
Structural and Kinetic Characterization of the SpeG Spermidine/Spermine N-acetyltransferase from Methicillin-Resistant Staphylococcus aureus USA300
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. SpeG Expression and Purification for Protein Crystallization and Initial Substrate Screening Assay
2.2. Crystallization and Structure Solution
2.3. Protein Expression and Purification for SaSpeG Steady-State Kinetic Characterization
2.4. SaSpeG Kinetic Characterization
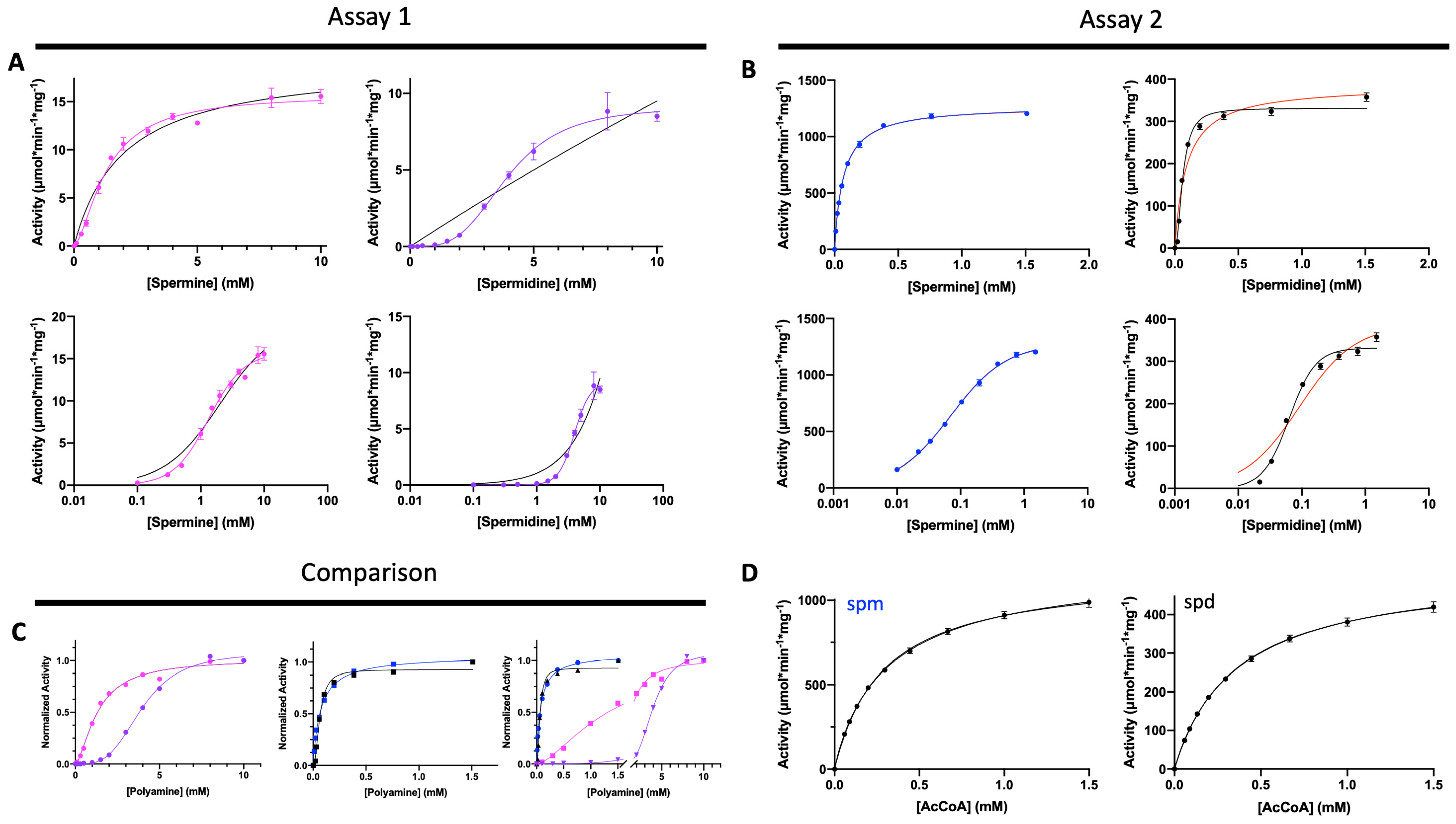
2.4.1. Assay 1: (Standard Assay) No Pre-Incubation of Enzyme with Polyamine
2.4.2. Assay 2: Pre-Incubation of Enzyme with Polyamine
2.4.3. Polyamine Substrate Saturation Curves via Assay 2
2.4.4. AcCoA Substrate Saturation Curves via Assay 2
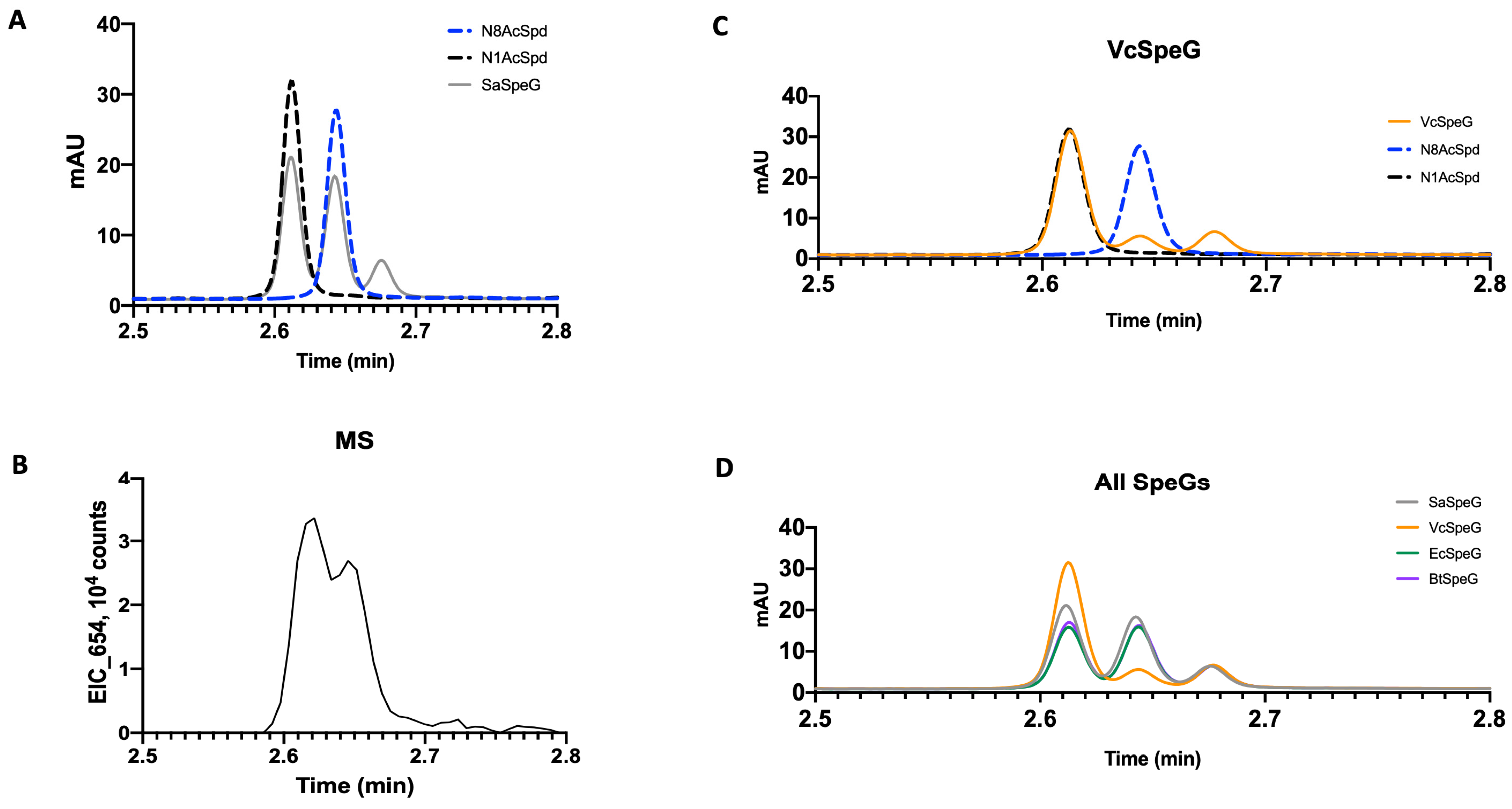
2.5. SpeG Spermidine Substrate Specificity LCMS Assays
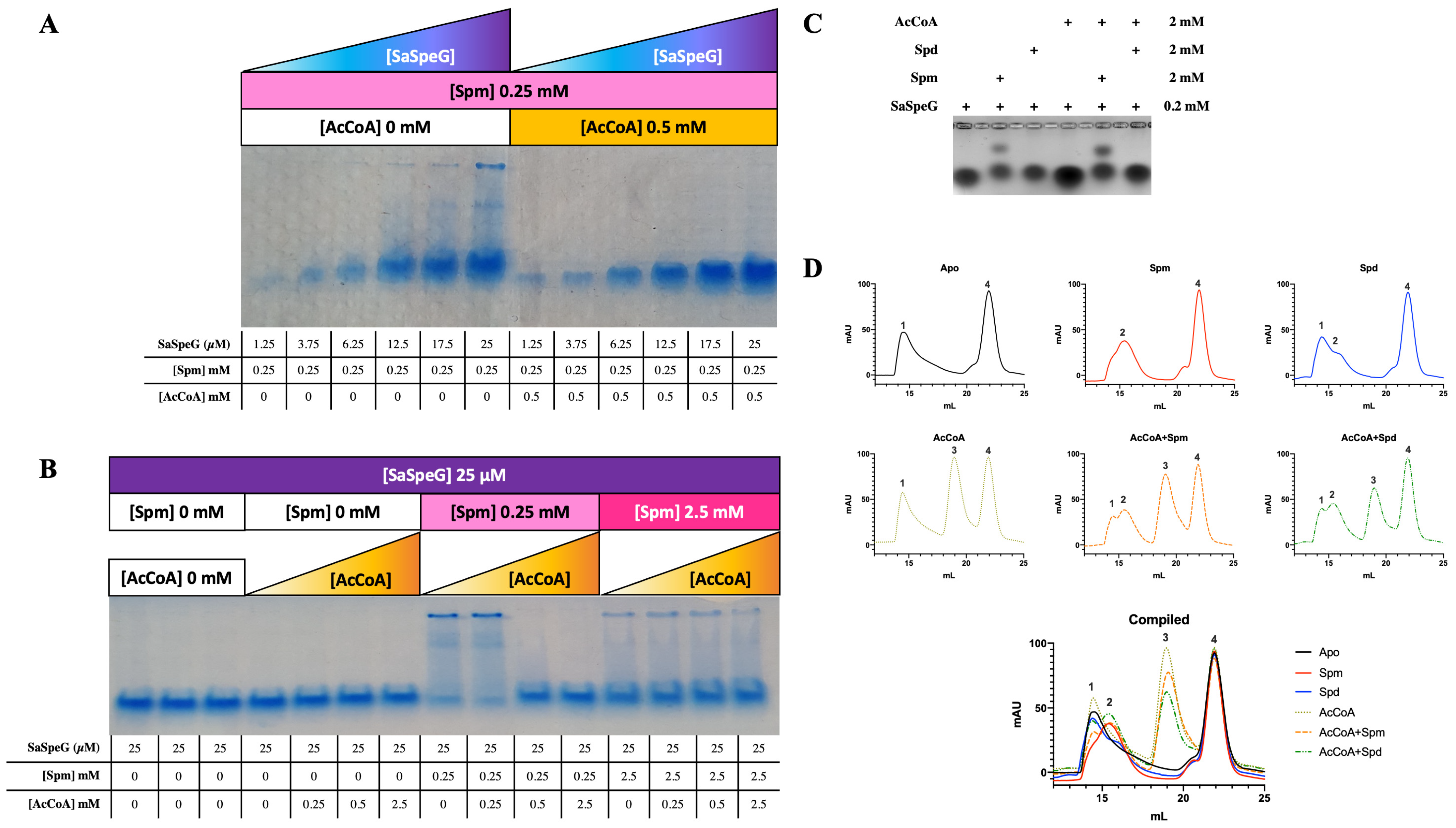
2.6. Native PAGE Assays
2.7. Electrophoretic Mobility Shift Assays (EMSAs)
2.8. Analytical Size-Exclusion Chromatography (SEC)
3. Results
3.1. SaSpeG Kinetic Characterization
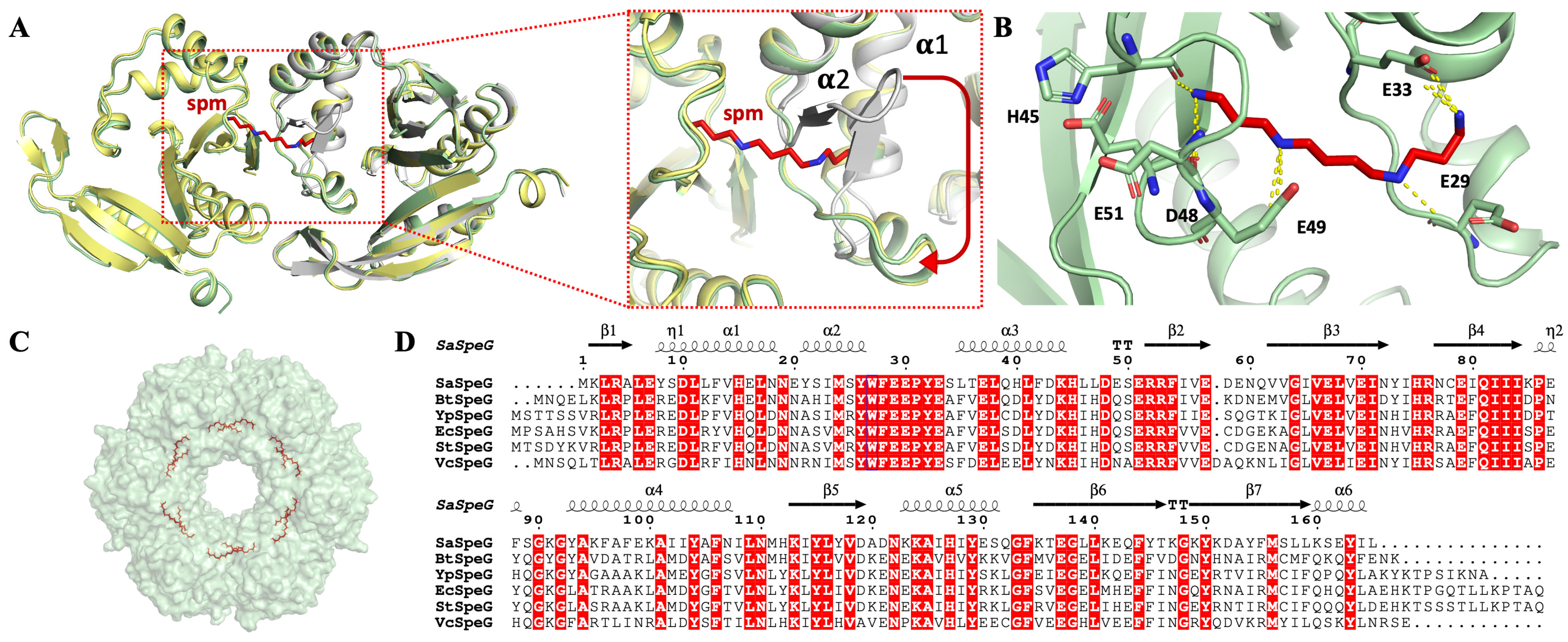
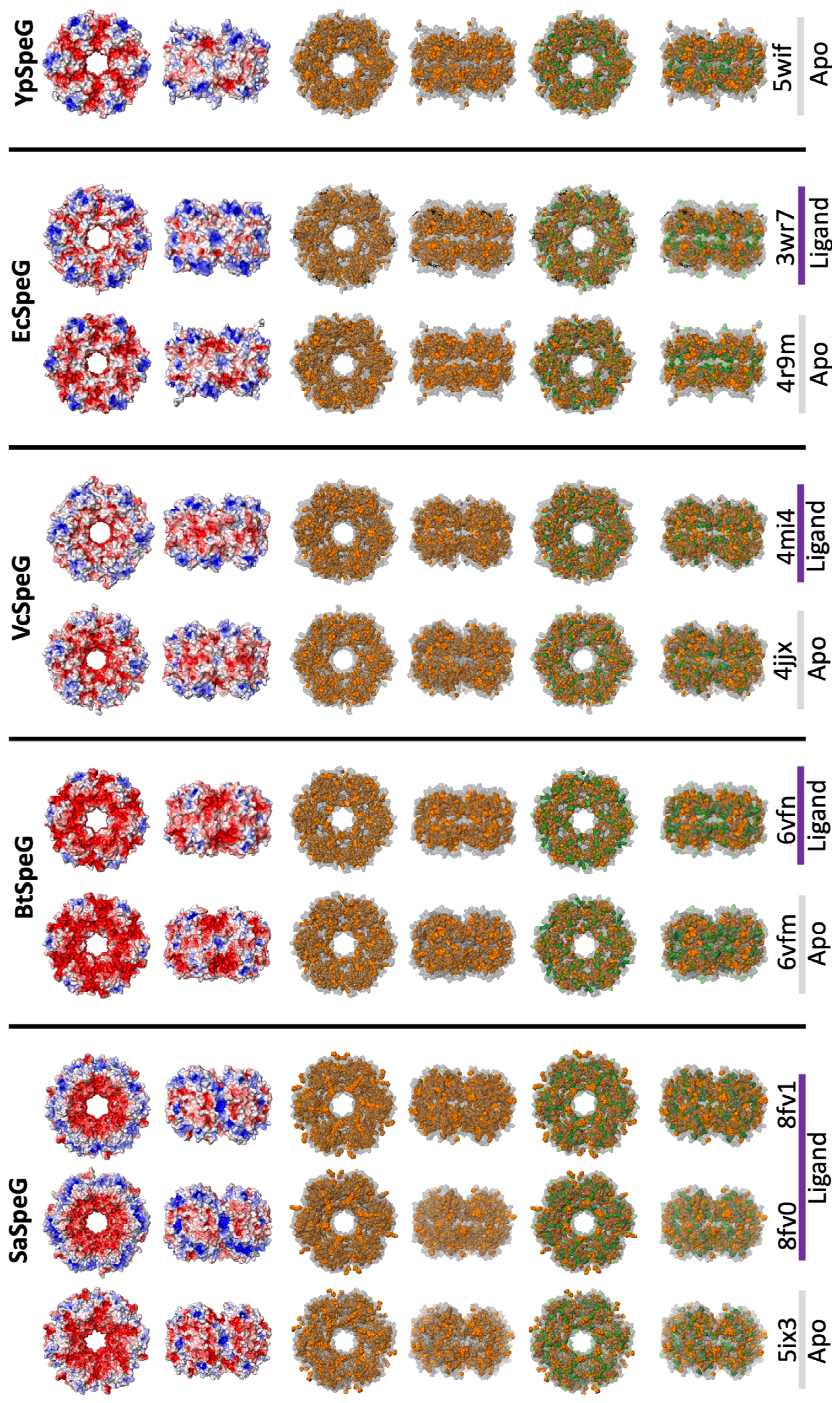
3.2. 3D Structure of SaSpeG
3.3. Native PAGE, EMSA, and SEC Analysis of SaSpeG
4. Discussion
4.1. Oligomeric State of SaSpeG
4.2. Differential Effects of Spermine and Spermidine on SaSpeG Cooperativity and Heterogeneity
4.3. Comparison of SaSpeG Kinetics
4.4. Dynamic Role of SaSpeG
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Planet, P.J. Life After USA300: The Rise and Fall of a Superbug. J. Infect. Dis. 2017, 215, S71–S77. [Google Scholar] [CrossRef] [PubMed]
- Diep, B.A.; Stone, G.G.; Basuino, L.; Graber, C.J.; Miller, A.; des Etages, S.A.; Jones, A.; Palazzolo-Ballance, A.M.; Perdreau-Remington, F.; Sensabaugh, G.F.; et al. The arginine catabolic mobile element and staphylococcal chromosomal cassette mec linkage: Convergence of virulence and resistance in the USA300 clone of methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 2008, 197, 1523–1530. [Google Scholar] [CrossRef] [Green Version]
- Schwendener, S.; Perreten, V. The bla and mec families of β-lactam resistance genes in the genera Macrococcus, Mammaliicoccus and Staphylococcus: An in-depth analysis with emphasis on Macrococcus. J. Antimicrob. Chemother. 2022, 77, 1796–1827. [Google Scholar] [CrossRef] [PubMed]
- Uehara, Y. Current Status of Staphylococcal Cassette Chromosome mec (SCCmec). Antibiotics 2022, 11, 86. [Google Scholar] [CrossRef]
- Joshi, G.S.; Spontak, J.S.; Klapper, D.G.; Richardson, A.R. Arginine catabolic mobile element encoded speG abrogates the unique hypersensitivity of Staphylococcus aureus to exogenous polyamines. Mol. Microbiol. 2011, 82, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Planet, P.J.; LaRussa, S.J.; Dana, A.; Smith, H.; Xu, A.; Ryan, C.; Uhlemann, A.C.; Boundy, S.; Goldberg, J.; Narechania, A.; et al. Emergence of the epidemic methicillin-resistant Staphylococcus aureus strain USA300 coincides with horizontal transfer of the arginine catabolic mobile element and speG-mediated adaptations for survival on skin. mBio 2013, 4, e00889-13. [Google Scholar] [CrossRef] [Green Version]
- Thurlow, L.R.; Joshi, G.S.; Clark, J.R.; Spontak, J.S.; Neely, C.J.; Maile, R.; Richardson, A.R. Functional modularity of the arginine catabolic mobile element contributes to the success of USA300 methicillin-resistant Staphylococcus aureus. Cell Host Microbe 2013, 13, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Maezato, Y.; Kim, S.H.; Kurihara, S.; Liang, J.; Michael, A.J. Polyamine-independent growth and biofilm formation, and functional spermidine/spermine N-acetyltransferases in Staphylococcus aureus and Enterococcus faecalis. Mol. Microbiol. 2019, 111, 159–175. [Google Scholar] [CrossRef] [Green Version]
- Michael, A.J. Biosynthesis of polyamines and polyamine-containing molecules. Biochem. J. 2016, 473, 2315–2329. [Google Scholar] [CrossRef]
- Rosenthal, S.M.; Dubin, D.T. Metabolism of polyamines by Staphylococcus. J. Bacteriol. 1962, 84, 859–863. [Google Scholar] [CrossRef] [Green Version]
- Rozansky, R.; Bachrach, U.; Grossowicz, N. Studies on the antibacterial action of spermine. J. Gen. Microbiol. 1954, 10, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Diep, B.A.; Gill, S.R.; Chang, R.F.; Phan, T.H.; Chen, J.H.; Davidson, M.G.; Lin, F.; Lin, J.; Carleton, H.A.; Mongodin, E.F.; et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 2006, 367, 731–739. [Google Scholar] [CrossRef]
- Aung, M.S.; Kawaguchiya, M.; Urushibara, N.; Sumi, A.; Ito, M.; Kudo, K.; Morimoto, S.; Hosoya, S.; Kobayashi, N. Molecular Characterization of Methicillin-Resistant Staphylococcus aureus from Outpatients in Northern Japan: Increasing Tendency of ST5/ST764 MRSA-IIa with Arginine Catabolic Mobile Element. Microb. Drug Resist. 2017, 23, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Tsai, J.C.; Chen, H.J.; Hung, W.C.; Hsueh, P.R.; Teng, L.J. A novel staphylococcal cassette chromosomal element, SCCfusC, carrying fusC and speG in fusidic acid-resistant methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 1224–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, C.P.; Boyle-Vavra, S.; Daum, R.S. The arginine catabolic mobile element is not associated with enhanced virulence in experimental invasive disease caused by the community-associated methicillin-resistant Staphylococcus aureus USA300 genetic background. Infect. Immun. 2009, 77, 2650–2656. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Conly, J.; McClure, J.-A.; Kurwa, H.A.; Zhang, K. Arginine Catabolic Mobile Element in Evolution and Pathogenicity of the Community-Associated Methicillin-Resistant Staphylococcus aureus Strain USA300. Microorganisms 2020, 8, 275. [Google Scholar] [CrossRef] [Green Version]
- Studier, F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Battye, T.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- McCoy, A.J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 32–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Le, V.T.B.; Tsimbalyuk, S.; Lim, E.Q.; Solis, A.; Gawat, D.; Boeck, P.; Lim, E.Q.; Renolo, R.; Forwood, J.K.; Kuhn, M.L. The Vibrio cholerae SpeG Spermidine/Spermine N-Acetyltransferase Allosteric Loop and β6-β7 Structural Elements Are Critical for Kinetic Activity. Front. Mol. Biosci. 2021, 8, 645768. [Google Scholar] [CrossRef]
- Kuhn, M.L.; Majorek, K.A.; Minor, W.; Anderson, W.F. Broad-substrate screen as a tool to identify substrates for bacterial Gcn5-related N-acetyltransferases with unknown substrate specificity. Protein Sci. 2013, 22, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Filippova, E.V.; Kuhn, M.L.; Osipiuk, J.; Kiryukhina, O.; Joachimiak, A.; Ballicora, M.A.; Anderson, W.F. A novel polyamine allosteric site of SpeG from Vibrio cholerae is revealed by its dodecameric structure. J. Mol. Biol. 2015, 427, 1316–1334. [Google Scholar] [CrossRef] [Green Version]
- Tsimbalyuk, S.; Shornikov, A.; Thi Bich Le, V.; Kuhn, M.L.; Forwood, J.K. SpeG polyamine acetyltransferase enzyme from Bacillus thuringiensis forms a dodecameric structure and exhibits high catalytic efficiency. J. Struct. Biol. 2020, 210, 107506. [Google Scholar] [CrossRef]
- Le, V.T.B.; Dang, J.; Lim, E.Q.; Kuhn, M.L. Criticality of a conserved tyrosine residue in the SpeG protein from Escherichia coli. Protein Sci. 2021, 30, 1264–1269. [Google Scholar] [CrossRef]
- Fukuchi, J.; Kashiwagi, K.; Takio, K.; Igarashi, K. Properties and structure of spermidine acetyltransferase in Escherichia coli. J. Biol. Chem. 1994, 269, 22581–22585. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, K.; Duarte, J.M.; Biyani, N.; Bliven, S.; Capitani, G. A PDB-wide, evolution-based assessment of protein-protein interfaces. BMC Struct. Biol. 2014, 14, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Filippova, E.V.; Weigand, S.; Osipiuk, J.; Kiryukhina, O.; Joachimiak, A.; Anderson, W.F. Substrate-Induced Allosteric Change in the Quaternary Structure of the Spermidine N-Acetyltransferase SpeG. J. Mol. Biol. 2015, 427, 3538–3553. [Google Scholar] [CrossRef] [Green Version]
- Filippova, E.V.; Weigand, S.; Kiryukhina, O.; Wolfe, A.J.; Anderson, W.F. Analysis of crystalline and solution states of ligand-free spermidine N-acetyltransferase (SpeG) from Escherichia coli. Acta Crystallogr. D Struct. Biol. 2019, 75, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Haglund, Å.C.; Marsden, N.V.B. Hydrophobic and polar contributions to solute affinity for a highly crosslinked water-swollen (sephadex) gel. J. Polym. Sci. Polym. Lett. Ed. 1980, 18, 271–279. [Google Scholar] [CrossRef]
- Hellberg, U.; Ivarsson, J.-P.; Johansson, B.-L. Characteristics of Superdex® prep grade media for gel filtration chromatography of proteins and peptides. Process Biochem. 1996, 31, 163–172. [Google Scholar] [CrossRef]
- Thao, S.; Escalante-Semerena, J.C. Biochemical and Thermodynamic Analyses of Salmonella enterica Pat, a Multidomain, Multimeric N-Lysine Acetyltransferase Involved in Carbon and Energy Metabolism. mBio 2011, 2, e00216-11. [Google Scholar] [CrossRef] [Green Version]
- Majorek, K.A.; Kuhn, M.L.; Chruszcz, M.; Anderson, W.F.; Minor, W. Double trouble-Buffer selection and His-tag presence may be responsible for nonreproducibility of biomedical experiments. Protein Sci. 2014, 23, 1359–1368. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.-B.; Huang, C.-J.; Huang, C.-H.; Wang, K.-C.; Chang, N.-W.; Pan, H.-Y.; Fang, H.-W.; Huang, M.-T.; Chen, C.-K. speG Is Required for Intracellular Replication of Salmonella in Various Human Cells and Affects Its Polyamine Metabolism and Global Transcriptomes. Front. Microbiol. 2017, 8, 02245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, D.-W.; Lee, B.; Heo, S.; Oh, Y.; Heo, G.; Lee, J.-H. Two genes involved in clindamycin resistance of Bacillus licheniformis and Bacillus paralicheniformis identified by comparative genomic analysis. PLoS ONE 2020, 15, e0231274. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Km or S0.5 (mM) | kcat (s−1) | Catalytic Efficiency (M−1s−1) | n | ||||
|---|---|---|---|---|---|---|---|
| Assay 1 | |||||||
| Spm | 1.3 | ± | 0.1 | 5.3 | 4.1 × 103 | 1.6 ± 0.2 | |
| Spd | 4.0 | ± | 0.1 | 3.0 | 7.5 × 102 | 3.5 ± 0.2 | |
| Assay 2 | |||||||
| Spm | 0.070 | ± | 0.001 | 420 | 6.0 × 106 | ||
| Spd | 0.063 | ± | 0.005 | 110 | 1.7 × 106 | 2.1 ± 0.3 | |
| AcCoA (spm) | 0.29 | ± | 0.01 | 390 | 1.3 × 106 | ||
| AcCoA (spd) | 0.35 | ± | 0.01 | 170 | 4.9 × 105 | ||
| Li et al. | |||||||
| Spm | 0.295 | ± | 0.005 | 3.52 | 1.2 × 104 | ||
| Spd | 1.33 | ± | 0.12 | 2.48 | 1.8 × 103 | ||
| pI | #AAs | #A, V, I, L | #F, W, Y | % Small Hydrophobic AAs | % Aromatic AAs | % Small Hydrophobic and Aromatic AAs | |
|---|---|---|---|---|---|---|---|
| SaSpeG | 5.28 | 165 | 8, 7, 16, 18 | 11, 1, 14 | 30 | 16 | 45 |
| BtSpeG | 5 | 171 | 9, 14, 10, 14 | 11, 1, 12 | 27 | 14 | 42 |
| VcSpeG | 5.58 | 173 | 9, 9, 15, 19 | 10, 1, 10 | 30 | 12 | 42 |
| EcSpeG | 6.2 | 186 | 13, 12, 11, 19 | 9, 1, 11 | 30 | 11 | 41 |
| YpSpeG | 5.99 | 181 | 11, 8, 18, 16 | 10, 1, 11 | 29 | 12 | 41 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsimbalyuk, S.; Shornikov, A.; Srivastava, P.; Le, V.T.B.; Warren, I.; Khandokar, Y.B.; Kuhn, M.L.; Forwood, J.K. Structural and Kinetic Characterization of the SpeG Spermidine/Spermine N-acetyltransferase from Methicillin-Resistant Staphylococcus aureus USA300. Cells 2023, 12, 1829. https://doi.org/10.3390/cells12141829
Tsimbalyuk S, Shornikov A, Srivastava P, Le VTB, Warren I, Khandokar YB, Kuhn ML, Forwood JK. Structural and Kinetic Characterization of the SpeG Spermidine/Spermine N-acetyltransferase from Methicillin-Resistant Staphylococcus aureus USA300. Cells. 2023; 12(14):1829. https://doi.org/10.3390/cells12141829
Chicago/Turabian StyleTsimbalyuk, Sofiya, Aleksander Shornikov, Parul Srivastava, Van Thi Bich Le, Imani Warren, Yogesh B. Khandokar, Misty L. Kuhn, and Jade K. Forwood. 2023. "Structural and Kinetic Characterization of the SpeG Spermidine/Spermine N-acetyltransferase from Methicillin-Resistant Staphylococcus aureus USA300" Cells 12, no. 14: 1829. https://doi.org/10.3390/cells12141829