

Epigenetic Modifications of MiRNAs in Osteoarthritis: A Systematic Review on Their Methylation Levels and Effects on Chondrocytes, Extracellular Matrix and Joint Inflammation

 , , ,
, , , 
Abstract
:
1. Introduction
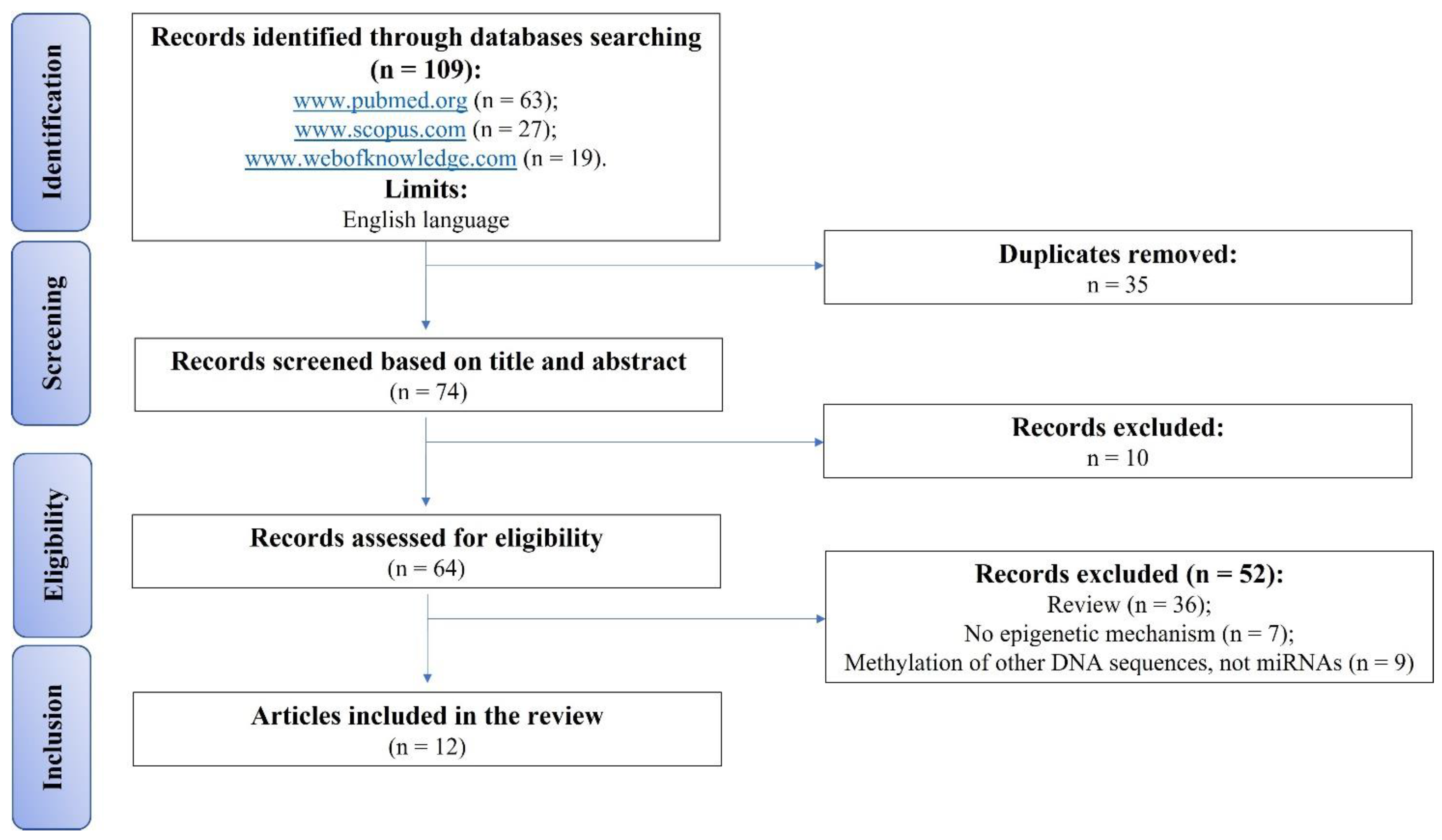
2. Materials and Methods
2.1. Eligibility Criteria
2.2. Search Strategy
2.3. Risk of Bias Assessments within Individual Studies
3. Results
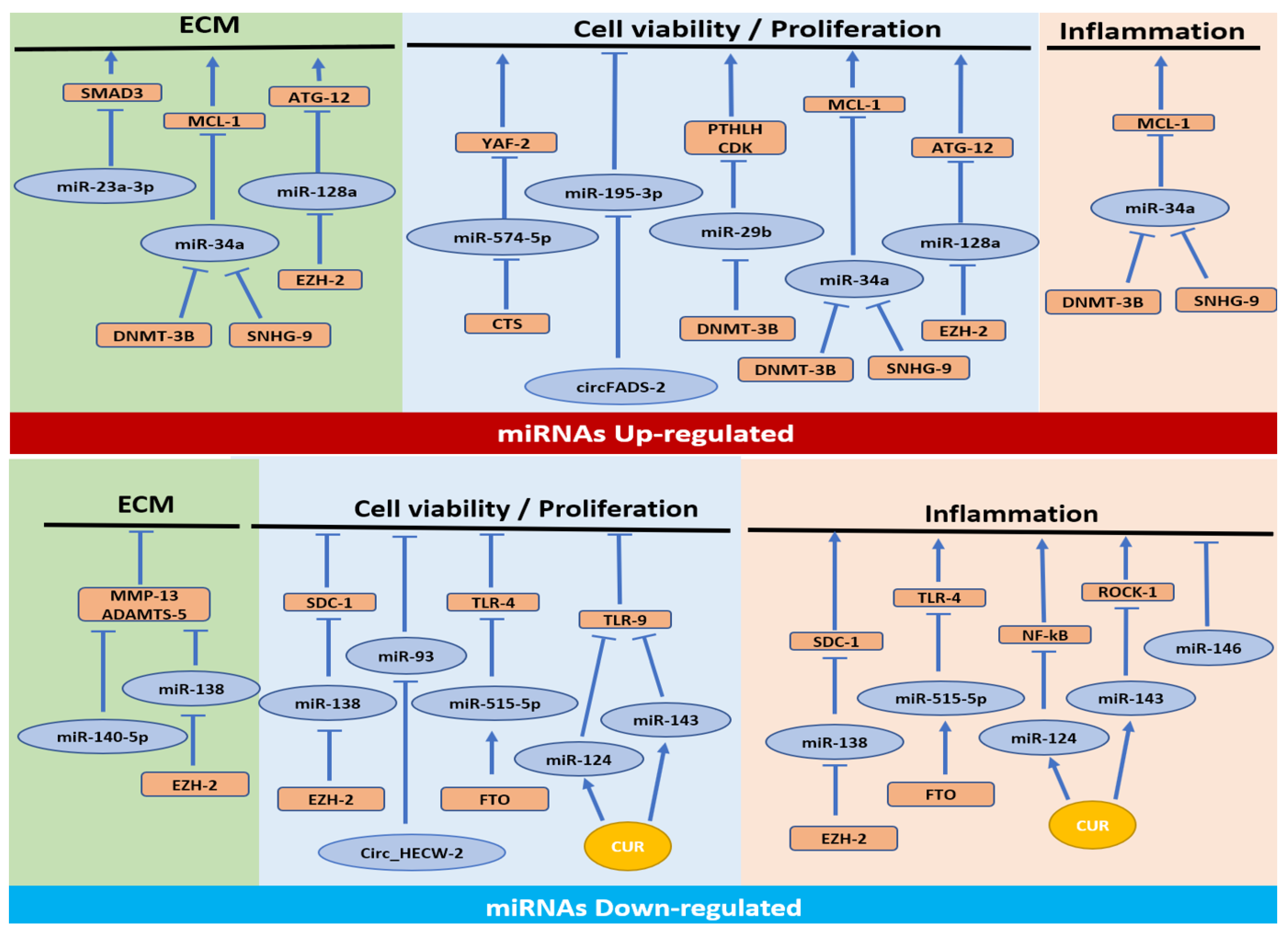
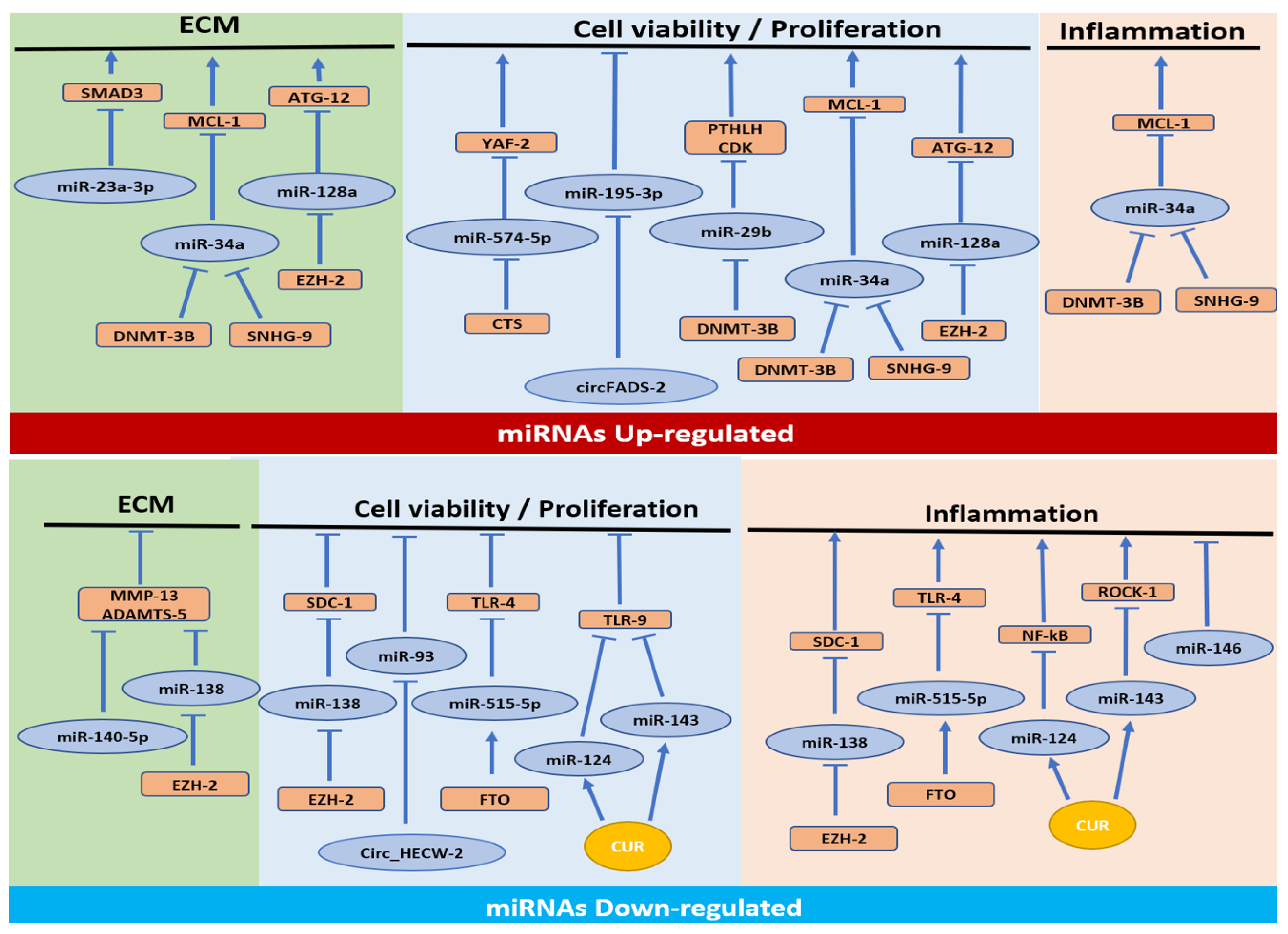
3.1. MiRNAs Upregulated in OA
3.1.1. Cell Proliferation/Apoptosis Pathway
3.1.2. Cell Proliferation/Apoptosis, ECM Synthesis and Inflammatory Pathways
3.1.3. Cell Proliferation/Apoptosis and ECM Synthesis Pathways
3.1.4. ECM Synthesis Pathway
3.2. MiRNAs Downregulated in OA
3.2.1. Cell Proliferation/Apoptosis and Inflammation Pathways
3.2.2. Inflammation Pathway
3.2.3. Cell Proliferation/Apoptosis Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, M.; Shen, J.; Jin, H.; Im, H.J.; Sandy, J.; Chen, D. Recent progress in understanding molecular mechanisms of cartilage degeneration during osteoarthritis. Ann. N. Y. Acad. Sci. 2011, 1240, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, L.; Yang, C.; Song, Y.; Liu, W.; Wang, K.; Li, S.; Zhang, Y. MicroRNA-23a-3p promotes the development of osteoarthritis by directly targeting SMAD3 in chondrocytes. Biochem. Biophys. Res. Commun. 2016, 478, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Safiri, S.; Kolahi, A.-A.; Smith, E.; Hill, C.; Bettampadi, D.; Mansournia, M.A.; Hoy, D.; Ashrafi-Asgarabad, A.; Sepidarkish, M.; Almasi-Hashiani, A.; et al. Global, regional and national burden of osteoarthritis 1990–2017: A systematic analysis of the Global Burden of Disease Study 2017. Ann. Rheum. Dis. 2020, 79, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Grassel, S.; Zaucke, F.; Madry, H. Osteoarthritis: Novel molecular mechanisms increase our understanding of the disease pathology. J. Clin. Med. 2021, 10, 1938. [Google Scholar] [CrossRef]
- Maiese, K. Picking a bone with WISP1 (CCN4): New strategies against degenerative joint disease. J. Transl. Sci. 2016, 1, 83–85. [Google Scholar] [CrossRef] [Green Version]
- Wallis, J.A.; Barton, C.J.; Brusco, N.K.; Kemp, J.L.; Sherwood, J.; Young, K.; Jennings, S.; Trivett, A.; Ackerman, I.N. Exploring views of orthopaedic surgeons, rheumatologists and general practitioners about osteoarthritis management. Musculoskelet. Care 2021, 19, 524–532. [Google Scholar] [CrossRef]
- Carballo, C.B.; Nakagawa, Y.; Sekiya, I.; Rodeo, S.A. Basic Science of Articular Cartilage. Clin. Sports Med. 2017, 36, 413–425. [Google Scholar] [CrossRef]
- Sudirman, S.; Chang, H.W.; Chen, C.K.; Kong, Z.L. A dietary polysaccharide from Eucheuma cottonii downregulates proinflammatory cytokines and ameliorates osteoarthritis-associated cartilage degradation in obese rats. Food Funct. 2019, 10, 5697–5706. [Google Scholar] [CrossRef]
- Conaghan, P.G.; Cook, A.D.; Hamilton, J.A.; Tak, P.P. Therapeutic options for targeting inflammatory osteoarthritis pain. Nat. Rev. Rheumatol. 2019, 15, 355–363. [Google Scholar] [CrossRef]
- Watkins, L.R.; Chavez, R.A.; Landry, R.; Fry, M.; Green-Fulgham, S.M.; Coulson, J.D.; Collins, S.D.; Glover, D.K.; Rieger, J.; Forsayeth, J.R. Targeted interleukin-10 plasmid DNA therapy in the treatment of osteoarthritis: Toxicology and pain efficacy assessments. Brain Behav. Immun. 2020, 90, 155–166. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, X.; Xing, L.; Tian, F. Wnt signaling: A promising target for osteoarthritis therapy. Cell Commun. Signal. 2019, 17, 97. [Google Scholar] [CrossRef] [Green Version]
- Oliviero, A.; Della Porta, G.; Peretti, G.; Maffulli, N. MicroRNA in osteoarthritis: Physiopathology, diagnosis and therapeutic challenge. Br. Med. Bull. 2019, 130, 137–147. [Google Scholar] [CrossRef]
- Qatrun Nada, D.; Masniza, M.L.; Abdullah, N.; Marlini, M.; Elias, M.H.; Pathmanathan, S.G.; Hayati, A.R.; Fadlul Azim, F.; Hamid, A.A.; Nur Fariha, M.M. Distinct microRNA expression pattern in breast cancer cells following anti-neoplastic treatment: A systematic review and functional analysis of microRNA target genes. Malays. J. Pathol. 2022, 44, 367–385. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayed, D.; Abdellatif, M.; Oikawa, S.; Lee, M.; Motohashi, N.; Maeda, S.; Akimoto, T.; Syed, M.; Ball, J.P.; Mathis, K.W.; et al. MicroRNAs in Development and Disease. Physiol. Rev. 2011, 91, 827–887. [Google Scholar] [CrossRef] [PubMed]
- Fathollahi, A.; Aslani, S.; Jamshidi, A.; Mahmoudi, M. Epigenetics in osteoarthritis: Novel spotlight. J. Cell. Physiol. 2019, 234, 12309–12324. [Google Scholar] [CrossRef] [PubMed]
- Swingler, T.E.; Niu, L.; Smith, P.; Paddy, P.; Le, L.; Barter, M.J.; Young, D.; Clark, I.M. The function of microRNAs in cartilage and osteoarthritis. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 120), 40–47. [Google Scholar] [PubMed]
- Díaz-Prado, S.; Cicione, C.; Muiños-López, E.; Hermida-Gómez, T.; Oreiro, N.; Fernández-López, C.; Blanco, F.J. Characterization of microRNA expression profiles in normal and osteoarthritic human chondrocytes. BMC Musculoskelet. Disord. 2012, 13, 144. [Google Scholar] [CrossRef] [Green Version]
- Trachana, V.; Ntoumou, E.; Anastasopoulou, L.; Tsezou, A. Studying microRNAs in osteoarthritis: Critical overview of different analytical approaches. Mech. Ageing Dev. 2018, 171, 15–23. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Króliczewski, J.; Sobolewska, A.; Lejnowski, D.; Collawn, J.F.; Bartoszewski, R. microRNA single polynucleotide polymorphism influences on microRNA biogenesis and mRNA target specificity. Gene 2018, 640, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef]
- Simon, T.C.; Jeffries, M.A. The epigenomic landscape in osteoarthritis. Curr. Rheumatol. Rep. 2017, 19, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; de Andrés, M.C.; Hashimoto, K.; Itoi, E.; Otero, M.; Goldring, M.B.; Oreffo, R.O.C. DNA methylation of the RUNX2 P1 promoter mediates MMP13 transcription in chondrocytes. Sci. Rep. 2017, 7, 7771. [Google Scholar] [CrossRef] [PubMed]
- van Wijnen, A.J.; Westendorf, J.J. Epigenetics as a New Frontier in Orthopedic Regenerative Medicine and Oncology. J. Orthop. Res. 2019, 37, 1465–1474. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, M.; O’Keefe, R.J.; Shen, J.; Li, Z.; Zhou, J.; Zhou, X.; Mao, J.J. Epigenetic and therapeutic implications of dnmt3b in temporomandibular joint osteoarthritis. Am. J. Transl. Res. 2019, 11, 1736–1747. [Google Scholar]
- Yue, S.; Su, X.; Teng, J.; Wang, J.; Guo, M. Cryptotanshinone interferes with chondrocyte apoptosis in osteoarthritis by inhibiting the expression of miR-574-5p. Mol. Med. Rep. 2021, 23, 424. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Shao, W.; Shen, N. LncRNA SNHG9 is downregulated in osteoarthritis and inhibits chondrocyte apoptosis by downregulating miR-34a through methylation. BMC Musculoskelet. Disord. 2020, 21, 511. [Google Scholar] [CrossRef]
- Zhang, H.; Ge, J.; Lu, X. CircFADS2 is downregulated in osteoarthritis and suppresses LPS-induced apoptosis of chondrocytes by regulating miR-195-5p methylation. Arch. Gerontol. Geriatr. 2021, 96, 104477. [Google Scholar] [CrossRef]
- Dou, P.; He, Y.; Yu, B.; Duan, J. Downregulation of microRNA-29b by DNMT3B decelerates chondrocyte apoptosis and the progression of osteoarthritis via PTHLH/CDK4/RUNX2 axis. Aging 2020, 13, 7676–7690. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Zhao, Y.; Xu, T. DNA methyltransferase 3 beta mediates the methylation of the microRNA-34a promoter and enhances chondrocyte viability in osteoarthritis. Bioengineered 2021, 12, 11138–11155. [Google Scholar] [CrossRef] [PubMed]
- Lian, W.S.; Ko, J.Y.; Wu, R.W.; Sun, Y.C.; Chen, Y.S.; Wu, S.L.; Weng, L.H.; Jahr, H.; Wang, F.S. MicroRNA-128a represses chondrocyte autophagy and exacerbates knee osteoarthritis by disrupting Atg12. Cell Death Dis. 2018, 9, 919. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, X.; Ding, X.; Huang, T.; Song, D.; Tao, H. EZH2 is associated with cartilage degeneration in osteoarthritis by promoting SDC1 expression via histone methylation of the microRNA-138 promoter. Lab. Investig. 2021, 101, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Zhang, J.; Yang, J.; Lv, Q.; Zhong, C. Overexpression of FTO alleviates osteoarthritis by regulating the processing of miR-515-5p and the TLR4/MyD88/NF-κB axis. Int. Immunopharmacol. 2023, 114, 109524. [Google Scholar] [CrossRef]
- Qiu, B.; Xu, X.; Yi, P.; Hao, Y. Curcumin reinforces MSC-derived exosomes in attenuating osteoarthritis via modulating the miR-124/NF-kB and miR-143/ROCK1/TLR9 signalling pathways. J. Cell Mol. Med. 2020, 24, 10855–10865. [Google Scholar] [CrossRef]
- Papathanasiou, I.; Trachana, V.; Mourmoura, E.; Tsezou, A. DNA methylation regulates miR-140-5p and miR-146a expression in osteoarthritis. Life Sci. 2019, 228, 274–284. [Google Scholar] [CrossRef]
- Zuo, J.; Chen, C.; Zhang, X.; Wu, J.; Li, C.; Huang, S.; He, P.; Wa, Q.; Zhang, W. Circ_HECW2 regulates LPS-induced apoptosis of chondrocytes via miR-93 methylation. Immunity Inflamm. Dis. 2021, 9, 943–949. [Google Scholar] [CrossRef]
- Nugent, M. MicroRNAs: Exploring new horizons in osteoarthritis. Osteoarthr. Cartil. 2016, 24, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.L.; Esa, M.S.; Li, K.H.C.; Krishnan, S.R.G.; Elgallab, G.M.; Pearce, M.S.; Young, D.A.; Birrell, F.N. Osteoporosis, fracture, osteoarthritis & sarcopenia: A systematic review of circulating microRNA association. Bone 2021, 152, 116068. [Google Scholar]
- Yan, Z.; Xiong, J.; Zhao, C.; Qin, C.; He, C. Decreasing cartilage damage in a rat model of osteoarthritis by intra-articular injection of deoxycholic acid. Int. J. Clin. Exp. Med. 2015, 8, 9038–9045. [Google Scholar] [PubMed]
- Wu, C.-J.; Liu, R.-X.; Huan, S.-W.; Tang, W.; Zeng, Y.-K.; Zhang, J.-C.; Yang, J.; Li, Z.-Y.; Zhou, Y.; Zha, Z.-G.; et al. Senescent skeletal cells cross-talk with synovial cells plays a key role in the pathogenesis of osteoarthritis. Arthritis Res. Ther. 2022, 24, 59. [Google Scholar] [CrossRef] [PubMed]
- Di Nicola, V. Degenerative osteoarthritis a reversible chronic disease. Regen. Ther. 2020, 15, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Kim, H.A. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 2001, 153, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Thielen, N.; Neefjes, M.; Wiegertjes, R.; van den Akker, G.; Vitters, E.; van Beuningen, H.; Davidson, E.B.; Koenders, M.; van Lent, P.; van de Loo, F.; et al. Osteoarthritis-Related Inflammation Blocks TGF-β’s Protective Effect on Chondrocyte Hypertrophy via (de)Phosphorylation of the SMAD2/3 Linker Region. Int. J. Mol. Sci. 2021, 22, 8124. [Google Scholar] [CrossRef]
- Valdes, A.M.; Spector, T.D.; Tamm, A.; Kisand, K.; Doherty, S.A.; Dennison, E.M.; Mangino, M.; Tamm, A.; Kerna, I.; Hart, D.J.; et al. Genetic variation in the SMAD3 gene is associated with hip and knee osteoarthritis. Arthritis Rheum. 2010, 62, 2347–2352. [Google Scholar] [CrossRef]
- Sun, K.; Luo, J.; Guo, J.; Yao, X.; Jing, X.; Guo, F. The PI3K/AKT/mTOR signaling pathway in osteoarthritis: A narrative review. Osteoarthr. Cartil. 2020, 28, 400–409. [Google Scholar] [CrossRef]
- Li, Z.; Dai, A.; Yang, M.; Chen, S.; Deng, Z.; Li, L. p38MAPK Signaling Pathway in Osteoarthritis: Pathological and Therapeutic Aspects. J. Inflamm. Res. 2022, 15, 723–734. [Google Scholar] [CrossRef]
- Rajagopal, K.; Ramesh, S.; Madhuri, V. Early Addition of Parathyroid Hormone–Related Peptide Regulates the Hypertrophic Differentiation of Mesenchymal Stem Cells. Cartilage 2021, 13, 143S–152S. [Google Scholar] [CrossRef] [PubMed]
- Gambari, L.; Cellamare, A.; Grassi, F.; Grigolo, B.; Panciera, A.; Ruffilli, A.; Faldini, C.; Desando, G. Overview of Anti-Inflammatory and Anti-Nociceptive Effects of Polyphenols to Halt Osteoarthritis: From Preclinical Studies to New Clinical Insights. Int. J. Mol. Sci. 2022, 23, 15861. [Google Scholar] [CrossRef] [PubMed]
- Tew, S.R.; Hardingham, T.E. Regulation of SOX9 mRNA in human articular chondrocytes involving p38 MAPK activation and mRNA stabilization. J. Biol. Chem. 2006, 281, 39471–39479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Z.; Jia, Y.; Liu, H.; He, M.; Yang, Y.; Xiao, W.; Li, Y. RhoA/ROCK pathway: Implication in osteoarthritis and therapeutic targets. Am. J. Transl. Res. 2019, 11, 5324–5331. [Google Scholar]
- Segarra-Queralt, M.; Piella, G.; Noailly, J. Network-based modelling of mechano-inflammatory, chondrocyte regulation in early osteoarthritis. Front. Bioeng. Biotechnol. 2023, 11, 1006066. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Aim | In Vitro Model | Analyses | Results | Conclusions in OA | Ref. |
|---|---|---|---|---|---|
| Evaluation of CTS on miR-574-5p methylation in chondrocyte apoptosis during OA | Chondrocytes from mice (4 days) treated with IL1β. (1) Cells+CTS; (2) Cells+CTS+5-aza-CdR; (3) Cells+CTS and transfected with: miR-574-5p mimic; oe-YAF2 | RT-qPCR (miR-574-5p and Yaf2); WB (Bax, Bcl-2); Flow cytometry (cell apoptosis); CCK-8 assay (cell proliferation); Dual-luciferase reporter assay (relationship between miR-574-5p and Yaf2); MSP (miR-574-5p methylation) | Cells: ↑ miR-574-5p, Bax, apoptosis; ↓ Yaf2, Bcl-2, cell proliferation, ↓miR-574-5p promoter methylation. Cells+CTS: ↓ miR-574-5p, Bax, apoptosis; ↑ Yaf2, Bcl-2, cell proliferation, miR-574-5p promoter methylation. Cells+CTS+5-aza-CdR: ↓ miR-574-5p promoter methylation. Cells+CTS+miR-574-5p mimic: ↑ miR-574-5p, Bax, apoptosis; ↓ Yaf2, Bcl2, cell proliferation. Cells+CTS+miR-574-5p mimic +oe-YAF2: ↑ Yaf2, cell proliferation; ↓ Bax, cell apoptosis | - miR-574-5p upregulated. - Bax is upregulated, while Yaf2 and Bcl-2 are downregulated. - Negative correlation between miR-574-5p and Yaf2 and Bcl-2. - Positive correlation between miR-574-5p and Bax. - CTS reduces miR-574-5p and increases miR-574-5p promoter methylation. - CTS could affect chondrocyte proliferation and apoptosis by regulating the expression of miR-574-5p and then interfering with Yaf2. | Yue 2021 [28] |
| Evaluation of the interaction between SNHG9 and miR-34a methylation in chondrocyte apoptosis during OA | (1)SF from OA pz (n = 60). (2) Chondrocytes from OA (n = 60) or healthy donors (n = 60) and transfected with: oe-SNHG9; miR-34a mimic | RT-qPCR (SNHG9, miR-34a); ELISA (CASPASE-3); Flow cytometry (cell apoptosis); MSP (miR-34a methylation) | OA SF: ↓ SNHG9; ↑ miR-34a, CASPASE-3, cell apoptosis. OA cells+oe-SNHG9: ↓ miR-34a, apoptosis, CASPASE-3; ↑ miR-34a methylation. OA cells+miR-34a mimic: ↑ apoptosis, CASPASE-3. Healthy cells+oe-SNHG9: no changes in miR-34a, miR-34a methylation, apoptosis, CASPASE-3. Healthy cells+miR-34a mimic: no changes in SNHG9, apoptosis, CASPASE-3. | - miR-34a upregulated. - Negative correlation between SNHG9 and miR-34a. - Overexpression of SNHG9 decreases miR-34a through methylation. - Overexpression of SNHG9 decreases chondrocyte apoptosis through miR-34a | Zhang 2020 [29] |
| Evaluation of the interaction between circFADS2 and miR-195-5p methylation in chondrocyte apoptosis during OA | (1) SF from OA pz (n = 63); (2) Purchased human OA chondrocytes transfected with: oe-circFADS2; miR-195-5p mimic | RT-qPCR (circFADS2, miR-195-5p); Flow cytometry (cell apoptosis); MSP (miR-195-5p methylation). | OA SF: ↓ circFADS2; ↑ miR-195-5p. Cells+oe-circFADS2: ↓ miR-195-5p, apoptosis; ↑ miR-195-5p methylation. Cells+miR-195-5p mimic: ↑ apoptosis; no changes in circFADS2 expression | - miR-195-5p upregulated. - Negative correlation between CircFADS2 and miR-195-5p. - Overexpression of circFADS2 decreases miR-195-5p expression through methylation. - Overexpression of circFADS2 decrases chondrocytes apoptosis through miR-195-5p | Zhang 2021 [30] |
| Evaluation of the relationship between DNMT3B and miR-29b methylation in cell apoptosis, ECM synthesis and inflammation in OA | (1) Cartilage or chondrocytes from OA pz (n = 46) treated with IL1β and transfected with: sh-DNMT3B; sh-CDK4; oe-DNMT3B; oe-PTHLH; miR-29b mimic; miR-29b inhibitor | RT-qPCR (DNMT3B, miR-29b, PTHLH, CDK4, RUNX2); WB (DNMT3B, PTHLH, CDK4, Ki67, Aggrecan, MMP3, MMP13); MTT (cell viability); Flow cytometry (cell apoptosis); IHF (Ki67, Aggrecan); MSP (miR-29b promoter methylation); Dual-luciferase reporter gene assay (relationship between miR-29b and PTHLH). | OA cartilage: ↓ DNMT3B, PTHLH, CDK4, miR-29b promoter methylation; ↑ miR-29b. Cells: ↓ miR-29b methylation; ↑ miR-29b expression. Cells+oe-DNMT3B: ↑ miR-29b methylation, PTHLH, CDK4, ubiquitinated RUNX2, cell viability, Ki67, Aggrecan; ↓ miR-29b expression, apoptosis, MMP3, MMP13. Cells+miR-29b mimic: ↓ luciferase activity of PTHLH. Cells+miR-29b inhibitor: ↑ PTHLH. Cells+sh-DNMT3B: ↓ PTHLH. Cells+oe-PTHLH: ↑ CDK4, cell viability, Ki67 and Aggrecan; ↓ apoptosis, MMP3 and MMP13. Cells+oe-PTHLH+sh-CDK4: ↓ cell viability, Ki67, Aggrecan; ↑ apoptosis, MMP3 and MMP13 | - miR-29b upregulated. - Negative correlation between DNMT3B and miR-29b. - Overexpression of DNMT3B decreases miR-29b through methylation of the miR-29b promoter. - DNMT3B increases PTHLH expression by inhibiting miR-29b. - PTHLH impedes the apoptosis of OA chondrocytes by elevating CDK4 | Dou 2021 [31] |
| Evaluation of the role of miR-34a epigenetic mechanism in the degradation of ECM, cell viability and inflammatory response in OA | (1) Cartilage or chondrocytes from OA pz (n = 55) transfected or not with: miR-34a inhibitor; sh-DNMT3B; sh-MCL1. | RT-qPCR (miR-34a, DNMT3B, MCL1, MMP3, MMP13, COL2A1, iNOS, COX2, TNFα, IL6, PG); In situ hybridization (miR-34a expression in tissues); CCK-8 (cell viability); Flow cytometry (Cell apoptosis); WB (PI3K/AKT); MSP (miR-34a methylation); Dual-luciferase reporter gene assay (relationship between MCL1 and miR-34a). | OA cartilage and cells: ↑ miR-34a, MMP3, MMP13, iNOS, COX2, TNFα, IL6; ↓ COL2A1, MCL1, DNMT3B, miR-34a methylation. Cells+sh-DNMT3B: ↑ miR-34a; ↓ MCL1, miR-34a methylation. Cells+miR-34a inhibitor: ↓ MMP3, MMP13, apoptosis, iNOS, COX2, TNFα, IL6; ↑ cell viability, COL2A1, PG, PI3K/AKT pathway activity, MCL1 luciferase activity. Cells+sh-DNMT3B+sh-MCL1: ↓ cell viability, PG, PI3K/AKT pathway activity; ↑ apoptosis, iNOS, COX2, TNFα, IL6. | - miR-34a upregulated. - Positive correlation between miR-34a and MMP3, MMP13, iNOS, COX2, TNFα and IL6. - Negative correlation between miR-34a and COL2A1, MCL1 and DNMT3B. - DNMT3B suppresses miR-34 through an epigenetic mechanism. - miR-34a targets and inhibits MCL1 expression. - DNMT3B/miR-34a/MCL1 axis regulates chondrocyte viability. - DNMT3B/miR-34a/MCL1 axis regulates ECM degradation. - DNMT3B/miR-34a/MCL1 axis regulates the inflammatory response. - DNMT3B/miR-34a/MCL1 axis affects chondrocyte viability and OA progression via the PI3K/AKT pathway. | Xiong 2021 [32] |
| Evaluation of the miR-128a methylation effects on chondrocyte survival and articular cartilage anabolism during OA | (1) Cartilage from OA pz (n = 28). 2) 293T cells treated with IL1β transfected with: oe-miR-128; Sh-miR-128. (3) Chondrocytes from 7 days old rats and transfected with: oe-EZH2; EZH2 Rnai; (4) Chondrocytes from 7 days old rats, cultured in micromasses and transfected with: oe-miR-128a; sh-miR-128a | TUNEL staining and flow cytometry (Cell apoptosis); qRT-PCR (ATG4, ATG12, p62, BECLIN, COL2A1, AGGRECAN, SOX9, IL1β, CXCL9); WB (LC3, BAX, BCL2,CASPASE-3, EZH2, H3K27me1, H3K27me2, H3K27me3); Dual-Luciferase reporter assay (relationship between ATG12 and miR-128a); ChIP (EZH2 and H3K27me3 binding on miR-138 promoter). | OA cartilage and cells: ↑ miR-128a expression; ↓ ATG12 expression, LC3, H3K27. Cells+oe-miR-128: ↓ 3′-UTR luciferase activity of ATG12, ATG12 expression, LC3 concentration. Cells+sh-miR-128: ↑ 3′-UTR luciferase activity of ATG12, ATG12 expression, LC3 concentration. Cells+oe-EZH2: ↑ methylated H3K27 and H3K27me2 enrichment in the proximal region of the miR-128a promoter, ATG12 and LC3; ↓ miR-128a expression. Cells+EZH2 Rnai: ↓ methylated H3K27, H3K27me2 enrichment to the miR-128a promoter region, ATG12, LC3; ↑ miR-128a expression. Cell micromasses+oe-miR-128a: ↑ BAX, BCL2, and cleaved caspase-3, annexin-V, apoptosis; ↓ Alcian blue staining, SOX9, COL2A1, and AGGRECAN. Cell micromasses+sh-miR-128a: ↓ Bax, Bcl-2, cleaved caspase-3, apoptosis; ↑SOX9, COL2A1, and AGGRECAN. | - miR-128a upregulated. - Negative correlation between miR-128a and ATG12, EZH2 and H3K27. - Overexpression of EZH2 decreases miR-128a through methylation of histone H3K27. - miR-128a binds directly to the 3′-UTR of ATG12. - miR-128a reduces survival and cartilage formation capacity of chondrocytes | Lian 2018 [33] |
| Evaluation of the biological effects of miR-23a-3p methylation in ECM synthesis in OA | (1) Cartilage from OA pz (n = 10). (2) Purchased SW1353 cells transfected with: miR-23a-3p mimic; miR-23a-3p inhibitor; sh-SMAD3 | qRT-PCR (SMAD3, COL2A1, AGGRECAN, miR-23a-3p); WB (SMAD3, COL2A1, AGGRECAN); MSP (miR-23a-3p promoter methylation); Dual-luciferase reporter assay (relationship between SMAD3 and miR-23a-3p) | OA cartilage: ↑ miR-23a-3p; ↓ SMAD3, miR-23a-3p promoter methylation. Cells+miR-23a-3p mimics: ↓ luciferase activity of SMAD3,, COL2A1, AGGRECAN. Cells+miR-23a-3p inhibitor: ↑ SMAD3, COL2A1, AGGRECAN. Cells+sh-SMAD3: ↓ SMAD3. Cells+ miR-23a-3p inhibitor+sh-SMAD3: ↓ SMAD3, COL2A1, AGGRECAN. | - miR-23a-3p upregulated. - Negative correlation between miR-23a-3p and SMAD3. - Hypomethylation of the miR-23a-3p promoter explains the increased expression of miR-23a-3p. - SMAD3 is a target of miR-23a-3p. - miR-23a-3p overexpression suppresses ECM synthesis. - SMAD3 is essential in the miR-23a-3p-induced downregulation of AGGRECAN and COL2A1. | Kang 2016 [2] |
| Evaluation of the molecular mechanism of EZH2/miR-138/SDC1 and epigenetic regulation in cell apoptosis and inflammation in OA | (1) Cartilage and chondrocytes from OA pz (n = 25). (2) Chondrocytes from healthy donors treated with IL1β and transfected or not with: sh-EZH2; miR-138 mimic; oe-EZH2; oe-EZH2 | RT-qPCR (EZH2, miR-138, SDC1, MMP13, ADAMTS4, ADAMTS5); WB (EZH2, SDC1); Flow Cytometry (cell apoptosis); Dual-luciferase reporter assay (relationship between SDC1 and miR-138); ChIP (EZH2 and H3K27me3 binding on miR-138 promoter). | OA cartilage and chondrocytes: ↑ EZH2, SDC1; ↓ miR-138. Cells from healthy donors+IL1β: ↑ EZH2, SDC1, apoptosis, MMP13, ADAMTS4, ADAMTS5, miR-138 promoter histone methylation; ↓ miR-138. Cells+sh-EZH2: ↓ EZH2, apoptosis, MMP13, ADAMTS-4, ADAMTS-5, miR-138 promoter histone methylation; ↑ miR-138. Cells+oe-EZH2: ↑ EZH2, SDC1, apoptosis, MMP13, ADAMTS4, ADAMTS5, miR-138 promoter histone methylation; ↓ miR-138. Cells+miR-138 mimic: ↓ SDC1, SDC1 luciferase activity. Cells+oe-EZH2+mir-138 mimic: ↑ miR-138; ↓ SDC1, apoptosis, MMP13, ADAMTS4, ADAMTS5. | - miR-138 deregulated. - EZH2 is upregulated and promotes OA progression. - Negative correlation between EZH2 and SDC1 with miR-138. - EZH2 inhibits miR-138 expression by increasing histone methylation at its promoter. - SDC1 is a target of miR-138. - EZH2 induces cartilage catabolism-related factors by regulating miR-138/SDC1 signalling | Wang 2021 [34] |
| Evaluation of the relationship between FTO and miR-515-5p in m6A-dependent manner on cell apoptosis and inflammation in OA | (1) Purchased human OA C28/I2 chondrocytes treated with LPS transfected or not with: oe-FTO; sh-FTO; miR-515-5p inhibitor; oe-TLR4 | CCK-8 (cell viability); Flow cytometry (cell apoptosis); WB (BCL-2, BAX, CLEAVED-CASPASE-3, FTO, TLR4, MYD88, P/T-P65, P/T-IΚBα); ELISA (IL-6, IL-1β, and TNF-α); m6A RNA methylation level; RT-qPCR (COX-2, iNOS, FTO, miR-515-5p and pri-miR-515-5p); MeRIP (m6A RNA immunoprecipitation); Dual-luciferase reporter assay (relationship between pri-miR-515-5p and DGCR8). | Cells: ↓ cell viability, BCL-2, miR-515-5p expression, FTO; ↑ apoptosis, BAX, CLEAVED-CASPASE-3, COX-2 and iNOS expression, IL-6, IL-1β, and TNF-α, COX-2, pri-miR-515-5p m6A methylation level, TLR4, MyD88, p/t-p65, and p/t-IκBα. Cells+oe-FTO: ↓ cell apotosis, BAX, CLEAVED-CASPASE-3, IL-6, IL-1β, and TNF-α, COX-2, iNOS, pri-miR-515-5p m6A methylation level, TLR4, MYD88, P/T-P65, AND P/T-IΚBα; ↑ cell viability, BCL-2, miR-515-5p expression, pri-miR-515-5p binding level to DGCR8. Cells+sh-FTO: ↓ FTO expression, miR-515-5p expression; ↑ pri-miR-515-5p m6A methylation level. Cells+oe-FTO+miR-515-5p inhibitor: ↓ miR-515-5p, cell viability, Bcl-2; ↑ apoptosis, Bax, cleaved-caspase-3, IL-6, IL-1β, TNF-α, COX-2, iNOS, TLR4. Cells+oe-FTO+oe-TLR4: ↑ MyD88, p/t-p65, and p/t-IκBα | - miR-515-5p deregulated. - FTO reduced. - FTO interacts with DGCR8 and modulates the pri-miR-515-5p processing in an m6A dependent manner. - FTO alleviates OA injury by regulating miR-515-5p. - miR-515-5p inhibits the MyD88/NF-κB pathway by targeting TLR4. - FTO might inhibit TLR4 levels by targeting miR-515-5p. | Cai 2023 [35] |
| Evaluation of the miR-143 methylation underlying the role of CUR in OA treatment | (1) Purchased primary chondrocytes treated with IL1β with or without: BMSC-Exos; BMSC-Exos+CUR. (2) Primary chondrocytes+CUR. (3)Primary chondrocytes transfected with: miR-143 mimics; miR-124 mimics | qRT-PCR (miR-124, miR-143, Rock1, NF-kB); CCK-8 (cell proliferation); Annexin V assay (Cell apoptosis); WB (Rock1, Tlr9, Nf-kB); Bisulphite sequencing; Dual-luciferase reporter assay (relationship between NF-kB and miR-124 or ROCK1 and miR-143) | Cells: ↓ cell viability, miR-124, miR-143; ↑ cell apoptosis, Rock1, Tlr9, Nf-kB. Cells+BMSC-Exos and cells+BMSC-Exos+CUR: ↑ cell viability, miR-124, miR-143; ↓ cell apoptosis, Rock1, Tlr9, Nf-kB. Cells+CUR: ↓ methylation of miR-143 and miR-124 promoters; ↑ miR-143 and miR-124. Cells+miR-143 mimics: ↓ 3′-UTR luciferase activity of Nf-kB. Cells+miR-124 mimics: ↓ 3′-UTR luciferase activity of Rock1. | - miR-124 and miR-143 deregulated. - Exosomes derived from CUR treated MSCs maintain the viability of chondrocytes and protects chondrocytes against IL-1β-induced apoptosis. - Exosomes derived from CUR treated MSCs restore the expression of miRNAs and genes related to OA. - CUR up-regulates miR-143 and miR-124 expression by reducing the DNA methylation of their promoters. - miR-143 and miR-124 act by inhibiting Rock1 and Nf-kB gene target. | Qiu 2020 [36] |
| Evaluation of the role of miR-140-5p and miR-146a methylation in OA | (1)Chondrocytes and synoviocytes from OA pz (n = 20) treated or not with 5-AzadC and trasfected or not with: sh-SMAD3; miR-140-5p inhibitor; sh-NF-kB; miR-146a inhibitor. | RT-qPCR (miR-140, miR-146a, MMP13, ADAMTS5, TRAF-6, IRAK-1, IL6, IL1β, TNFA); MSP (specific regions of miR-140 and miR-146a promoter methylation); Bisulfite sequencing (miR-146a promoter methylation); ChIP (NF-kB or SMAD3 binding on miR-146a promoter). | Chondrocytes: ↓ miR-140-5p binding affinity of SMAD3; ↑ miR-140 regulatory region methylation. Synoviocytes: ↓ miR-146a, NF-kB binding on miR-146a promoter; ↑ miR-146a promoter methylation. Chondrocytes+5-AzadC: ↓ miR-140 regulatory region methylation, MMP13, ADAMTS5; ↑ miR-140-5p. Synoviocytes+5-AzadC: ↓ miR-146a promoter methylation, IRAK-1, IL1β, IL6; ↑ miR-146a. Chondrocytes+5-AzadC+sh-SMAD3: ↓ miR-140-5p. Chondrocytes+5-AzadC+miR-140-5p inhibitor: ↑ MMP13, ADAMTS5. Synoviocytes+5-AzadC+sh-NF-kB: ↓ miR-146a. Synoviocytes+5-AzadC+miR-146a inhibitor: ↑ IRAK-1, IL1β, IL6. Synoviocytes+miR-146a: ↓ IRAK-1, IL1β, IL6. | - miR-140-5p and miR-146a deregulated. - miR-140 regulatory region methylation in chondrocytes. - Negative correlation between miR-140-5p expression and miR-140 regulatory region methylation in chondrocytes. - miR-146a promoter methylation in synoviocytes. - Negative correlation between miR-146a expression and miR-146a promoter methylation in synoviocytes. - Methylation impairs SMAD3 binding affinity on miR-140 regulatory region. - MiR-146a promoter methylation impairs the binding affinity of NF-kB. - Methylation mediates downregulation of miR-140-5p on MMP-13 and ADAMTS5 expression levels. - Methylation mediates miR-146a on the expression of inflammatory factors involved in OA | Papathanasiou 2019 [37] |
| Evaluation of the role of Circ_HECW2 and miR-93 methylation in cell apoptosis in OA | (1) SF from OA pz (n = 64). (2) Purchased human OA chondrocytes transfected with: oe-Circ_HECW2; miR-93 mimic | RT-qPCR (Circ_HECW2, miR-93); Flow cytometry (cell apoptosis); MSP (miR-93 methylation) | OA SF and cells: ↑ Circ_HECW2; ↓ miR-93. Cells+oe-Circ_HECW2: ↓ miR-93; ↑ miR-93 methylation, apoptosis. Cells+miR-93 mimic: ↓ apoptosis; no changes in Circ_HECW2 | - miR-93 deregulated. - Circ_HECW2 upregulated. - Negative correlation between Circ_HECW2 and miR-93. - Circ_HECW2 overexpression decreases miR-93 expression through methylation. - Circ_HECW2 overexpression increases chondrocyte apoptosis via miR-93 | Zuo 2021 [38] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veronesi, F.; Costa, V.; Bellavia, D.; Basoli, V.; Giavaresi, G. Epigenetic Modifications of MiRNAs in Osteoarthritis: A Systematic Review on Their Methylation Levels and Effects on Chondrocytes, Extracellular Matrix and Joint Inflammation. Cells 2023, 12, 1821. https://doi.org/10.3390/cells12141821
Veronesi F, Costa V, Bellavia D, Basoli V, Giavaresi G. Epigenetic Modifications of MiRNAs in Osteoarthritis: A Systematic Review on Their Methylation Levels and Effects on Chondrocytes, Extracellular Matrix and Joint Inflammation. Cells. 2023; 12(14):1821. https://doi.org/10.3390/cells12141821
Chicago/Turabian StyleVeronesi, Francesca, Viviana Costa, Daniele Bellavia, Valentina Basoli, and Gianluca Giavaresi. 2023. "Epigenetic Modifications of MiRNAs in Osteoarthritis: A Systematic Review on Their Methylation Levels and Effects on Chondrocytes, Extracellular Matrix and Joint Inflammation" Cells 12, no. 14: 1821. https://doi.org/10.3390/cells12141821
APA StyleVeronesi, F., Costa, V., Bellavia, D., Basoli, V., & Giavaresi, G. (2023). Epigenetic Modifications of MiRNAs in Osteoarthritis: A Systematic Review on Their Methylation Levels and Effects on Chondrocytes, Extracellular Matrix and Joint Inflammation. Cells, 12(14), 1821. https://doi.org/10.3390/cells12141821