
Platelet Metabolic Flexibility: A Matter of Substrate and Location




{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Platelets in Diseases
1.2. Platelet Metabolism
1.3. Platelets’ Metabolic Plasticity: Adaptation to Energy Demands
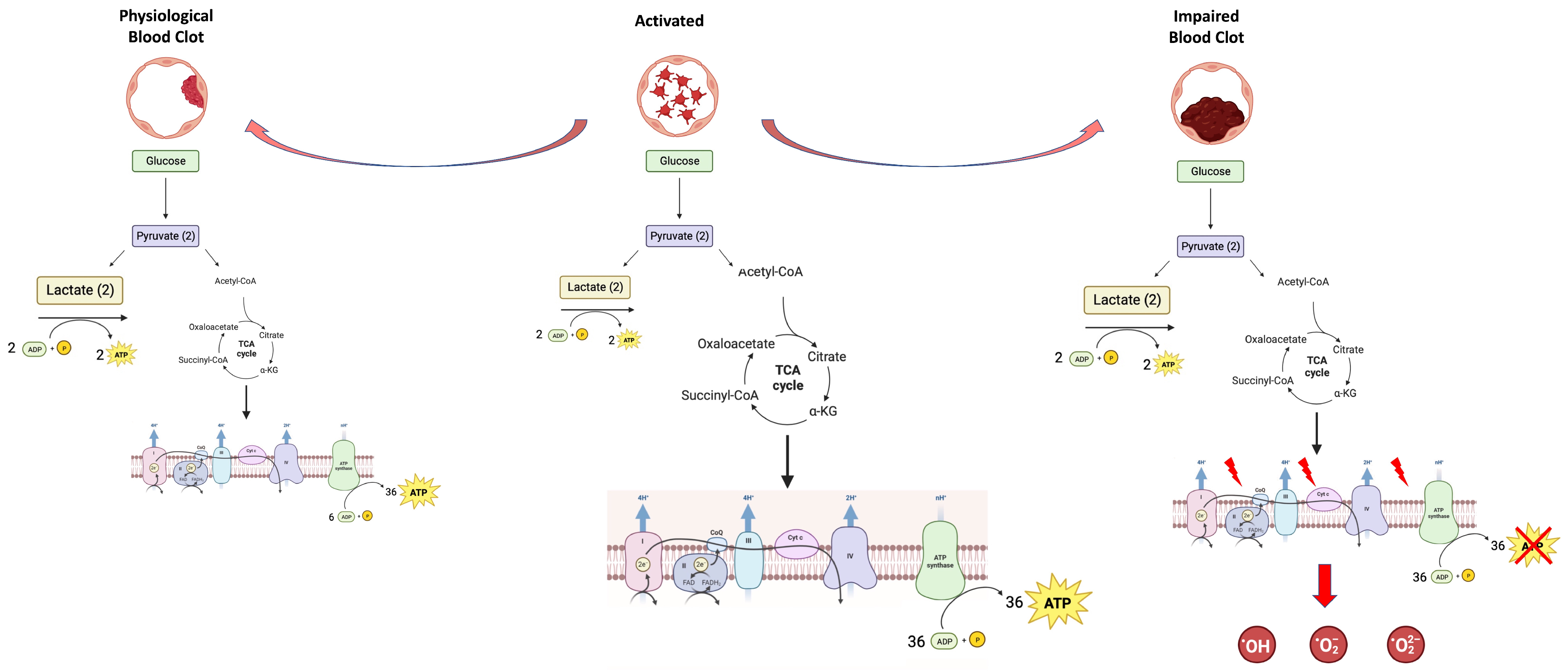
1.4. Platelets’ Metabolic Plasticity—The Other Side of the Coin: Production of Oxidative Stress
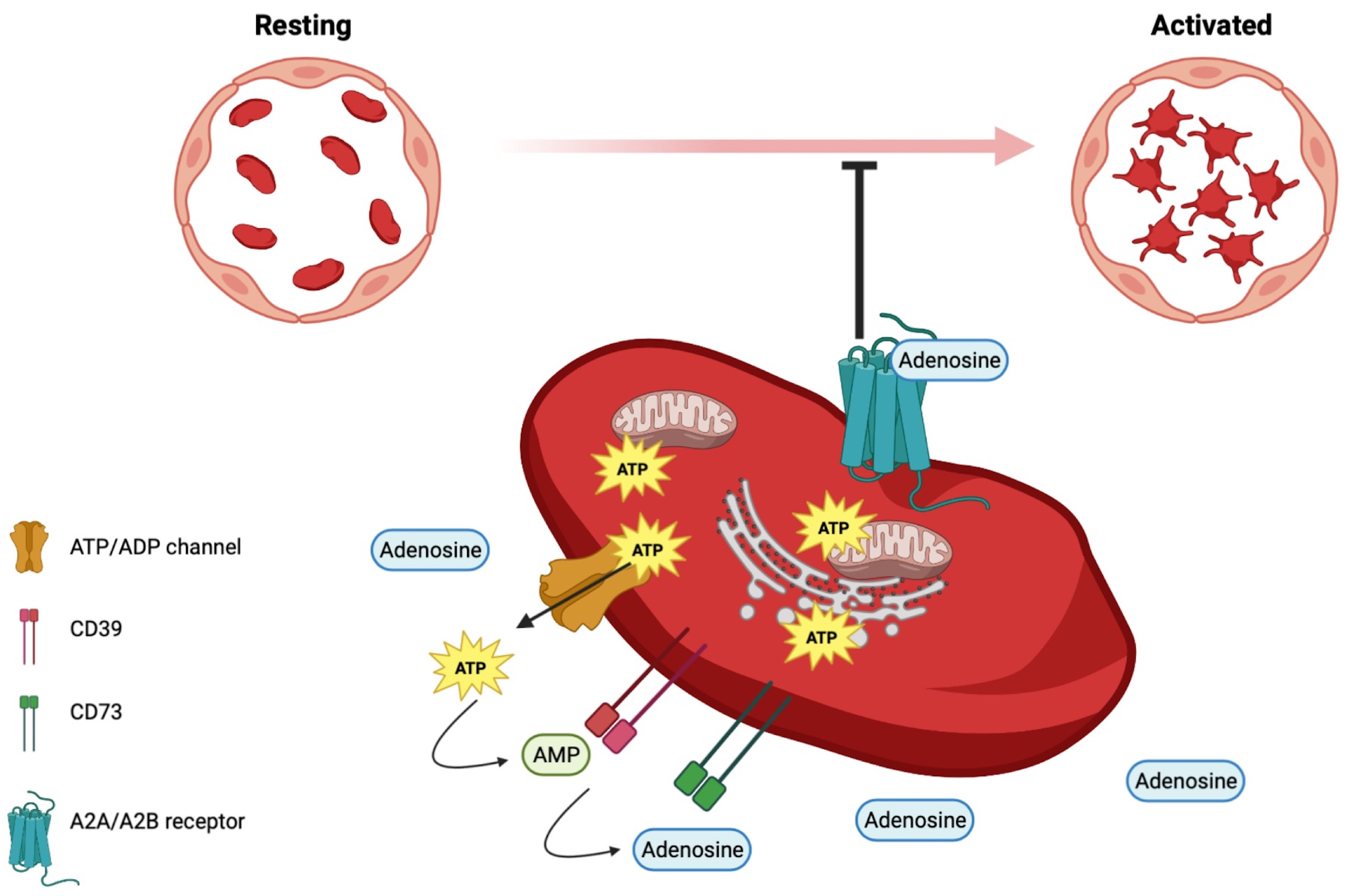
1.5. Adenyl Nucleotides beyond Energetics in Platelets
1.6. Omics Approach: A New Strategy for Studying Platelet Biology
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Machlus, K.R.; Italiano, J.E. The Incredible Journey: From Megakaryocyte Development to Platelet Formation. J. Cell. Biol. 2013, 201, 785–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bizzozero, J. Ueber Einen Neuen Formbestandtheil Des Blutes Und Dessen Rolle Bei Der Thrombose Und Der Blutgerinnung-Untersuchungen. Arch. Pathol. Anat. Physiol. Klin. Med. 1882, 90, 261–332. [Google Scholar] [CrossRef]
- Broos, K.; Feys, H.B.; De Meyer, S.F.; Vanhoorelbeke, K.; Deckmyn, H. Platelets at Work in Primary Hemostasis. Blood Rev. 2011, 25, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Franco, A.T.; Corken, A.; Ware, J. Platelets at the Interface of Thrombosis, Inflammation, and Cancer. Blood 2015, 126, 582–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorchheimer, D.A.; Becker, R. Platelets in Atherothrombosis. Mayo Clin. Proc. 2006, 81, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Yan, Z.; Meng, Z.; Li, X.; Liu, M.; Ren, X.; Zhu, M.; He, Q.; Zhang, Q.; Song, K.; et al. Relationship between Mean Platelet Volume and Metabolic Syndrome in Chinese Patients. Sci. Rep. 2018, 8, 14574. [Google Scholar] [CrossRef] [Green Version]
- Braun, A.; Anders, H.J.; Gudermann, T.; Mammadova-Bach, E. Platelet-Cancer Interplay: Molecular Mechanisms and New Therapeutic Avenues. Front. Oncol. 2021, 11, 665534. [Google Scholar] [CrossRef]
- Santilli, F.; Vazzana, N.; Liani, R.; Guagnano, M.T.; Davì, G. Platelet Activation in Obesity and Metabolic Syndrome. Obes. Rev. 2012, 13, 27–42. [Google Scholar] [CrossRef]
- Yang, X.J.; Zhang, L.Y.; Ma, Q.H.; Sun, H.P.; Xu, Y.; Chen, X.; Pan, C.W. Platelet Parameters in Chinese Older Adults with Metabolic Syndrome. Endocr. Connect. 2020, 9, 696–704. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, C.; Pan, F.; Chen, Y.; Xiong, L.; Li, Y.; Chu, X.; Huang, G. Platelets in the Tumor Microenvironment and Their Biological Effects on Cancer Hallmarks. Front. Oncol. 2023, 13, 1121401. [Google Scholar] [CrossRef] [PubMed]
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging Roles for Platelets as Immune and Inflammatory Cells. Blood 2014, 123, 2759–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, C.; Pitchford, S. Platelets and Allergic Inflammation. Clin. Exp. Allergy 2014, 44, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Ripoche, J. Blood Platelets and Inflammation: Their Relationship with Liver and Digestive Diseases. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 353–357. [Google Scholar] [CrossRef]
- Li, L.; Xu, P.; Zhang, Z.; Zhou, X.; Chen, C.; Lu, C. Platelets Can Reflect the Severity of Crohn’s Disease without the Effect of Anemia. Clinics 2020, 75, e1596. [Google Scholar] [CrossRef]
- Plantureux, L.; Mege, D.; Crescence, L.; Carminita, E.; Robert, S.; Cointe, S.; Brouilly, N.; Ezzedine, W.; Dignat-George, F.; Dubois, C.; et al. The Interaction of Platelets with Colorectal Cancer Cells Inhibits Tumor Growth but Promotes Metastasis. Cancer Res. 2020, 80, 291–303. [Google Scholar] [CrossRef]
- Lambert, M.P. Platelets in Liver and Renal Disease. Hematology 2016, 2016, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Herster, F.; Karbach, S.; Chatterjee, M.; Weber, A.N.R. Platelets: Underestimated Regulators of Autoinflammation in Psoriasis. J. Investig. Dermatol. 2021, 141, 1395–1403. [Google Scholar] [CrossRef]
- Singh, A.; Bisht, P.; Bhattacharya, S.; Guchhait, P. Role of Platelet Cytokines in Dengue Virus Infection. Front. Cell. Infect. Microbiol. 2020, 10, 561366. [Google Scholar] [CrossRef]
- Tokarz-Deptuła, B.; Palma, J.; Baraniecki, Ł.; Stosik, M.; Kołacz, R.; Deptuła, W. What Function Do Platelets Play in Inflammation and Bacterial and Viral Infections? Front. Immunol. 2021, 12, 770436. [Google Scholar] [CrossRef]
- Da’Dara, A.A.; Skelly, P.J. Schistosomes versus Platelets. Thromb. Res. 2014, 134, 1176–1181. [Google Scholar] [CrossRef] [PubMed]
- Perzborn, E.; Heitmeier, S.; Laux, V. Platelets and Their Role in Hemostasis and Thrombosis—From Physiology to Pathophysiology and Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 12772. [Google Scholar] [CrossRef]
- Amelirad, A.; Shamsasanjan, K.; Akbarzadehlaleh, P.; Sarvar, D.P. Signaling Pathways of Receptors Involved in Platelet Activation and Shedding of These Receptors in Stored Platelets. Adv. Pharm. Bull. 2019, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Bergmeier, W.; Piffath, C.L.; Goerge, T.; Cifuni, S.M.; Ruggeri, Z.M.; Ware, J.; Wagner, D.D. The Role of Platelet Adhesion Receptor GPIbα Far Exceeds That of Its Main Ligand, von Willebrand Factor, in Arterial Thrombosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16900–16905. [Google Scholar] [CrossRef]
- Manke, M.C.; Ahrends, R.; Borst, O. Platelet Lipid Metabolism in Vascular Thrombo-Inflammation. Pharm. Ther. 2022, 237, 108258. [Google Scholar] [CrossRef]
- Wagner, D.D.; Burger, P.C. Platelets in Inflammation and Thrombosis. Arter. Thromb. Vasc. Biol. 2003, 23, 2131–2137. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Tyagi, T.; Antoniak, S. Platelet in Thrombo-Inflammation: Unraveling New Therapeutic Targets. Front. Immunol. 2022, 13, 1039843. [Google Scholar] [CrossRef]
- Colicchia, M.; Perrella, G.; Gant, P.; Rayes, J. Novel Mechanisms of Thrombo-Inflammation during Infection: Spotlight on Neutrophil Extracellular Trap-Mediated Platelet Activation. Res. Pract. Thromb. Haemost. 2023, 7, 100116. [Google Scholar] [CrossRef]
- Projahn, D.; Koenen, R.R. Platelets: Key Players in Vascular Inflammation. J. Leukoc. Biol. 2012, 92, 1167–1175. [Google Scholar] [CrossRef]
- Ludwig, N.; Hilger, A.; Zarbock, A.; Rossaint, J. Platelets at the Crossroads of Pro-Inflammatory and Resolution Pathways during Inflammation. Cells 2022, 11, 1957. [Google Scholar] [CrossRef]
- Maiocchi, S.; Alwis, I.; Wu, M.C.L.; Yuan, Y.; Jackson, S.P. Thromboinflammatory Functions of Platelets in Ischemia-Reperfusion Injury and Its Dysregulation in Diabetes. Semin. Thromb. Hemost. 2018, 44, 102–113. [Google Scholar] [CrossRef]
- McFadyen, J.D.; Stevens, H.; Peter, K. The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications. Circ. Res. 2020, 127, 571–587. [Google Scholar] [CrossRef]
- Barrett, T.J.; Bilaloglu, S.; Cornwell, M.; Burgess, H.M.; Virginio, V.W.; Drenkova, K.; Ibrahim, H.; Yuriditsky, E.; Aphinyanaphongs, Y.; Lifshitz, M.; et al. Platelets Contribute to Disease Severity in COVID-19. J. Thromb. Haemost. 2021, 19, 3139. [Google Scholar] [CrossRef]
- Chaudhary, P.K.; Kim, S.; Kim, S. An Insight into Recent Advances on Platelet Function in Health and Disease. Int. J. Mol. Sci. 2022, 23, 6022. [Google Scholar] [CrossRef]
- Aibibula, M.; Naseem, K.M.; Sturmey, R.G. Glucose Metabolism and Metabolic Flexibility in Blood Platelets. J. Thromb. Haemost. 2018, 16, 2300–2314. [Google Scholar] [CrossRef] [Green Version]
- Ravi, S.; Chacko, B.; Sawada, H.; Kramer, P.A.; Johnson, M.S.; Benavides, G.A.; O’Donnell, V.; Marques, M.B.; Darley-Usmar, V.M. Metabolic Plasticity in Resting and Thrombin Activated Platelets. PLoS ONE 2015, 10, e0123597. [Google Scholar] [CrossRef] [Green Version]
- Verhoeven, A.J.M.; Gorter, G.; Mommersteeg, M.E.; Akkerman, J.W.N. The Energetics of Early Platelet Responses. Energy Consumption during Shape Change and Aggregation with Special Reference to Protein Phosphorylation and the Polyphosphoinositide Cycle. Biochem. J. 1985, 228, 451–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, P.P.; Ekhlak, M.; Dash, D. Energy Metabolism in Platelets Fuels Thrombus Formation: Halting the Thrombosis Engine with Small-Molecule Modulators of Platelet Metabolism. Metabolism 2023, 145, 155596. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.P.; Tiwari, A.; Singh, N.; Gautam, D.; Sonkar, V.K.; Agarwal, V.; Dash, D. Aerobic Glycolysis Fuels Platelet Activation: Small-Molecule Modulators of Platelet Metabolism as Anti-Thrombotic Agents. Haematologica 2019, 104, 806–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sake, C.L.; Metcalf, A.J.; Meagher, M.; Di Paola, J.; Neeves, K.B.; Boyle, N.R. Isotopically Nonstationary 13C Metabolic Flux Analysis in Resting and Activated Human Platelets. Metab. Eng. 2022, 69, 313–322. [Google Scholar] [CrossRef]
- Kulkarni, P.P.; Ekhlak, M.; Singh, V.; Kailashiya, V.; Singh, N.; Dash, D. Fatty Acid Oxidation Fuels Agonist-Induced Platelet Activation and Thrombus Formation: Targeting β-Oxidation of Fatty Acids as an Effective Anti-Platelet Strategy. FASEB J. 2023, 37, e22768. [Google Scholar] [CrossRef]
- Heijnen, H.F.; Oorschot, V.; Sixma, J.J.; Slot, J.W.; James, D.E. Thrombin stimulates Glucose Transport in Human Platelets via the Translocation of the Glucose Transporter GLUT-3 from alpha-Granules to the Cell Surface. J. Cell. Biol. 1997, 138, 323–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchinger, H.; Jain, K.; Tyagi, T.; Hwa, J. Role of Platelet Mitochondria: Life in a Nucleus-Free Zone. Front. Cardiovasc. Med. 2019, 6, 495633. [Google Scholar] [CrossRef] [Green Version]
- Fidler, T.P.; Campbell, R.A.; Funari, T.; Dunne, N.; Balderas Angeles, E.; Middleton, E.A.; Chaudhuri, D.; Weyrch, A.S.; Abel, E.D. Deletion of GLUT1 and GLUT3 Reveals Multiple Roles for Glucose Metabolism in Platelet and Megakaryocyte Function. Cell. Rep. 2017, 20, 881–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidler, T.P.; Middleton, E.A.; Rowley, J.W.; Boudreau, L.H.; Campbell, R.A.; Souvenir, R.; Funari, T.; Tessandier, N.; Boilard, E.; Weyrich, A.S.; et al. Glucose Transporter 3 Potentiates Degranulation and Is Required for Platelet Activation. Arter. Thromb. Vasc. Biol. 2017, 37, 1628–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakhya, K.S.; Vekaria, H.; Coenen, D.M.; Omali, L.; Lykins, J.; Joshi, S.; Alfar, H.R.; Wang, Q.J.; Sullivan, P.; Whiteheart, S.W. Platelet Glycogenolysis Is Important for Energy Production and Function. Platelets 2023, 34, 2222184. [Google Scholar] [CrossRef]
- Prakhya, S.; Omali, L.; Lykins, J.; Coenen, D.; Vekaria, H.; Whiteheart, S.W. Role of Glycogen Mobilization and Mitochondrial Bioenergetics in Platelet Function, Hemostasis, and Thrombosis. Blood 2022, 140, 1965–1966. [Google Scholar] [CrossRef]
- Litvinov, R.I.; Weisel, J.W. Blood Clot Contraction: Mechanisms, Pathophysiology, and Disease. Res. Pract. Thromb. Haemost. 2022, 7, 100023. [Google Scholar] [CrossRef]
- Tomasiak-Lozowska, M.M.; Misztal, T.; Rusak, T.; Branska-Januszewska, J.; Bodzenta-Lukaszyk, A.; Tomasiak, M. Asthma Is Associated with Reduced Fibrinolytic Activity, Abnormal Clot Architecture, and Decreased Clot Retraction Rate. Allergy 2017, 72, 314–319. [Google Scholar] [CrossRef]
- Ravera, S.; Signorello, M.G.; Bartolucci, M.; Ferrando, S.; Manni, L.; Caicci, F.; Calzia, D.; Panfoli, I.; Morelli, A.; Leoncini, G. Extramitochondrial Energy Production in Platelets. Biol. Cell. 2018, 110, 97–108. [Google Scholar] [CrossRef]
- Tomaiuolo, M.; Stalker, T.J.; Welsh, J.D.; Diamond, S.L.; Sinno, T.; Brass, L.F. A Systems Approach to Hemostasis: 2. Computational Analysis of Molecular Transport in the Thrombus Microenvironment. Blood 2014, 124, 1816–1823. [Google Scholar] [CrossRef] [Green Version]
- Akkerman, J.W.N.; Gorter, G.; Schrama, L.; Holmsen, H. A Novel Technique for Rapid Determination of Energy Consumption in Platelets. Demonstration of Different Energy Consumption Associated with Three Secretory Responses. Biochem. J. 1983, 210, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Duyvené de Wit, L.J.; Badenhorst, P.N.; Heyns, A.D. Ultrastructural Morphometric Observations on Serial Sectioned Human Blood Platelet Subpopulations. Eur. J. Cell. Biol. 1987, 43, 408–411. [Google Scholar]
- Zharikov, S.; Shiva, S. Platelet Mitochondrial Function: From Regulation of Thrombosis to Biomarker of Disease. Biochem. Soc. Trans. 2013, 41, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Li, X.; Yu, D.; Li, X.; Li, Y.; Long, Y.; Yuan, Y.; Ji, Z.; Zhang, M.; Wen, J.G.; et al. Application of Mitochondrial Pyruvate Carrier Blocker UK5099 Creates Metabolic Reprogram and Greater Stem-like Properties in LnCap Prostate Cancer Cells In Vitro. Oncotarget 2015, 6, 37758. [Google Scholar] [CrossRef] [Green Version]
- McDowell, R.E.; Aulak, K.S.; Almoushref, A.; Melillo, C.A.; Brauer, B.E.; Newman, J.E.; Tonelli, A.R.; Dweik, R.A. Platelet Glycolytic Metabolism Correlates with Hemodynamic Severity in Pulmonary Arterial Hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L562–L569. [Google Scholar] [CrossRef]
- Chen, A.F.; Chen, D.D.; Daiber, A.; Faraci, F.M.; Li, H.; Rembold, C.M.; Laher, I. Free Radical Biology of the Cardiovascular System. Clin. Sci. 2012, 123, 73–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madamanchi, N.R.; Vendrov, A.; Runge, M.S. Oxidative Stress and Vascular Disease. Arter. Thromb. Vasc. Biol. 2005, 25, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, E.; Gori, T.; Münzel, T. Oxidative Stress and Endothelial Dysfunction in Hypertension. Hypertens. Res. 2011, 34, 665–673. [Google Scholar] [CrossRef]
- Lum, H.; Roebuck, K.A. Oxidant Stress and Endothelial Cell Dysfunction. Am. J. Physiol. Cell. Physiol. 2001, 280, C719–C741. [Google Scholar] [CrossRef] [Green Version]
- Lüscher, T.F.; Barton, M. Biology of the Endothelium. Clin. Cardiol. 1997, 20, II-3–II-10. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.E. Oxidative Stress and Platelets. Arter. Thromb. Vasc. Biol. 2008, 28, s11–s16. [Google Scholar] [CrossRef] [Green Version]
- Singh, U.; Jialal, I. Oxidative Stress and Atherosclerosis. Pathophysiology 2006, 13, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.R.; Zhu, Y.; Halushka, P.V.; Lincoln, T.M.; Mendelsohn, M.E. Mechanism of Platelet Inhibition by Nitric Oxide: In Vivo Phosphorylation of Thromboxane Receptor by Cyclic GMP-Dependent Protein Kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 4888–4893. [Google Scholar] [CrossRef] [PubMed]
- Lupidi, G.; Angeletti, M.; Eleuteri, A.M.; Tacconi, L.; Coletta, M.; Fioretti, E. Peroxynitrite-Mediated Oxidation of Fibrinogen Inhibits Clot Formation. FEBS Lett. 1999, 462, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Rusak, T.; Tomasiak, M.; Ciborowski, M. Peroxynitrite Can Affect Platelet Responses by Inhibiting Energy Production. Acta Biochim. Pol. 2006, 53, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Tomasiak, M.; Stelmach, H.; Rusak, T.; Wysocka, J. Nitric Oxide and Platelet Energy Metabolism. Acta Biochim. Pol. 2004, 51, 789–803. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, E.; Davies, K.J.A. Mitochondrial Free Radical Generation, Oxidative Stress, and Aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Delaney, M.K.; Kim, K.; Estevez, B.; Xu, Z.; Stojanovic-Terpo, A.; Shen, B.; Ushio-Fukai, M.; Cho, J.; Du, X. Differential Roles of the NADPH-Oxidase 1 and 2 in Platelet Activation and Thrombosis. Arter. Thromb. Vasc. Biol. 2016, 36, 846–854. [Google Scholar] [CrossRef] [Green Version]
- Arbiser, J.L.; Petros, J.; Klafter, R.; Govindajaran, B.; McLaughlin, E.R.; Brown, L.F.; Cohen, C.; Moses, M.; Kilroy, S.; Arnold, R.S.; et al. Reactive Oxygen Generated by Nox1 Triggers the Angiogenic Switch. Proc. Natl. Acad. Sci. USA 2002, 99, 715–720. [Google Scholar] [CrossRef]
- Signorello, M.G.; Leoncini, G. The Molecular Mechanisms Involved in Lectin-Induced Human Platelet Aggregation. Biol. Chem. 2017, 398, 1335–1346. [Google Scholar] [CrossRef]
- Leoncini, G.; Maresca, M.; Colao, C. Oxidative Metabolism of Human Platelets. Biochem. Int. 1991, 25, 647–655. [Google Scholar]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H Oxidase: Role in Cardiovascular Biology and Disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef]
- Passacquale, G.; Sharma, P.; Perera, D.; Ferro, A. Antiplatelet Therapy in Cardiovascular Disease: Current Status and Future Directions. Br. J. Clin. Pharm. 2022, 88, 2686–2699. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, D.; Tan, P.; Xian, B.; Jiang, H.; Wu, Q.; Huang, X.; Zhang, P.; Xiao, X.; Pei, J. Mechanism of Platelet Activation and Potential Therapeutic Effects of Natural Drugs. Phytomedicine 2023, 108, 154463. [Google Scholar] [CrossRef] [PubMed]
- Coenen, D.M.; Mastenbroek, T.G.; Cosemans, J.M.E.M. Platelet Interaction with Activated Endothelium: Mechanistic Insights from Microfluidics. Blood 2017, 130, 2819–2828. [Google Scholar] [CrossRef]
- Hamilos, M.; Petousis, S.; Parthenakis, F. Interaction between Platelets and Endothelium: From Pathophysiology to New Therapeutic Options. Cardiovasc. Diagn. Ther. 2018, 8, 568. [Google Scholar] [CrossRef]
- Gachet, C.; Hechler, B. Platelet Purinergic Receptors in Thrombosis and Inflammation. Hamostaseologie 2020, 40, 145–152. [Google Scholar] [CrossRef]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in Immunity and Inflammation. Trends Mol. Med. 2013, 19, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panfoli, I.; Cassanello, M.; Bruschettini, M.; Colella, M.; Cerone, R.; Ravera, S.; Calzia, D.; Candiano, G.; Ramenghi, L. Why Do Premature Newborn Infants Display Elevated Blood Adenosine Levels? Med. Hypotheses 2016, 90, 53–56. [Google Scholar] [CrossRef] [Green Version]
- Colella, M.; Panfoli, I.; Doglio, M.; Cassanello, M.; Bruschi, M.; Angelis, L.C.D.; Candiano, G.; Parodi, A.; Malova, M.; Petretto, A.; et al. Adenosine Blood Level: A Biomarker of White Matter Damage in Very Low Birth Weight Infants. Curr. Pediatr. Rev. 2022, 18, 153–163. [Google Scholar] [CrossRef]
- van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet Biology and Functions: New Concepts and Clinical Perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Clemetson, K.J.; Capitanio, A.; Lüscher, E.F. High Resolution Two-Dimensional Gel Electrophoresis of the Proteins and Glycoproteins of Human Blood Platelets and Platelet Membranes. Biochim. Biophys. Acta (BBA)-Biomembr. 1979, 553, 11–24. [Google Scholar] [CrossRef]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The First Comprehensive and Quantitative Analysis of Human Platelet Protein Composition Allows the Comparative Analysis of Structural and Functional Pathways. Blood 2012, 120, e73–e82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solari, F.A.; Krahn, D.; Swieringa, F.; Verhelst, S.; Rassaf, T.; Tasdogan, A.; Zahedi, R.P.; Lorenz, K.; Renné, T.; Heemskerk, J.W.M.; et al. Multi-Omics Approaches to Study Platelet Mechanisms. Curr. Opin. Chem. Biol. 2023, 73, 102253. [Google Scholar] [CrossRef]
- Gutmann, C.; Joshi, A.; Mayr, M. Platelet “-Omics” in Health and Cardiovascular Disease. Atherosclerosis 2020, 307, 87–96. [Google Scholar] [CrossRef]
- Babur, Ö.; Melrose, A.R.; Cunliffe, J.M.; Klimek, J.; Pang, J.; Sepp, A.L.I.; Zilberman-Rudenko, J.; Yunga, S.T.; Zheng, T.; Parra-Izquierdo, I.; et al. Phosphoproteomic Quantitation and Causal Analysis Reveal Pathways in GPVI/ITAM-Mediated Platelet Activation Programs. Blood 2020, 136, 2346–2358. [Google Scholar] [CrossRef]
- Toonstra, C.; Hu, Y.; Zhang, H. Deciphering the Roles of N-Glycans on Collagen-Platelet Interactions. J. Proteome Res. 2019, 18, 2467–2477. [Google Scholar] [CrossRef]
- Unsworth, A.J.; Bombik, I.; Pinto-Fernandez, A.; McGouran, J.F.; Konietzny, R.; Zahedi, R.P.; Watson, S.P.; Kessler, B.M.; Pears, C.J. Human Platelet Protein Ubiquitylation and Changes Following GPVI Activation. Thromb. Haemost. 2019, 119, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denis, M.M.; Tolley, N.D.; Bunting, M.; Schwertz, H.; Jiang, H.; Lindemann, S.; Yost, C.C.; Rubner, F.J.; Albertine, K.H.; Swoboda, K.J.; et al. Escaping the Nuclear Confines: Signal-Dependent Pre-MRNA Splicing in Anucleate Platelets. Cell 2005, 122, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Davizon-Castillo, P.; Rowley, J.W.; Rondina, M.T. Megakaryocyte and Platelet Transcriptomics for Discoveries in Human Health and Disease. Arter. Thromb. Vasc. Biol. 2020, 40, 1432–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Swieringa, F.; Solari, F.A.; Provenzale, I.; Grassi, L.; De Simone, I.; Baaten, C.C.F.M.J.; Cavill, R.; Sickmann, A.; Frontini, M.; et al. Assessment of a Complete and Classified Platelet Proteome from Genome-Wide Transcripts of Human Platelets and Megakaryocytes Covering Platelet Functions. Sci. Rep. 2021, 11, 12358. [Google Scholar] [CrossRef]
- Keramati, A.R.; Chen, M.H.; Rodriguez, B.A.T.; Yanek, L.R.; Bhan, A.; Gaynor, B.J.; Ryan, K.; Brody, J.A.; Zhong, X.; Wei, Q.; et al. Genome Sequencing Unveils a Regulatory Landscape of Platelet Reactivity. Nat. Commun. 2021, 12, 3626. [Google Scholar] [CrossRef]
- Petersen, R.; Lambourne, J.J.; Javierre, B.M.; Grassi, L.; Kreuzhuber, R.; Ruklisa, D.; Rosa, I.M.; Tomé, A.R.; Elding, H.; Van Geffen, J.P.; et al. Platelet Function Is Modified by Common Sequence Variation in Megakaryocyte Super Enhancers. Nat. Commun. 2017, 8, 16058. [Google Scholar] [CrossRef] [Green Version]
- Simon, L.M.; Edelstein, L.C.; Nagalla, S.; Woodley, A.B.; Chen, E.S.; Kong, X.; Ma, L.; Fortina, P.; Kunapuli, S.; Holinstat, M.; et al. Human Platelet MicroRNA-MRNA Networks Associated with Age and Gender Revealed by Integrated Plateletomics. Blood 2014, 123, e37–e45. [Google Scholar] [CrossRef] [Green Version]
- Mantini, G.; Meijer, L.L.; Glogovitis, I.; In ‘T Veld, S.G.J.G.; Paleckyte, R.; Capula, M.; Le Large, T.Y.S.; Morelli, L.; Pham, T.V.; Piersma, S.R.; et al. Omics Analysis of Educated Platelets in Cancer and Benign Disease of the Pancreas. Cancers 2020, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, V.B.; Murphy, R.C.; Watson, S.P. Platelet Lipidomics: Modern Day Perspective on Lipid Discovery and Characterization in Platelets. Circ. Res. 2014, 114, 1185–1203. [Google Scholar] [CrossRef]
- Zeiler, M.; Moser, M.; Mann, M. Copy Number Analysis of the Murine Platelet Proteome Spanning the Complete Abundance Range. Mol. Cell. Proteom. 2014, 13, 3435–3445. [Google Scholar] [CrossRef] [Green Version]
- Peng, B.; Geue, S.; Coman, C.; Münzer, P.; Kopczynski, D.; Has, C.; Hoffmann, N.; Manke, M.C.; Lang, F.; Sickmann, A.; et al. Identification of Key Lipids Critical for Platelet Activation by Comprehensive Analysis of the Platelet Lipidome. Blood 2018, 132, e1–e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slatter, D.A.; Aldrovandi, M.; O’Connor, A.; Allen, S.M.; Brasher, C.J.; Murphy, R.C.; Mecklemann, S.; Ravi, S.; Darley-Usmar, V.; O’Donnell, V.B. Mapping the Human Platelet Lipidome Reveals Cytosolic Phospholipase A2 as a Regulator of Mitochondrial Bioenergetics during Activation. Cell Metab. 2016, 23, 930–944. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, M.; Rath, D.; Schlotterbeck, J.; Rheinlaender, J.; Walker-Allgaier, B.; Alnaggar, N.; Zdanyte, M.; Müller, I.; Borst, O.; Geisler, T.; et al. Regulation of Oxidized Platelet Lipidome: Implications for Coronary Artery Disease. Eur. Heart J. 2017, 38, 1993–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, A.P.; Byrnes, J.R.; Mackman, N. Hyperlipidemia, Tissue Factor, Coagulation and Simvastatin. Trends Cardiovasc. Med. 2014, 24, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goracci, L.; Petito, E.; Di Veroli, A.; Falcinelli, E.; Bencivenga, C.; Giglio, E.; Becattini, C.; De Robertis, E.; Vaudo, G.; Gresele, P. A Platelet Lipidomics Signature in Patients with COVID-19. Platelets 2023, 34, 2200847. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravera, S.; Signorello, M.G.; Panfoli, I. Platelet Metabolic Flexibility: A Matter of Substrate and Location. Cells 2023, 12, 1802. https://doi.org/10.3390/cells12131802
Ravera S, Signorello MG, Panfoli I. Platelet Metabolic Flexibility: A Matter of Substrate and Location. Cells. 2023; 12(13):1802. https://doi.org/10.3390/cells12131802
Chicago/Turabian StyleRavera, Silvia, Maria Grazia Signorello, and Isabella Panfoli. 2023. "Platelet Metabolic Flexibility: A Matter of Substrate and Location" Cells 12, no. 13: 1802. https://doi.org/10.3390/cells12131802
APA StyleRavera, S., Signorello, M. G., & Panfoli, I. (2023). Platelet Metabolic Flexibility: A Matter of Substrate and Location. Cells, 12(13), 1802. https://doi.org/10.3390/cells12131802