
3D-Printed Microfluidic Perfusion System for Parallel Monitoring of Hydrogel-Embedded Cell Cultures

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. 3D Printing and Post-Processing of Cultivation Device and Perfusion System Parts
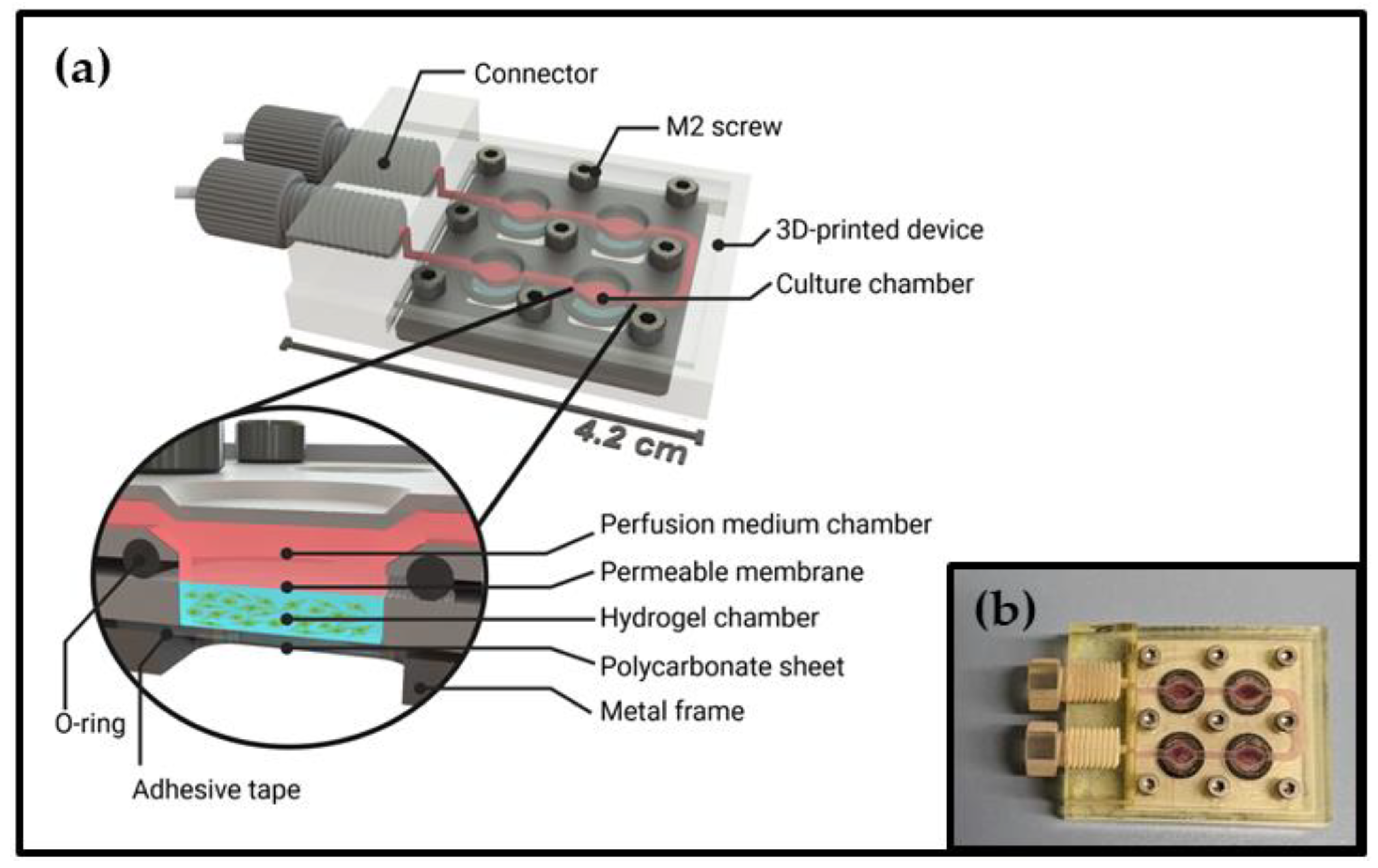
2.2. Assembly of the Cell Cultivation Device
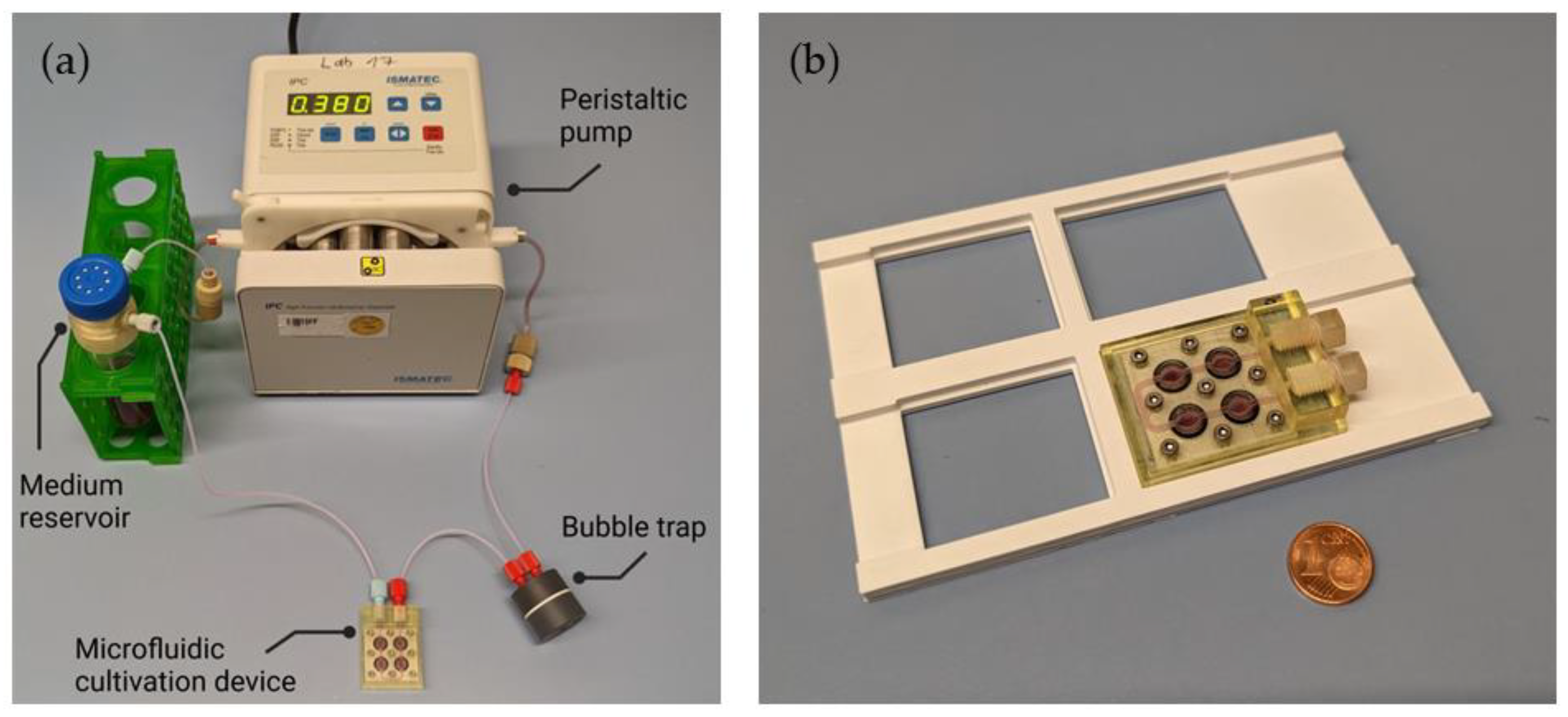
2.3. Assembly of the Perfusion System
2.4. Cell Line and Cell Culture Conditions
2.5. Hydrogel Preperation
2.6. Microscopic Analysis and Live/Dead Staining
2.7. Perfusion Experiments
3. Results
3.1. Design of the Microfluidic Cell Cultivation Device and Perfusion System
3.2. Cultivation and Imaging of Hydrogel-Embedded Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Castiaux, A.D.; Spence, D.M.; Martin, R.S. Review of 3D cell culture with analysis in microfluidic systems. Anal. Methods 2019, 11, 4220–4232. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.Y.C.; Hung, P.J.; Lee, P.J. Microfluidic array for three-dimensional perfusion culture of human mammary epithelial cells. Biomed. Microdevices 2011, 13, 753–758. [Google Scholar] [CrossRef]
- Rosser, J.; Olmos Calvo, I.; Schlager, M.; Purtscher, M.; Jenner, F.; Ertl, P. Recent Advances of Biologically Inspired 3D Microfluidic Hydrogel Cell Culture Systems. Cell Biol. Cell Metab. 2015, 2, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Terrell, J.A.; Jones, C.G.; Kabandana, G.K.M.; Chen, C. From cells-on-a-chip to organs-on-a-chip: Scaffolding materials for 3D cell culture in microfluidics. J. Mater. Chem. B 2020, 8, 6667–6685. [Google Scholar] [CrossRef]
- Azizipour, N.; Avazpour, R.; Rosenzweig, D.H.; Sawan, M.; Ajji, A. Evolution of biochip technology: A review from lab-on-a-chip to organ-on-a-chip. Micromachines 2020, 11, 599. [Google Scholar] [CrossRef]
- Ruedinger, F.; Lavrentieva, A.; Blume, C.; Pepelanova, I.; Scheper, T. Hydrogels for 3D mammalian cell culture: A starting guide for laboratory practice. Appl. Microbiol. Biotechnol. 2015, 99, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Saydé, T.; El Hamoui, O.; Alies, B.; Gaudin, K.; Lespes, G.; Battu, S. Biomaterials for three-dimensional cell culture: From applications in oncology to nanotechnology. Nanomaterials 2021, 11, 481. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Dahlmann, J.; Krause, A.; Möller, L.; Kensah, G.; Möwes, M.; Diekmann, A.; Martin, U.; Kirschning, A.; Gruh, I.; Dräger, G. Fully defined in situ cross-linkable alginate and hyaluronic acid hydrogels for myocardial tissue engineering. Biomaterials 2013, 34, 940–951. [Google Scholar] [CrossRef]
- Jacob, S.; Nair, A.B.; Shah, J.; Sreeharsha, N.; Gupta, S.; Shinu, P. Emerging Role of Hydrogels in Drug Delivery Systems, Tissue Engineering and Wound Management. Pharmaceutics 2021, 13, 357. [Google Scholar] [CrossRef] [PubMed]
- Correa, S.; Grosskopf, A.K.; Lopez Hernandez, H.; Chan, D.; Yu, A.C.; Stapleton, L.M.; Appel, E.A. Translational Applications of Hydrogels. Chem. Rev. 2021, 121, 11385–11457. [Google Scholar] [CrossRef]
- Young, E.W.K.; Beebe, D.J. Fundamentals of microfluidic cell culture in controlled microenvironments. Chem. Soc. Rev. 2010, 39, 1036. [Google Scholar] [CrossRef] [PubMed]
- Lavrentieva, A.; Fleischhammer, T.; Enders, A.; Pirmahboub, H.; Bahnemann, J.; Pepelanova, I. Fabrication of Stiffness Gradients of GelMA Hydrogels Using a 3D Printed Micromixer. Macromol. Biosci. 2020, 20, 2000107. [Google Scholar] [CrossRef] [PubMed]
- Maschmeyer, I.; Kakava, S. Organ-on-a-Chip. In Advances in Biochemical Engineering/Biotechnology; Bahnemann, J., Grünberger, A., Eds.; Springer: Cham, Switzerland, 2022; Volume 179, pp. 311–342. [Google Scholar]
- Moradi, E.; Jalili-Firoozinezhad, S.; Solati-Hashjin, M. Microfluidic organ-on-a-chip models of human liver tissue. Acta Biomater. 2020, 116, 67–83. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, J.; Wang, X.; Feng, L.; Wu, J.; Zhu, X.; Wen, W.; Gong, X. Organ-on-a-chip: Recent breakthroughs and future prospects. Biomed. Eng. Online 2020, 19, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahnemann, J.; Enders, A.; Winkler, S. Microfluidic Systems and Organ (Human) on a Chip. In Basic Concepts on 3D Cell Culture: Learning Materials in Biosciences; Kasper, C., Egger, D., Lavrentieva, A., Eds.; Springer: Cham, Switzerland, 2021; pp. 175–200. [Google Scholar]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting Organ-Level Lung Functions on a Chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, C.; Xu, N.; Liu, Z.-F.; Pang, D.-W.; Zhang, Z.-L. A virus-induced kidney disease model based on organ-on-a-chip: Pathogenesis exploration of virus-related renal dysfunctions. Biomaterials 2019, 219, 119367. [Google Scholar] [CrossRef]
- Jahanshahi, M.; Hamdi, D.; Godau, B.; Samiei, E.; Sanchez-Lafuente, C.; Neale, K.; Hadisi, Z.; Dabiri, S.; Pagan, E.; Christie, B.; et al. An Engineered Infected Epidermis Model for In Vitro Study of the Skin’s Pro-Inflammatory Response. Micromachines 2020, 11, 227. [Google Scholar] [CrossRef] [Green Version]
- Heuer, C.; Preuß, J.; Habib, T.; Enders, A.; Bahnemann, J. 3D printing in biotechnology—An insight into miniaturized and microfluidic systems for applications from cell culture to bioanalytics. Eng. Life Sci. 2021, 22, 744–759. [Google Scholar] [CrossRef]
- Winkler, S.; Grünberger, A.; Bahnemann, J. Microfluidics in Biotechnology: Quo Vadis. In Microfluidics in Biotechnology: Advances in Biochemical Engineering/Biotechnology; Bahnemann, J., Grünberger, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2021; pp. 355–380. [Google Scholar]
- Habib, T.; Brämer, C.; Heuer, C.; Ebbecke, J.; Beutel, S.; Bahnemann, J. 3D-Printed microfluidic device for protein purification in batch chromatography. Lab Chip 2022, 22, 986–993. [Google Scholar] [CrossRef]
- Siller, I.G.; Enders, A.; Gellermann, P.; Winkler, S.; Lavrentieva, A.; Scheper, T.; Bahnemann, J. Characterization of a customized 3D-printed cell culture system using clear, translucent acrylate that enables optical online monitoring. Biomed. Mater. 2020, 15, 055007. [Google Scholar] [CrossRef] [PubMed]
- Winkler, S.; Meyer, K.V.; Heuer, C.; Kortmann, C.; Dehne, M.; Bahnemann, J. In vitro biocompatibility evaluation of a heat-resistant 3D printing material for use in customized cell culture devices. Eng. Life Sci. 2022, 22, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, M.J.; Nordin, G.P.; Woolley, A.T. Moving from millifluidic to truly microfluidic sub-100-μm cross-section 3D printed devices. Anal. Bioanal. Chem. 2017, 409, 4311–4319. [Google Scholar] [CrossRef] [PubMed]
- Keyence Corporation. Hochauflösender 3D-Drucker Modellreihe AGILISTA-3000, 2020, Brochure 2020. Available online: https://www.keyence.de/products/3d-printers/3d-printers/agilista-3100/models/agilista-3200w/ (accessed on 28 February 2023).
- Bernard, M.; Jubeli, E.; Pungente, M.D.; Yagoubi, N. Biocompatibility of polymer-based biomaterials and medical devices-regulations,: In vitro screening and risk-management. Biomater. Sci. 2018, 6, 2025–2053. [Google Scholar] [CrossRef]
- ISO_10993-12; Biological Evaluation of Medical Devices—Part 12: Sample Preparation and Reference Materials. International Organization for Standardization: Geneva, Switzerland, 2013.
- Schmitz, C.; Pepelanova, I.; Seliktar, D.; Potekhina, E.; Belousov, V.V.; Scheper, T.; Lavrentieva, A. Live reporting for hypoxia: Hypoxia sensor–modified mesenchymal stem cells as in vitro reporters. Biotechnol. Bioeng. 2020, 117, 3265–3276. [Google Scholar] [CrossRef]
- Beckwith, A.L.; Borenstein, J.T.; Velasquez-Garcia, L.F. Monolithic, 3D-Printed microfluidic platform for recapitulation of dynamic tumor microenvironments. J. Microelectromech. Syst. 2018, 27, 1009–1022. [Google Scholar] [CrossRef]
- Joseph, X.; Akhil, V.; Arathi, A.; Mohanan, P. Comprehensive Development in Organ-On-A-Chip Technology. J. Pharm. Sci. 2022, 111, 18–31. [Google Scholar] [CrossRef]
- Schneider, S.; Gruner, D.; Richter, A.; Loskill, P. Membrane integration into PDMS-free microfluidic platforms for organ-on-chip and analytical chemistry applications. Lab Chip 2021, 21, 1866–1885. [Google Scholar] [CrossRef]
- Leung, C.M.; de Haan, P.; Ronaldson-Bouchard, K.; Kim, G.-A.; Ko, J.; Rho, H.S.; Chen, Z.; Habibovic, P.; Jeon, N.L.; Takayama, S.; et al. A guide to the organ-on-a-chip. Nat. Rev. Methods Prim. 2022, 2, 33. [Google Scholar] [CrossRef]
- Siller, I.G.; Preuss, J.-A.; Urmann, K.; Hoffmann, M.R.; Scheper, T.; Bahnemann, J. 3D-Printed Flow Cells for Aptamer-Based Impedimetric Detection of E. coli Crooks Strain. Sensors 2020, 20, 4421. [Google Scholar] [CrossRef] [PubMed]
- Arshavsky-Graham, S.; Enders, A.; Ackerman, S.; Bahnemann, J.; Segal, E. 3D-printed microfluidics integrated with optical nanostructured porous aptasensors for protein detection. Microchim. Acta 2021, 188, 67. [Google Scholar] [CrossRef] [PubMed]
- Winkler, S.; Menke, J.; Meyer, K.V.; Kortmann, C.; Bahnemann, J. Automation of cell culture assays using a 3D-printed servomotor-controlled microfluidic valve system. Lab Chip 2022, 22, 4656–4665. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyer, K.V.; Winkler, S.; Lienig, P.; Dräger, G.; Bahnemann, J. 3D-Printed Microfluidic Perfusion System for Parallel Monitoring of Hydrogel-Embedded Cell Cultures. Cells 2023, 12, 1816. https://doi.org/10.3390/cells12141816
Meyer KV, Winkler S, Lienig P, Dräger G, Bahnemann J. 3D-Printed Microfluidic Perfusion System for Parallel Monitoring of Hydrogel-Embedded Cell Cultures. Cells. 2023; 12(14):1816. https://doi.org/10.3390/cells12141816
Chicago/Turabian StyleMeyer, Katharina V., Steffen Winkler, Pascal Lienig, Gerald Dräger, and Janina Bahnemann. 2023. "3D-Printed Microfluidic Perfusion System for Parallel Monitoring of Hydrogel-Embedded Cell Cultures" Cells 12, no. 14: 1816. https://doi.org/10.3390/cells12141816
APA StyleMeyer, K. V., Winkler, S., Lienig, P., Dräger, G., & Bahnemann, J. (2023). 3D-Printed Microfluidic Perfusion System for Parallel Monitoring of Hydrogel-Embedded Cell Cultures. Cells, 12(14), 1816. https://doi.org/10.3390/cells12141816