealed by Direct RNA Sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. A Polarizing Question: Do M1 and M2 Microglia Exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Gabriela Sánchez, M.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gómez-Nicola, D.; Luheshi, G.; Vallières, L.; et al. Dark Microglia: A New Phenotype Predominantly Associated with Pathological States. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [Green Version]
- Walker, D.G.; Lue, L.-F. Immune Phenotypes of Microglia in Human Neurodegenerative Disease: Challenges to Detecting Microglial Polarization in Human Brains. Alzheimers Res. Ther. 2015, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Osman, A.M.; Sun, Y.; Burns, T.C.; He, L.; Kee, N.; Oliva-Vilarnau, N.; Alevyzaki, A.; Zhou, K.; Louhivuori, L.; Uhlén, P.; et al. Radiation Triggers a Dynamic Sequence of Transient Microglial Alterations in Juvenile Brain. Cell. Rep. 2020, 31, 107699. [Google Scholar] [CrossRef]
- Linkermann, A.; Stockwell, B.R.; Krautwald, S.; Anders, H.-J. Regulated Cell Death and Inflammation: An Auto-Amplification Loop Causes Organ Failure. Nat. Rev. Immunol. 2014, 14, 759–767. [Google Scholar] [CrossRef]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxid. Med. Cell. Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, X.; Xu, Y.; He, M.; Yang, J.; Li, J.; Li, Y.; Ao, G.; Cheng, J.; Jia, J. The Cystathionine β-Synthase/Hydrogen Sulfide Pathway Contributes to Microglia-Mediated Neuroinflammation Following Cerebral Ischemia. Brain Behav. Immun. 2017, 66, 332–346. [Google Scholar] [CrossRef]
- Tian, Q.; Tang, H.-L.; Tang, Y.-Y.; Zhang, P.; Kang, X.; Zou, W.; Tang, X.-Q. Hydrogen Sulfide Attenuates the Cognitive Dysfunction in Parkinson’s Disease Rats via Promoting Hippocampal Microglia M2 Polarization by Enhancement of Hippocampal Warburg Effect. Oxid. Med. Cell. Longev. 2022, 2022, 2792348. [Google Scholar] [CrossRef]
- Zhang, H.; Zhi, L.; Moochhala, S.; Moore, P.K.; Bhatia, M. Hydrogen Sulfide Acts as an Inflammatory Mediator in Cecal Ligation and Puncture-Induced Sepsis in Mice by Upregulating the Production of Cytokines and Chemokines via NF-ΚB. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L960–L971. [Google Scholar] [CrossRef]
- Hu, L.-F.; Wong, P.T.-H.; Moore, P.K.; Bian, J.-S. Hydrogen Sulfide Attenuates Lipopolysaccharide-Induced Inflammation by Inhibition of P38 Mitogen-Activated Protein Kinase in Microglia. J. Neurochem. 2007, 100, 1121–1128. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Bian, J.-S. Hydrogen Sulfide Protects Amyloid-β Induced Cell Toxicity in Microglia. J. Alzheimer’s Dis. 2011, 22, 1189–1200. [Google Scholar] [CrossRef]
- Giuliani, D.; Ottani, A.; Zaffe, D.; Galantucci, M.; Strinati, F.; Lodi, R.; Guarini, S. Hydrogen Sulfide Slows down Progression of Experimental Alzheimer’s Disease by Targeting Multiple Pathophysiological Mechanisms. Neurobiol. Learn. Mem. 2013, 104, 82–91. [Google Scholar] [CrossRef]
- Campolo, M.; Esposito, E.; Ahmad, A.; Di Paola, R.; Paterniti, I.; Cordaro, M.; Bruschetta, G.; Wallace, J.L.; Cuzzocrea, S. Hydrogen Sulfide-Releasing Cyclooxygenase Inhibitor ATB-346 Enhances Motor Function and Reduces Cortical Lesion Volume Following Traumatic Brain Injury in Mice. J. Neuroinflammation 2014, 11, 196. [Google Scholar] [CrossRef] [Green Version]
- Baumann, N.; Pham-Dinh, D. Biology of Oligodendrocyte and Myelin in the Mammalian Central Nervous System. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Herman, P.; Rothman, D.L.; Agarwal, D.; Hyder, F. Evaluating the Gray and White Matter Energy Budgets of Human Brain Function. J. Cereb. Blood Flow Metab. 2018, 38, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Attwell, D. The Energetics of CNS White Matter. J. Neurosci. 2012, 32, 356–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic Oligodendrocytes Maintain Myelin and Long-Term Axonal Integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.; Nave, K.-A. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Biol. 2016, 8, a020479. [Google Scholar] [CrossRef]
- Oka, A.; Belliveau, M.; Rosenberg, P.; Volpe, J. Vulnerability of Oligodendroglia to Glutamate: Pharmacology, Mechanisms, and Prevention. J. Neurosci. 1993, 13, 1441–1453. [Google Scholar] [CrossRef] [Green Version]
- Matute, C.; Domercq, M.; Sánchez-Gómez, M.-V. Glutamate-Mediated Glial Injury: Mechanisms and Clinical Importance. Glia 2006, 53, 212–224. [Google Scholar] [CrossRef]
- Matute, C.; Alberdi, E.; Domercq, M.; Sánchez-Gómez, M.-V.; Pérez-Samartín, A.; Rodríguez-Antigüedad, A.; Pérez-Cerdá, F. Excitotoxic Damage to White Matter. J. Anat. 2007, 210, 693–702. [Google Scholar] [CrossRef]
- Back, S.A.; Gan, X.; Li, Y.; Rosenberg, P.A.; Volpe, J.J. Maturation-Dependent Vulnerability of Oligodendrocytes to Oxidative Stress-Induced Death Caused by Glutathione Depletion. J. Neurosci. 1998, 18, 6241–6253. [Google Scholar] [CrossRef]
- Rosin, C.; Bates, T.E.; Skaper, S.D. Excitatory Amino Acid Induced Oligodendrocyte Cell Death Invitro: Receptor-Dependent and -Independent Mechanisms. J. Neurochem. 2004, 90, 1173–1185. [Google Scholar] [CrossRef]
- Bridges, R.; Lutgen, V.; Lobner, D.; Baker, D.A. Thinking Outside the Cleft to Understand Synaptic Activity: Contribution of the Cystine-Glutamate Antiporter (System x c −) to Normal and Pathological Glutamatergic Signaling. Pharmacol. Rev. 2012, 64, 780–802. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, V. Redox Regulation of Cellular Stress Response in Neurodegenerative Disorders. Ital. J. Biochem. 2006, 55, 263–282. [Google Scholar]
- Seydi, E.; Irandoost, Z.; Khansari, M.G.; Naserzadeh, P.; Tanbakosazan, F.; Pourahmad, J. Toxicity of Hydrogen Sulfide on Rat Brain Neurons. Drug. Res. 2022, 72, 197–202. [Google Scholar] [CrossRef]
- Butt, A.M. Neurotransmitter-mediated Calcium Signalling in Oligodendrocyte Physiology and Pathology. Glia 2006, 54, 666–675. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Meredith, D. The SLC16 Gene Family?From Monocarboxylate Transporters (MCTs) to Aromatic Amino Acid Transporters and Beyond. Pflgers Arch. Eur. J. Physiol. 2004, 447, 619–628. [Google Scholar] [CrossRef]
- Nijland, P.G.; Michailidou, I.; Witte, M.E.; Mizee, M.R.; van der Pol, S.M.A.; van het Hof, B.; Reijerkerk, A.; Pellerin, L.; van der Valk, P.; de Vries, H.E.; et al. Cellular Distribution of Glucose and Monocarboxylate Transporters in Human Brain White Matter and Multiple Sclerosis Lesions. Glia 2014, 62, 1125–1141. [Google Scholar] [CrossRef]
- Alberini, C.M.; Cruz, E.; Descalzi, G.; Bessières, B.; Gao, V. Astrocyte Glycogen and Lactate: New Insights into Learning and Memory Mechanisms. Glia 2018, 66, 1244–1262. [Google Scholar] [CrossRef]
- Marquet, P.; Duncan, S.H.; Chassard, C.; Bernalier-Donadille, A.; Flint, H.J. Lactate Has the Potential to Promote Hydrogen Sulphide Formation in the Human Colon. FEMS Microbiol. Lett. 2009, 299, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Guan, T.; Shafiq, K.; Xing, Y.; Sun, B.; Huang, Q.; Kong, J. Compromised Lactate Efflux Renders Vulnerability of Oligodendrocyte Precursor Cells to Metabolic Stresses. ACS Chem. Neurosci. 2020, 11, 2717–2727. [Google Scholar] [CrossRef]
- Headland, S.E.; Norling, L.V. The Resolution of Inflammation: Principles and Challenges. Semin. Immunol. 2015, 27, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Pro-Resolving Lipid Mediators Are Leads for Resolution Physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N. Proresolving Lipid Mediators and Mechanisms in the Resolution of Acute Inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiurchiù, V.; Leuti, A.; Maccarrone, M. Bioactive Lipids and Chronic Inflammation: Managing the Fire Within. Front. Immunol. 2018, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Tiberi, M.; Chiurchiù, V. Specialized Pro-Resolving Lipid Mediators and Glial Cells: Emerging Candidates for Brain Homeostasis and Repair. Front. Cell. Neurosci. 2021, 15, 673549. [Google Scholar] [CrossRef]
- Perretti, M.; D’Acquisto, F. Annexin A1 and Glucocorticoids as Effectors of the Resolution of Inflammation. Nat. Rev. Immunol. 2009, 9, 62–70. [Google Scholar] [CrossRef]
- Perretti, M.; Leroy, X.; Bland, E.J.; Montero-Melendez, T. Resolution Pharmacology: Opportunities for Therapeutic Innovation in Inflammation. Trends Pharmacol. Sci. 2015, 36, 737–755. [Google Scholar] [CrossRef]
- Perretti, M.; Gavins, F.N.E. Annexin 1: An Endogenous Anti-Inflammatory Protein. Physiology 2003, 18, 60–64. [Google Scholar] [CrossRef]
- Brancaleone, V.; Mitidieri, E.; Flower, R.J.; Cirino, G.; Perretti, M. Annexin A1 Mediates Hydrogen Sulfide Properties in the Control of Inflammation. J. Pharmacol. Exp. Ther. 2014, 351, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Mariggiò, M.A.; Minunno, V.; Riccardi, S.; Santacroce, R.; De Rinaldis, P.; Fumarulo, R. Sulfide Enhancement of Pmn Apoptosis. Immunopharmacol. Immunotoxicol. 1998, 20, 399–408. [Google Scholar] [CrossRef]
- Zanardo, R.C.O.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L.; Zanardo, R.C.O.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; et al. Hydrogen Sulfide Is an Endogenous Modulator of Leukocyte-mediated Inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef]
- Wallace, J.L.; Ferraz, J.G.P.; Muscara, M.N. Hydrogen Sulfide: An Endogenous Mediator of Resolution of Inflammation and Injury. Antioxid. Redox Signal. 2012, 17, 58–67. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Dalli, J. New Pro-Resolving n-3 Mediators Bridge Resolution of Infectious Inflammation to Tissue Regeneration. Mol. Aspects Med. 2018, 64, 1–17. [Google Scholar] [CrossRef]
- Sung, J.-J.; Kim, H.-J.; Choi-Kwon, S.; Lee, J.; Kim, M.; Lee, K.-W. Homocysteine Induces Oxidative Cytotoxicity in Cu,Zn-Superoxide Dismutase Mutant Motor Neuronal Cell. Neuroreport 2002, 13, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Adalbert, R.; Engelhardt, J.; Siklós, L. DL -Homocysteic Acid Application Disrupts Calcium Homeostasis and Induces Degeneration of Spinal Motor Neurons in Vivo. Acta Neuropathol. 2002, 103, 428–436. [Google Scholar] [CrossRef]
- del Aguila, M.A.; Longstreth, W.T.; McGuire, V.; Koepsell, T.D.; van Belle, G. Prognosis in Amyotrophic Lateral Sclerosis: A Population-Based Study. Neurology 2003, 60, 813–819. [Google Scholar] [CrossRef]
- Lee, A.; Arachchige, B.J.; Henderson, R.; Aylward, J.; McCombe, P.A. Elevated Levels of Homocysteinesulfinic Acid in the Plasma of Patients with Amyotrophic Lateral Sclerosis: A Potential Source of Excitotoxicity? Neurodegener. Dis. 2020, 20, 200–206. [Google Scholar] [CrossRef]
- Kaji, R.; Imai, T.; Iwasaki, Y.; Okamoto, K.; Nakagawa, M.; Ohashi, Y.; Takase, T.; Hanada, T.; Shimizu, H.; Tashiro, K.; et al. Ultra-High-Dose Methylcobalamin in Amyotrophic Lateral Sclerosis: A Long-Term Phase II/III Randomised Controlled Study. J. Neurol. Neurosurg. Psychiatry 2019, 90, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Oki, R.; Izumi, Y.; Fujita, K.; Miyamoto, R.; Nodera, H.; Sato, Y.; Sakaguchi, S.; Nokihara, H.; Kanai, K.; Tsunemi, T.; et al. Efficacy and Safety of Ultrahigh-Dose Methylcobalamin in Early-Stage Amyotrophic Lateral Sclerosis. JAMA Neurol. 2022, 79, 575. [Google Scholar] [CrossRef]
- Pastore, A.; Federici, G.; Bertini, E.; Piemonte, F. Analysis of Glutathione: Implication in Redox and Detoxification. Clin. Chim. Acta 2003, 333, 19–39. [Google Scholar] [CrossRef] [PubMed]
- Kim, K. Glutathione in the Nervous System as a Potential Therapeutic Target to Control the Development and Progression of Amyotrophic Lateral Sclerosis. Antioxidants 2021, 10, 1011. [Google Scholar] [CrossRef] [PubMed]
- Switzer, C.H.; Kasamatsu, S.; Ihara, H.; Eaton, P. SOD1 Is an Essential H 2 S Detoxifying Enzyme. Proc. Natl. Acad. Sci. USA 2023, 120, e2205044120. [Google Scholar] [CrossRef] [PubMed]
- Månberg, A.; Skene, N.; Sanders, F.; Trusohamn, M.; Remnestål, J.; Szczepińska, A.; Aksoylu, I.S.; Lönnerberg, P.; Ebarasi, L.; Wouters, S.; et al. Altered Perivascular Fibroblast Activity Precedes ALS Disease Onset. Nat. Med. 2021, 27, 640–646. [Google Scholar] [CrossRef]
- Lee, S.-K.; Chung, J.-H.; Choi, S.-C.; Auh, Q.-S.; Lee, Y.-M.; Lee, S.-I.; Kim, E.-C. Sodium Hydrogen Sulfide Inhibits Nicotine and Lipopolysaccharide-Induced Osteoclastic Differentiation and Reversed Osteoblastic Differentiation in Human Periodontal Ligament Cells. J. Cell. Biochem. 2013, 114, 1183–1193. [Google Scholar] [CrossRef]
- Wu, S.; Pan, C.; Geng, B.; Zhao, J.; Yu, F.; Pang, Y.; Tang, C.; Qi, Y. Hydrogen Sulfide Ameliorates Vascular Calcification Induced by Vitamin D3 plus Nicotine in Rats1. Acta Pharmacol. Sin. 2006, 27, 299–306. [Google Scholar] [CrossRef]
- Davoli, A.; Greco, V.; Spalloni, A.; Guatteo, E.; Neri, C.; Rizzo, G.R.; Cordella, A.; Romigi, A.; Cortese, C.; Bernardini, S.; et al. Evidence of Hydrogen Sulfide Involvement in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2015, 77, 697–709. [Google Scholar] [CrossRef]
- Spalloni, A.; Greco, V.; Ciriminna, G.; Corasolla Carregari, V.; Marini, F.; Pieroni, L.; Mercuri, N.B.; Urbani, A.; Longone, P. Impact of Pharmacological Inhibition of Hydrogen Sulphide Production in the SOD1G93A-ALS Mouse Model. Int. J. Mol. Sci. 2019, 20, 2550. [Google Scholar] [CrossRef] [Green Version]
- Greco, V.; Spalloni, A.; Corasolla Carregari, V.; Pieroni, L.; Persichilli, S.; Mercuri, N.; Urbani, A.; Longone, P. Proteomics and Toxicity Analysis of Spinal-Cord Primary Cultures upon Hydrogen Sulfide Treatment. Antioxidants 2018, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Nagai, M.; Re, D.B.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes Expressing ALS-Linked Mutated SOD1 Release Factors Selectively Toxic to Motor Neurons. Nat. Neurosci. 2007, 10, 615–622. [Google Scholar] [CrossRef] [Green Version]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spalloni, A.; de Stefano, S.; Gimenez, J.; Greco, V.; Mercuri, N.B.; Chiurchiù, V.; Longone, P. The Ying and Yang of Hydrogen Sulfide as a Paracrine/Autocrine Agent in Neurodegeneration: Focus on Amyotrophic Lateral Sclerosis. Cells 2023, 12, 1691. https://doi.org/10.3390/cells12131691
Spalloni A, de Stefano S, Gimenez J, Greco V, Mercuri NB, Chiurchiù V, Longone P. The Ying and Yang of Hydrogen Sulfide as a Paracrine/Autocrine Agent in Neurodegeneration: Focus on Amyotrophic Lateral Sclerosis. Cells. 2023; 12(13):1691. https://doi.org/10.3390/cells12131691
Chicago/Turabian StyleSpalloni, Alida, Susanna de Stefano, Juliette Gimenez, Viviana Greco, Nicola B. Mercuri, Valerio Chiurchiù, and Patrizia Longone. 2023. "The Ying and Yang of Hydrogen Sulfide as a Paracrine/Autocrine Agent in Neurodegeneration: Focus on Amyotrophic Lateral Sclerosis" Cells 12, no. 13: 1691. https://doi.org/10.3390/cells12131691
APA StyleSpalloni, A., de Stefano, S., Gimenez, J., Greco, V., Mercuri, N. B., Chiurchiù, V., & Longone, P. (2023). The Ying and Yang of Hydrogen Sulfide as a Paracrine/Autocrine Agent in Neurodegeneration: Focus on Amyotrophic Lateral Sclerosis. Cells, 12(13), 1691. https://doi.org/10.3390/cells12131691