

Allyl Isothiocianate Induces Ca2+ Signals and Nitric Oxide Release by Inducing Reactive Oxygen Species Production in the Human Cerebrovascular Endothelial Cell Line hCMEC/D3
 , ,
, ,  ,
, 
Abstract
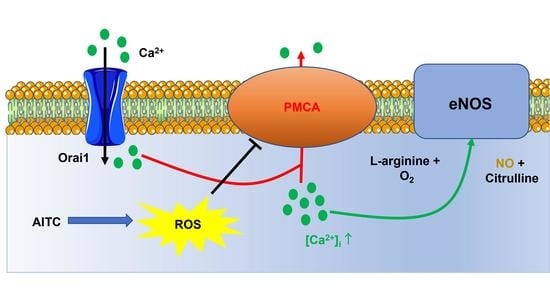
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Solutions
2.3. [Ca2+]i, NO and ROS Imaging
2.4. Electrophysiological Recordings
2.5. Immunoblotting
2.6. Statistics
2.7. Chemicals
3. Results
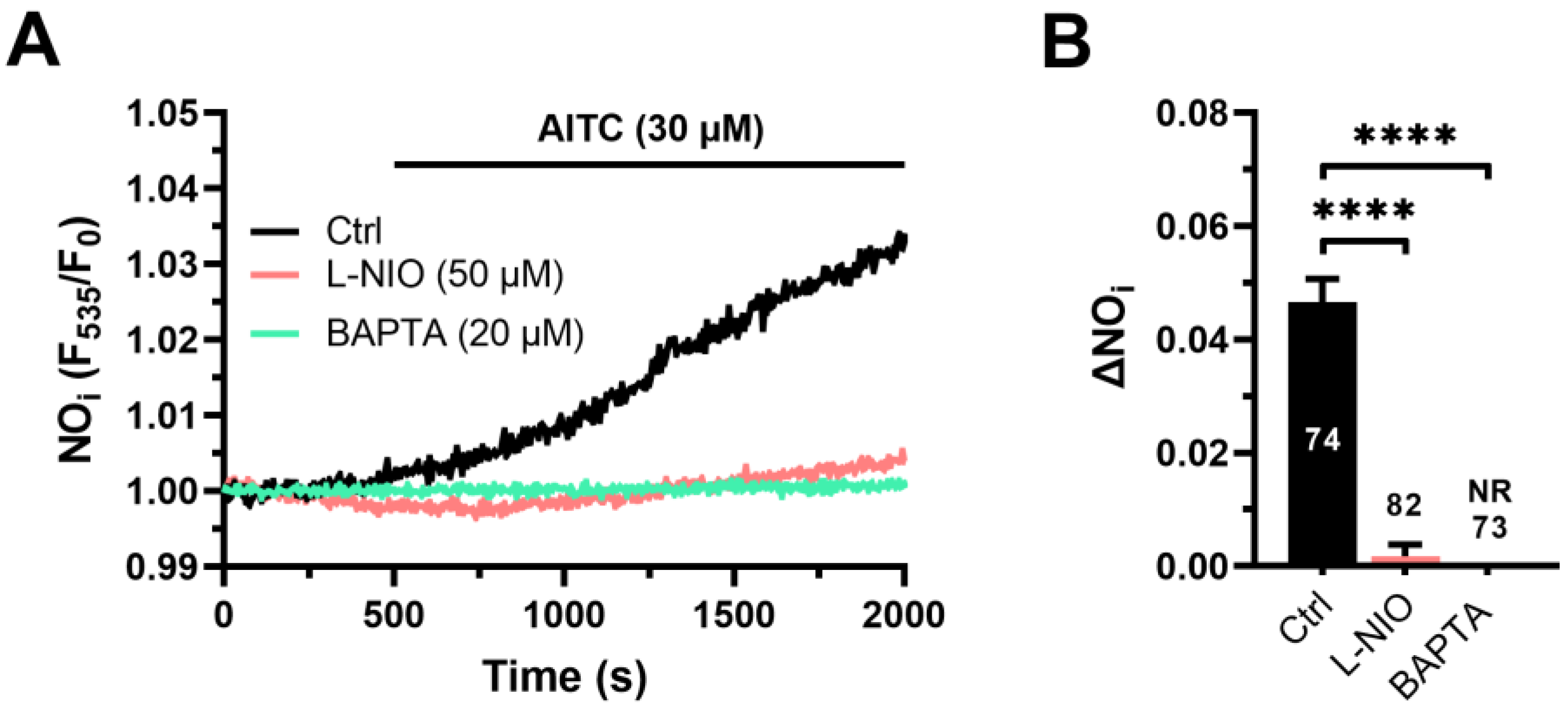
3.1. AITC Evokes Ca2+-Dependent NO Release in hCMEC/D3 Cells
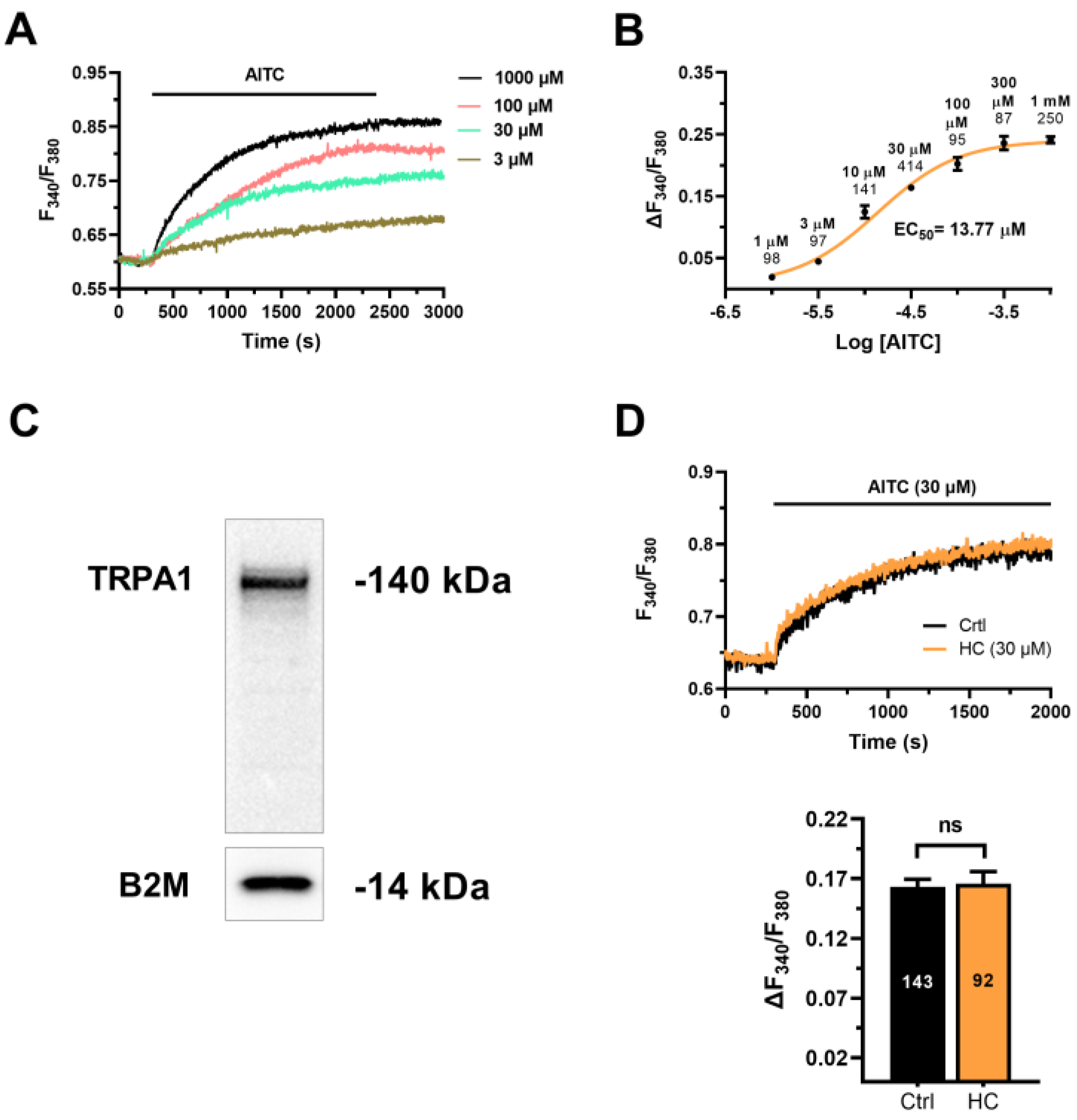
3.2. AITC Evokes TRPA1-Independent Ca2+ Signals in hCMEC/D3 Cells
3.3. Cytosolic ROS Mediate the Ca2+ Response to AITC in hCMEC/D3 Cells
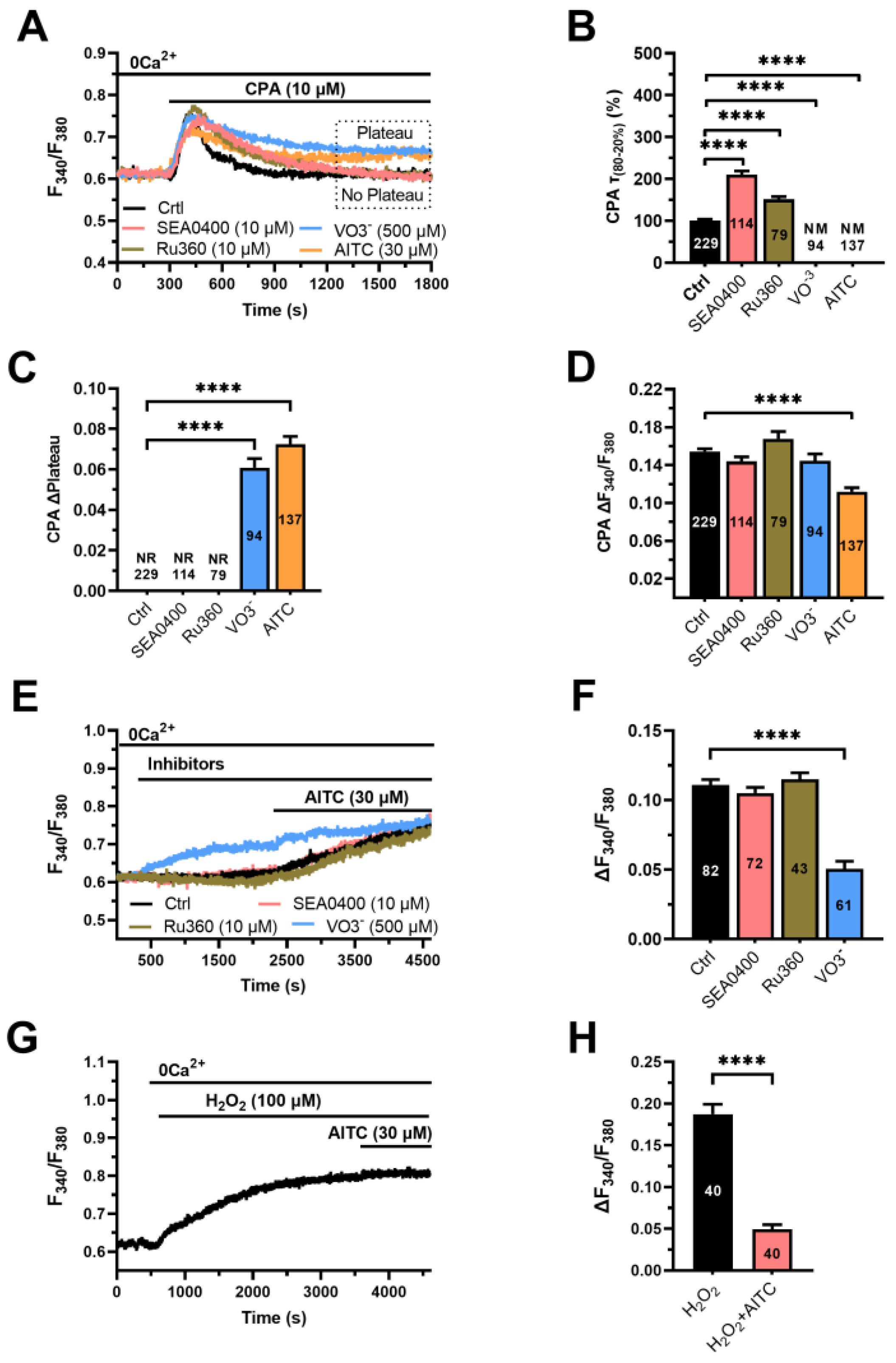
3.4. The Ca2+ Response to AITC Requires Cytosolic Ca2+ and Is Sustained by SOCE
3.5. AITC Inhibits PMCA Activity to Increase the [Ca2+]i in hCMEC/D3 Cells
3.6. AITC-Evoked NO Release Requires ROS-Dependent Inhibition of PMCA Activity and SOCE
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schaeffer, S.; Iadecola, C. Revisiting the neurovascular unit. Nat. Neurosci. 2021, 24, 1198–1209. [Google Scholar] [CrossRef]
- Longden, T.A.; Zhao, G.; Hariharan, A.; Lederer, W.J. Pericytes and the Control of Blood Flow in Brain and Heart. Annu. Rev. Physiol. 2023, 85, 137–164. [Google Scholar] [CrossRef]
- Longden, T.A.; Dabertrand, F.; Koide, M.; Gonzales, A.L.; Tykocki, N.R.; Brayden, J.E.; Hill-Eubanks, D.; Nelson, M.T. Capillary K(+)-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci. 2017, 20, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Longden, T.A.; Mughal, A.; Hennig, G.W.; Harraz, O.F.; Shui, B.; Lee, F.K.; Lee, J.C.; Reining, S.; Kotlikoff, M.I.; Konig, G.M.; et al. Local IP3 receptor-mediated Ca2+ signals compound to direct blood flow in brain capillaries. Sci. Adv. 2021, 7, eabh0101. [Google Scholar] [CrossRef]
- Thakore, P.; Alvarado, M.G.; Ali, S.; Mughal, A.; Pires, P.W.; Yamasaki, E.; Pritchard, H.A.; Isakson, B.E.; Tran, C.H.T.; Earley, S. Brain endothelial cell TRPA1 channels initiate neurovascular coupling. eLife 2021, 10, e63040. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Soda, T.; Moccia, F. Endothelial signaling at the core of neurovascular coupling: The emerging role of endothelial inward-rectifier K+ (Kir2.1) channels and N-methyl-d-aspartate receptors in the regulation of cerebral blood flow. Int. J. Biochem. Cell Biol. 2021, 135, 105983. [Google Scholar] [CrossRef]
- Harraz, O.F.; Longden, T.A.; Hill-Eubanks, D.; Nelson, M.T. PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. eLife 2018, 7, e38689. [Google Scholar] [CrossRef]
- Harraz, O.F.; Longden, T.A.; Dabertrand, F.; Hill-Eubanks, D.; Nelson, M.T. Endothelial GqPCR activity controls capillary electrical signaling and brain blood flow through PIP2 depletion. Proc. Natl. Acad. Sci. USA 2018, 115, E3569–E3577. [Google Scholar] [CrossRef] [Green Version]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef] [Green Version]
- Carter, K.J.; Ward, A.T.; Kellawan, J.M.; Eldridge, M.W.; Al-Subu, A.; Walker, B.J.; Lee, J.W.; Wieben, O.; Schrage, W.G. Nitric oxide synthase inhibition in healthy adults reduces regional and total cerebral macrovascular blood flow and microvascular perfusion. J. Physiol. 2021, 599, 4973–4989. [Google Scholar] [CrossRef]
- Hoiland, R.L.; Caldwell, H.G.; Howe, C.A.; Nowak-Fluck, D.; Stacey, B.S.; Bailey, D.M.; Paton, J.F.R.; Green, D.J.; Sekhon, M.S.; Macleod, D.B.; et al. Nitric oxide is fundamental to neurovascular coupling in humans. J. Physiol. 2020, 598, 4927–4939. [Google Scholar] [CrossRef]
- Zuccolo, E.; Laforenza, U.; Negri, S.; Botta, L.; Berra-Romani, R.; Faris, P.; Scarpellino, G.; Forcaia, G.; Pellavio, G.; Sancini, G.; et al. Muscarinic M5 receptors trigger acetylcholine-induced Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. J. Cell. Physiol. 2019, 234, 4540–4562. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Pellavio, G.; Botta, L.; Orgiu, M.; Forcaia, G.; Sancini, G.; Laforenza, U.; Moccia, F. Group 1 metabotropic glutamate receptors trigger glutamate-induced intracellular Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. Cell. Mol. Life Sci. 2020, 77, 2235–2253. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Faris, P.; Pellavio, G.; Orgiu, M.; Negri, S.; Forcaia, G.; Var-Gaz-Guadarrama, V.; Garcia-Carrasco, M.; Botta, L.; Sancini, G.; et al. Histamine induces intracellular Ca2+ oscillations and nitric oxide release in endothelial cells from brain microvascular circulation. J. Cell. Physiol. 2020, 235, 1515–1530. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Maniezzi, C.; Pellavio, G.; Spaiardi, P.; Botta, L.; Laforenza, U.; Biella, G.; Moccia, D.F. NMDA receptors elicit flux-independent intracellular Ca2+ signals via metabotropic glutamate receptors and flux-dependent nitric oxide release in human brain microvascular endothelial cells. Cell Calcium 2021, 99, 102454. [Google Scholar] [CrossRef]
- Negri, S.; Scolari, F.; Vismara, M.; Brunetti, V.; Faris, P.; Terribile, G.; Sancini, G.; Berra-Romani, R.; Moccia, F. GABA(A) and GABA(B) Receptors Mediate GABA-Induced Intracellular Ca2+ Signals in Human Brain Microvascular Endothelial Cells. Cells 2022, 11, 3860. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Berra-Romani, R. Targeting the endothelial Ca2+ tool kit to rescue endothelial dysfunction in obesity associated-hypertension. Curr. Med. Chem. 2019, 27, 240–257. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Angelone, T. Targeting endothelial ion signalling to rescue cerebral blood flow in cerebral disorders. Vascul. Pharmacol. 2022, 145, 106997. [Google Scholar] [CrossRef]
- Thakore, P.; Ali, S.; Earley, S. Regulation of vascular tone by transient receptor potential ankyrin 1 channels. Curr. Top. Membr. 2020, 85, 119–150. [Google Scholar] [CrossRef]
- Alvarado, M.G.; Thakore, P.; Earley, S. Transient Receptor Potential Channel Ankyrin 1: A Unique Regulator of Vascular Function. Cells 2021, 10, 1167. [Google Scholar] [CrossRef]
- Sullivan, M.N.; Gonzales, A.L.; Pires, P.W.; Bruhl, A.; Leo, M.D.; Li, W.; Oulidi, A.; Boop, F.A.; Feng, Y.; Jaggar, J.H.; et al. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci. Signal. 2015, 8, ra2. [Google Scholar] [CrossRef] [Green Version]
- Hansted, A.K.; Bhatt, D.K.; Olesen, J.; Jensen, L.J.; Jansen-Olesen, I. Effect of TRPA1 activator allyl isothiocyanate (AITC) on rat dural and pial arteries. Pharmacol. Rep. 2019, 71, 565–572. [Google Scholar] [CrossRef]
- Bo, P.; Lien, J.C.; Chen, Y.Y.; Yu, F.S.; Lu, H.F.; Yu, C.S.; Chou, Y.C.; Yu, C.C.; Chung, J.G. Allyl Isothiocyanate Induces Cell Toxicity by Multiple Pathways in Human Breast Cancer Cells. Am. J. Chin. Med. 2016, 44, 415–437. [Google Scholar] [CrossRef]
- Chiang, J.H.; Tsai, F.J.; Hsu, Y.M.; Yin, M.C.; Chiu, H.Y.; Yang, J.S. Sensitivity of allyl isothiocyanate to induce apoptosis via ER stress and the mitochondrial pathway upon ROS production in colorectal adenocarcinoma cells. Oncol. Rep. 2020, 44, 1415–1424. [Google Scholar] [CrossRef]
- Tusskorn, O.; Senggunprai, L.; Prawan, A.; Kukongviriyapan, U.; Kukongviriyapan, V. Phenethyl isothiocyanate induces calcium mobilization and mitochondrial cell death pathway in cholangiocarcinoma KKU-M214 cells. BMC Cancer 2013, 13, 571. [Google Scholar] [CrossRef] [Green Version]
- Weksler, B.; Romero, I.A.; Couraud, P.O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 2013, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Bintig, W.; Begandt, D.; Schlingmann, B.; Gerhard, L.; Pangalos, M.; Dreyer, L.; Hohnjec, N.; Couraud, P.O.; Romero, I.A.; Weksler, B.B.; et al. Purine receptors and Ca2+ signalling in the human blood-brain barrier endothelial cell line hCMEC/D3. Purinergic Signal. 2012, 8, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Bader, A.; Bintig, W.; Begandt, D.; Klett, A.; Siller, I.G.; Gregor, C.; Schaarschmidt, F.; Weksler, B.; Romero, I.; Couraud, P.O.; et al. Adenosine receptors regulate gap junction coupling of the human cerebral microvascular endothelial cells hCMEC/D3 by Ca2+ influx through cyclic nucleotide-gated channels. J. Physiol. 2017, 595, 2497–2517. [Google Scholar] [CrossRef] [Green Version]
- Berra-Romani, R.; Faris, P.; Negri, S.; Botta, L.; Genova, T.; Moccia, F. Arachidonic Acid Evokes an Increase in Intracellular Ca2+ Concentration and Nitric Oxide Production in Endothelial Cells from Human Brain Microcirculation. Cells 2019, 8, 689. [Google Scholar] [CrossRef] [Green Version]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Nezza, F.D.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; et al. Glutamate triggers intracellular Ca2+ oscillations and nitric oxide release by inducing NAADP- and InsP3 -dependent Ca2+ release in mouse brain endothelial cells. J. Cell. Physiol. 2019, 234, 3538–3554. [Google Scholar] [CrossRef]
- Zuccolo, E.; Laforenza, U.; Ferulli, F.; Pellavio, G.; Scarpellino, G.; Tanzi, M.; Turin, I.; Faris, P.; Lucariello, A.; Maestri, M.; et al. Stim and Orai mediate constitutive Ca2+ entry and control endoplasmic reticulum Ca2+ refilling in primary cultures of colorectal carcinoma cells. Oncotarget 2018, 9, 31098–31119. [Google Scholar] [CrossRef] [Green Version]
- Lodola, F.; Rosti, V.; Tullii, G.; Desii, A.; Tapella, L.; Catarsi, P.; Lim, D.; Moccia, F.; Antognazza, M.R. Conjugated polymers optically regulate the fate of endothelial colony-forming cells. Sci. Adv. 2019, 5, eaav4620. [Google Scholar] [CrossRef] [Green Version]
- Negri, S.; Faris, P.; Tullii, G.; Vismara, M.; Pellegata, A.F.; Lodola, F.; Guidetti, G.; Rosti, V.; Antognazza, M.R.; Moccia, F. Conjugated polymers mediate intracellular Ca2+ signals in circulating endothelial colony forming cells through the reactive oxygen species-dependent activation of Transient Receptor Potential Vanilloid 1 (TRPV1). Cell Calcium 2022, 101, 102502. [Google Scholar] [CrossRef]
- Moccia, F.; Frost, C.; Berra-Romani, R.; Tanzi, F.; Adams, D.J. Expression and function of neuronal nicotinic ACh receptors in rat microvascular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H486–H491. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Berra-Romani, R.; Baruffi, S.; Spaggiari, S.; Adams, D.J.; Taglietti, V.; Tanzi, F. Basal nonselective cation permeability in rat cardiac microvascular endothelial cells. Microvasc. Res. 2002, 64, 187–197. [Google Scholar]
- Moccia, F.; Villa, A.; Tanzi, F. Flow-activated Na+ and K+ current in cardiac microvascular endothelial cells. J. Mol. Cell. Cardiol. 2000, 32, 1589–1593. [Google Scholar] [CrossRef]
- Sobradillo, D.; Hernandez-Morales, M.; Ubierna, D.; Moyer, M.P.; Nunez, L.; Villalobos, C. A reciprocal shift in transient receptor potential channel 1 (TRPC1) and stromal interaction molecule 2 (STIM2) contributes to Ca2+ remodeling and cancer hallmarks in colorectal carcinoma cells. J. Biol. Chem. 2014, 289, 28765–28782. [Google Scholar] [CrossRef] [Green Version]
- Locatelli, F.; Soda, T.; Montagna, I.; Tritto, S.; Botta, L.; Prestori, F.; D’Angelo, E. Calcium Channel-Dependent Induction of Long-Term Synaptic Plasticity at Excitatory Golgi Cell Synapses of Cerebellum. J. Neurosci. 2021, 41, 3307–3319. [Google Scholar] [CrossRef]
- Remigante, A.; Morabito, R.; Spinelli, S.; Trichilo, V.; Loddo, S.; Sarikas, A.; Dossena, S.; Marino, A. d-Galactose Decreases Anion Exchange Capability through Band 3 Protein in Human Erythrocytes. Antioxidants 2020, 9, 689. [Google Scholar] [CrossRef]
- Moccia, F.; Berra-Romani, R.; Baruffi, S.; Spaggiari, S.; Signorelli, S.; Castelli, L.; Magistretti, J.; Taglietti, V.; Tanzi, F. Ca2+ uptake by the endoplasmic reticulum Ca2+-ATPase in rat microvascular endothelial cells. Biochem. J. 2002, 364, 235–244. [Google Scholar]
- Kim, M.J.; Choi, K.J.; Yoon, M.N.; Oh, S.H.; Kim, D.K.; Kim, S.H.; Park, H.S. Hydrogen peroxide inhibits Ca2+ efflux through plasma membrane Ca2+-ATPase in mouse parotid acinar cells. Korean J. Physiol. Pharmacol. 2018, 22, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Talavera, K.; Startek, J.B.; Alvarez-Collazo, J.; Boonen, B.; Alpizar, Y.A.; Sanchez, A.; Naert, R.; Nilius, B. Mammalian Transient Receptor Potential TRPA1 Channels: From Structure to Disease. Physiol. Rev. 2020, 100, 725–803. [Google Scholar] [CrossRef]
- Moccia, F.; Montagna, D. Transient Receptor Potential Ankyrin 1 (TRPA1) Channel as a Sensor of Oxidative Stress in Cancer Cells. Cells 2023, 12, 1261. [Google Scholar] [CrossRef]
- Earley, S.; Gonzales, A.L.; Crnich, R. Endothelium-dependent cerebral artery dilation mediated by TRPA1 and Ca2+-Activated K+ channels. Circ. Res. 2009, 104, 987–994. [Google Scholar] [CrossRef] [Green Version]
- Hakim, M.A.; Buchholz, J.N.; Behringer, E.J. Electrical dynamics of isolated cerebral and skeletal muscle endothelial tubes: Differential roles of G-protein-coupled receptors and K+ channels. Pharmacol. Res. Perspect. 2018, 6, e00391. [Google Scholar] [CrossRef]
- Longden, T.A.; Dunn, K.M.; Draheim, H.J.; Nelson, M.T.; Weston, A.H.; Edwards, G. Intermediate-conductance calcium-activated potassium channels participate in neurovascular coupling. Br. J. Pharmacol. 2011, 164, 922–933. [Google Scholar] [CrossRef] [Green Version]
- Bartekova, M.; Adameova, A.; Gorbe, A.; Ferenczyova, K.; Pechanova, O.; Lazou, A.; Dhalla, N.S.; Ferdinandy, P.; Giricz, Z. Natural and synthetic antioxidants targeting cardiac oxidative stress and redox signaling in cardiometabolic diseases. Free Radic. Biol. Med. 2021, 169, 446–477. [Google Scholar] [CrossRef]
- Moccia, F.; Brunetti, V.; Perna, A.; Guerra, G.; Soda, T.; Berra-Romani, R. The Molecular Heterogeneity of Store-Operated Ca2+ Entry in Vascular Endothelial Cells: The Different roles of Orai1 and TRPC1/TRPC4 Channels in the Transition from Ca2+-Selective to Non-Selective Cation Currents. Int. J. Mol. Sci. 2023, 24, 3259. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Perna, A.; De Luca, A.; Soda, T.; Romani, R.B.; Guerra, G. Targeting Endolysosomal Two-Pore Channels to Treat Cardiovascular Disorders in the Novel COronaVIrus Disease 2019. Front. Physiol. 2021, 12, 629119. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Endolysosomal Ca2+ signaling in cardiovascular health and disease. Int. Rev. Cell Mol. Biol. 2021, 363, 203–269. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Guzman-Silva, A.; Vargaz-Guadarrama, A.; Flores-Alonso, J.C.; Alonso-Romero, J.; Trevino, S.; Sanchez-Gomez, J.; Coyotl-Santiago, N.; Garcia-Carrasco, M.; Moccia, F. Type 2 Diabetes Alters Intracellular Ca2+ Handling in Native Endothelium of Excised Rat Aorta. Int. J. Mol. Sci. 2019, 21, 250. [Google Scholar] [CrossRef] [Green Version]
- Yanda, M.K.; Tomar, V.; Cole, R.; Guggino, W.B.; Cebotaru, L. The Mitochondrial Ca2+ import complex is altered in ADPKD. Cell Calcium 2022, 101, 102501. [Google Scholar] [CrossRef]
- Sheikh, A.Q.; Hurley, J.R.; Huang, W.; Taghian, T.; Kogan, A.; Cho, H.; Wang, Y.; Narmoneva, D.A. Diabetes alters intracellular calcium transients in cardiac endothelial cells. PLoS ONE 2012, 7, e36840. [Google Scholar] [CrossRef] [Green Version]
- Klishin, A.; Sedova, M.; Blatter, L.A. Time-dependent modulation of capacitative Ca2+ entry signals by plasma membrane Ca2+ pump in endothelium. Am. J. Physiol. 1998, 274, C1117–C1128. [Google Scholar]
- Snitsarev, V.A.; Taylor, C.W. Overshooting cytosolic Ca2+ signals evoked by capacitative Ca2+ entry result from delayed stimulation of a plasma membrane Ca2+ pump. Cell Calcium 1999, 25, 409–417. [Google Scholar] [CrossRef]
- Mapelli, L.; Gagliano, G.; Soda, T.; Laforenza, U.; Moccia, F.; D’Angelo, E.U. Granular Layer Neurons Control Cerebellar Neurovascular Coupling Through an NMDA Receptor/NO-Dependent System. J. Neurosci. 2017, 37, 1340–1351. [Google Scholar] [CrossRef] [Green Version]
- Andrei, S.R.; Ghosh, M.; Sinharoy, P.; Damron, D.S. Stimulation of TRPA1 attenuates ischemia-induced cardiomyocyte cell death through an eNOS-mediated mechanism. Channels 2019, 13, 192–206. [Google Scholar] [CrossRef] [Green Version]
- Negri, S.; Faris, P.; Moccia, F. Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime? Int. J. Mol. Sci. 2021, 22, 9821. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef] [Green Version]
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A. Mitochondria and endothelial function. Circ. Res. 2013, 112, 1171–1188. [Google Scholar] [CrossRef] [Green Version]
- Bruce, J.I.; Elliott, A.C. Oxidant-impaired intracellular Ca2+ signaling in pancreatic acinar cells: Role of the plasma membrane Ca2+-ATPase. Am. J. Physiol. Cell Physiol. 2007, 293, C938–C950. [Google Scholar] [CrossRef] [Green Version]
- Szewczyk, M.M.; Pande, J.; Akolkar, G.; Grover, A.K. Caloxin 1b3: A novel plasma membrane Ca2+-pump isoform 1 selective inhibitor that increases cytosolic Ca2+ in endothelial cells. Cell Calcium 2010, 48, 352–357. [Google Scholar] [CrossRef]
- Long, Y.; Chen, S.W.; Gao, C.L.; He, X.M.; Liang, G.N.; Wu, J.; Jiang, C.X.; Liu, X.; Wang, F.; Chen, F. ATP2B1 Gene Silencing Increases NO Production Under Basal Conditions Through the Ca2+/calmodulin/eNOS Signaling Pathway in Endothelial Cells. Hypertens. Res. 2018, 41, 246–252. [Google Scholar] [CrossRef]
- Zaidi, A. Plasma membrane Ca-ATPases: Targets of oxidative stress in brain aging and neurodegeneration. World J. Biol. Chem. 2010, 1, 271–280. [Google Scholar] [CrossRef]
- Holton, M.; Mohamed, T.M.; Oceandy, D.; Wang, W.; Lamas, S.; Emerson, M.; Neyses, L.; Armesilla, A.L. Endothelial nitric oxide synthase activity is inhibited by the plasma membrane calcium ATPase in human endothelial cells. Cardiovasc. Res. 2010, 87, 440–448. [Google Scholar] [CrossRef]
- Guo, X.; Liu, R.; Jia, M.; Wang, Q.; Wu, J. Ischemia Reperfusion Injury Induced Blood Brain Barrier Dysfunction and the Involved Molecular Mechanism. Neurochem. Res. 2023, 48, 2320–2334. [Google Scholar] [CrossRef]
- Shaw, R.L.; Norton, C.E.; Segal, S.S. Apoptosis in resistance arteries induced by hydrogen peroxide: Greater resilience of endothelium versus smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1625–H1633. [Google Scholar] [CrossRef]
- Akki, R.; Siracusa, R.; Morabito, R.; Remigante, A.; Campolo, M.; Errami, M.; La Spada, G.; Cuzzocrea, S.; Marino, A. Neuronal-like differentiated SH-SY5Y cells adaptation to a mild and transient H2O2-induced oxidative stress. Cell Biochem. Funct. 2018, 36, 56–64. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS signaling and redox biology in endothelial cells. Cell. Mol. Life Sci. 2015, 72, 3281–3303. [Google Scholar] [CrossRef]
- Santoro, M.M. Fashioning blood vessels by ROS signalling and metabolism. Semin. Cell Dev. Biol. 2018, 80, 35–42. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berra-Romani, R.; Brunetti, V.; Pellavio, G.; Soda, T.; Laforenza, U.; Scarpellino, G.; Moccia, F. Allyl Isothiocianate Induces Ca2+ Signals and Nitric Oxide Release by Inducing Reactive Oxygen Species Production in the Human Cerebrovascular Endothelial Cell Line hCMEC/D3. Cells 2023, 12, 1732. https://doi.org/10.3390/cells12131732
Berra-Romani R, Brunetti V, Pellavio G, Soda T, Laforenza U, Scarpellino G, Moccia F. Allyl Isothiocianate Induces Ca2+ Signals and Nitric Oxide Release by Inducing Reactive Oxygen Species Production in the Human Cerebrovascular Endothelial Cell Line hCMEC/D3. Cells. 2023; 12(13):1732. https://doi.org/10.3390/cells12131732
Chicago/Turabian StyleBerra-Romani, Roberto, Valentina Brunetti, Giorgia Pellavio, Teresa Soda, Umberto Laforenza, Giorgia Scarpellino, and Francesco Moccia. 2023. "Allyl Isothiocianate Induces Ca2+ Signals and Nitric Oxide Release by Inducing Reactive Oxygen Species Production in the Human Cerebrovascular Endothelial Cell Line hCMEC/D3" Cells 12, no. 13: 1732. https://doi.org/10.3390/cells12131732