
S1P Lyase Deficiency in the Brain Promotes Astrogliosis and NLRP3 Inflammasome Activation via Purinergic Signaling
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. List of Abbreviations
2.2. Antibodies and Chemicals
2.3. Animals
2.4. Cell Culture
2.5. Western Immunoblotting
2.6. RNA Isolation and Real-Time PCR
2.7. Immunocytochemistry
2.8. Immunohistochemistry
2.9. ADP and ATP Measurement
2.10. Enzyme-Linked Immunosorbent Assay (ELISA)
2.11. Measurement of Intracellular Calcium Concentration ([Ca2+]i)
2.12. RNA-Seq
2.13. Active Motif CUT&Tag
2.14. Treatment of Cells
2.14.1. S1P and S1PR2,4 Agonist Treatment
2.14.2. P2Y1R Antagonist Treatment
2.14.3. P2Y1R Agonist Treatment
2.15. Statistical Analysis
3. Results
3.1. Neural Ablation of SGPL1 Triggers Astrogliosis in Murine Brains via the P2Y1R
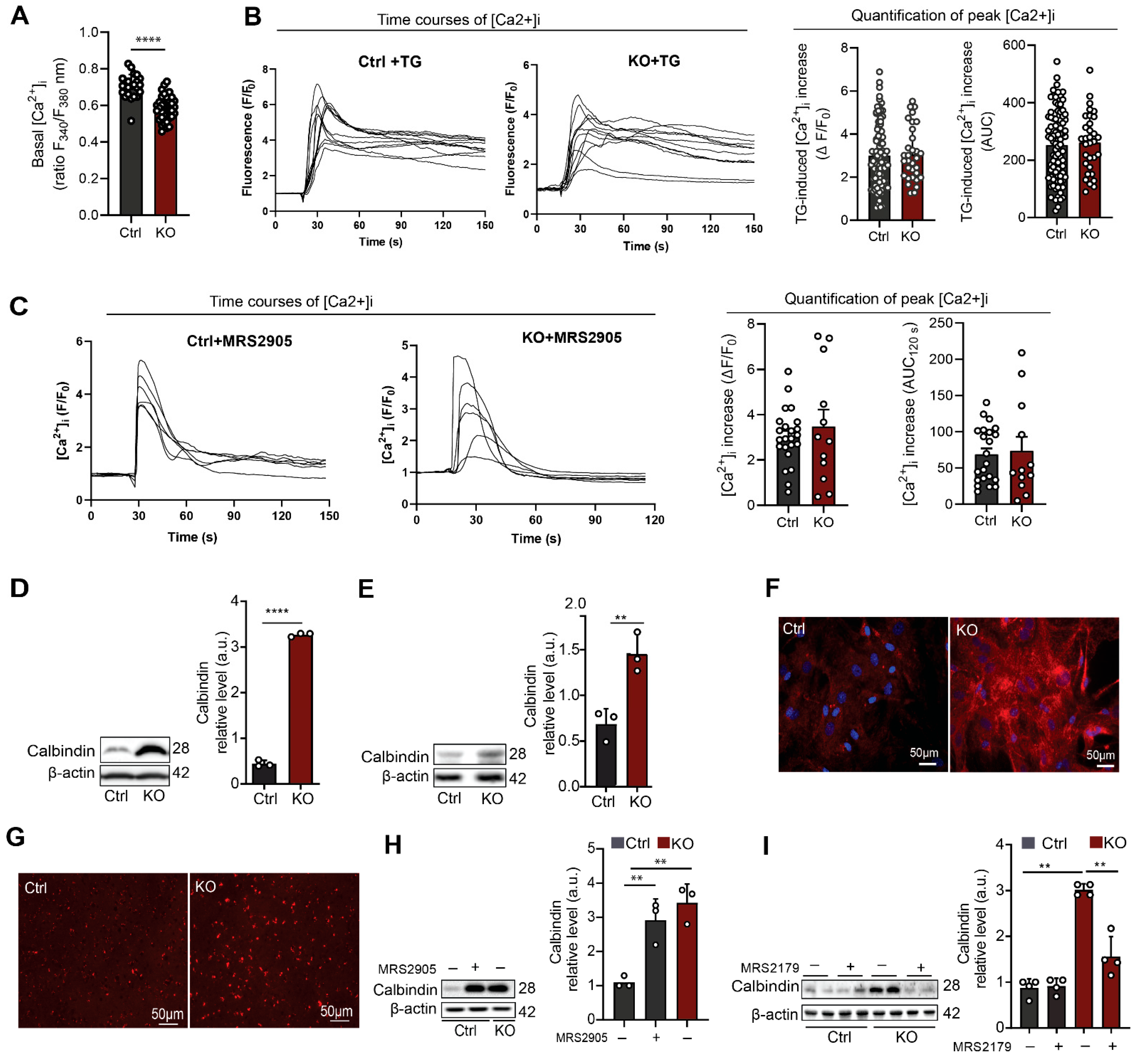
3.2. SGPL1-Deficient Astrocytes Exhibit Decreased Basal [Ca2+]i and Elevated Calbindin Expression Mediated by P2Y1R
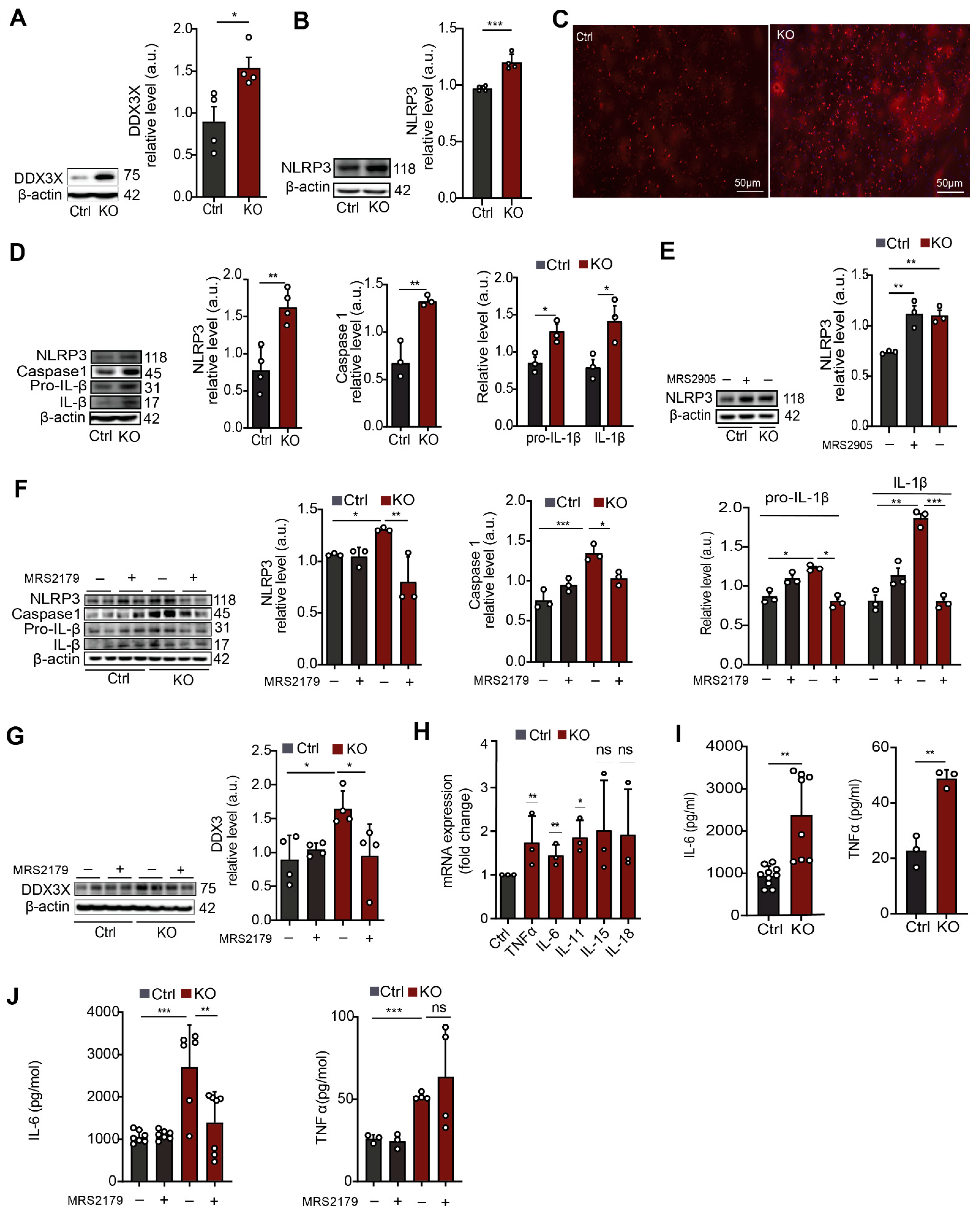
3.3. In SGPL1-Deficient Astrocytes Transcriptional and Epigenetic Alterations Lead to Activation of the NLRP3 Inflammasome via a P2Y1R-Mediated Mechanism
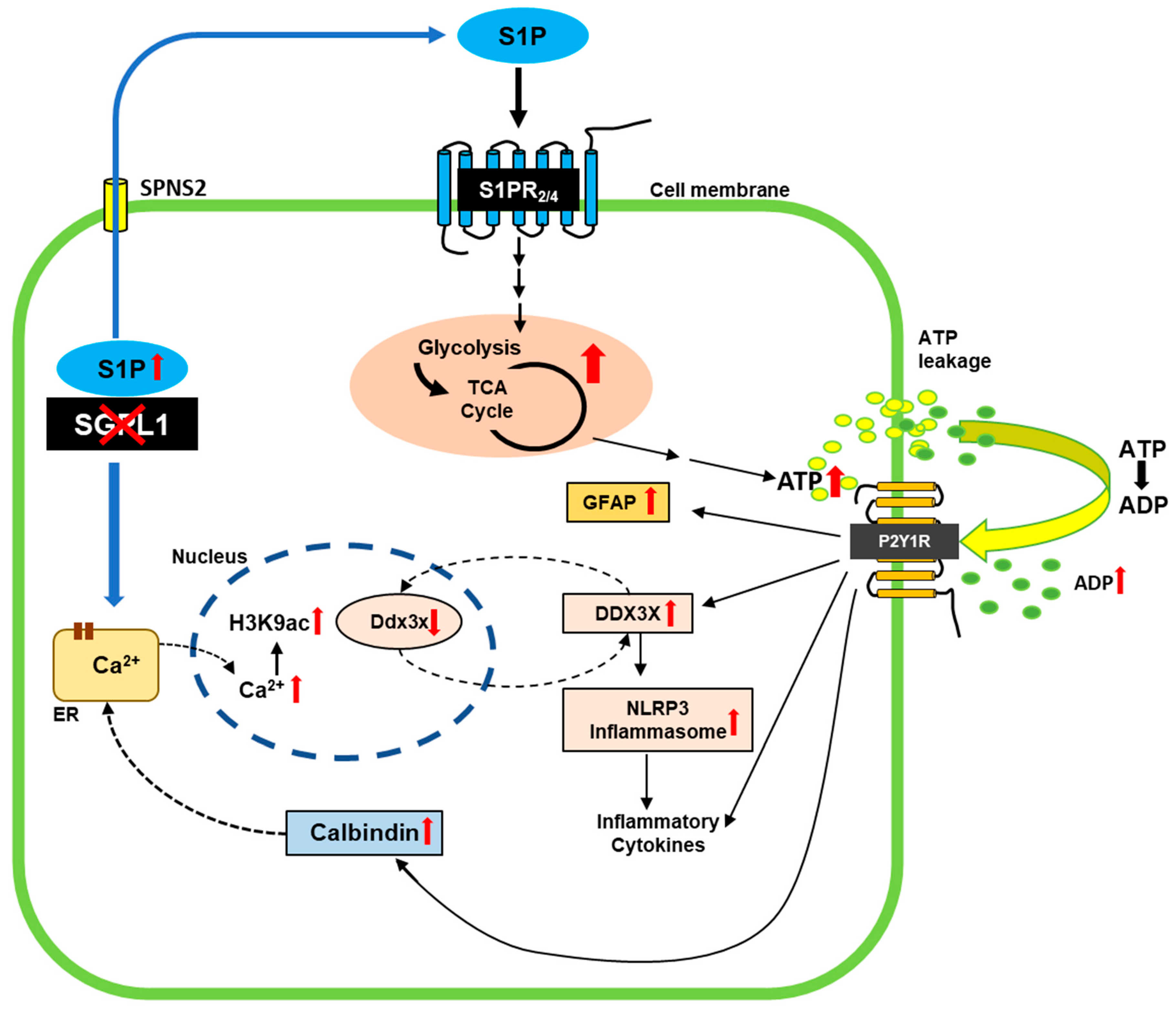
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, I.; van Echten-Deckert, G. Sphingosine 1-phosphate—A double edged sword in the brain. Biochim. Biophys. Acta 2017, 1859, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Grassi, S.; Mauri, L.; Prioni, S.; Cabitta, L.; Sonnino, S.; Prinetti, A.; Giussani, P. Sphingosine 1-Phosphate Receptors and Metabolic Enzymes as Druggable Targets for Brain Diseases. Front. Pharmacol. 2019, 10, 807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edsall, L.C.; Spiegel, S. Enzymatic measurement of sphingosine 1-phosphate. Anal. Biochem. 1999, 272, 80–86. [Google Scholar] [CrossRef]
- Futerman, A.H. Sphingolipids. In Biochemistry of Lipids and Membranes, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 297–326. [Google Scholar]
- Blondeau, N.; Lai, Y.; Tyndall, S.; Popolo, M.; Topalkara, K.; Pru, J.K.; Zhang, L.; Kim, H.; Liao, J.K.; Ding, K.; et al. Distribution of sphingosine kinase activity and mRNA in rodent brain. J. Neurochem. 2007, 103, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Saba, J.D. Fifty years of lyase and a moment of truth: Sphingosine phosphate lyase from discovery to disease. J. Lipid. Res. 2019, 60, 456–463. [Google Scholar] [CrossRef] [Green Version]
- Afsar, S.Y.; Alam, S.; Fernandez Gonzalez, C.; van Echten-Deckert, G. Sphingosine-1-phosphate-lyase deficiency affects glucose metabolism in a way that abets oncogenesis. Mol. Oncol. 2022, 16, 3642–3653. [Google Scholar] [CrossRef]
- Choi, Y.J.; Saba, J.D. Sphingosine phosphate lyase insufficiency syndrome (SPLIS): A novel inborn error of sphingolipid metabolism. Adv. Biol. Regul. 2019, 71, 128–140. [Google Scholar] [CrossRef]
- Bamborschke, D.; Pergande, M.; Becker, K.; Koerber, F.; Dotsch, J.; Vierzig, A.; Weber, L.T.; Cirak, S. A novel mutation in sphingosine-1-phosphate lyase causing congenital brain malformation. Brain Dev. 2018, 40, 480–483. [Google Scholar] [CrossRef]
- Liu, Y.; Wada, R.; Yamashita, T.; Mi, Y.; Deng, C.X.; Hobson, J.P.; Rosenfeldt, H.M.; Nava, V.E.; Chae, S.S.; Lee, M.J.; et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J. Clin. Investig. 2000, 106, 951–961. [Google Scholar] [CrossRef] [Green Version]
- Mizugishi, K.; Yamashita, T.; Olivera, A.; Miller, G.F.; Spiegel, S.; Proia, R.L. Essential role for sphingosine kinases in neural and vascular development. Mol. Cell Biol. 2005, 25, 11113–11121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Echten-Deckert, G.; Hagen-Euteneuer, N.; Karaca, I.; Walter, J. Sphingosine-1-phosphate: Boon and bane for the brain. Cell Physiol. Biochem. 2014, 34, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Couttas, T.A.; Kain, N.; Daniels, B.; Lim, X.Y.; Shepherd, C.; Kril, J.; Pickford, R.; Li, H.; Garner, B.; Don, A.S. Loss of the neuroprotective factor Sphingosine 1-phosphate early in Alzheimer’s disease pathogenesis. Acta Neuropathol. Commun. 2014, 2, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccom, J.; Loukh, N.; Lauwers-Cances, V.; Touriol, C.; Nicaise, Y.; Gentil, C.; Uro-Coste, E.; Pitson, S.; Maurage, C.A.; Duyckaerts, C.; et al. Reduced sphingosine kinase-1 and enhanced sphingosine 1-phosphate lyase expression demonstrate deregulated sphingosine 1-phosphate signaling in Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 12. [Google Scholar] [CrossRef]
- Soliven, B.; Miron, V.; Chun, J. The neurobiology of sphingosine 1-phosphate signaling and sphingosine 1-phosphate receptor modulators. Neurology 2011, 76, S9–S14. [Google Scholar] [CrossRef]
- Takasugi, N.; Sasaki, T.; Suzuki, K.; Osawa, S.; Isshiki, H.; Hori, Y.; Shimada, N.; Higo, T.; Yokoshima, S.; Fukuyama, T.; et al. BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. J. Neurosci. 2011, 31, 6850–6857. [Google Scholar] [CrossRef] [Green Version]
- Hagen, N.; Hans, M.; Hartmann, D.; Swandulla, D.; van Echten-Deckert, G. Sphingosine-1-phosphate links glycosphingolipid metabolism to neurodegeneration via a calpain-mediated mechanism. Cell Death Differ. 2011, 18, 1356–1365. [Google Scholar] [CrossRef] [Green Version]
- Ibanez, C.; Simo, C.; Barupal, D.K.; Fiehn, O.; Kivipelto, M.; Cedazo-Minguez, A.; Cifuentes, A. A new metabolomic workflow for early detection of Alzheimer’s disease. J. Chromatogr. A 2013, 1302, 65–71. [Google Scholar] [CrossRef]
- Mitroi, D.N.; Deutschmann, A.U.; Raucamp, M.; Karunakaran, I.; Glebov, K.; Hans, M.; Walter, J.; Saba, J.; Graler, M.; Ehninger, D.; et al. Sphingosine 1-phosphate lyase ablation disrupts presynaptic architecture and function via an ubiquitin-proteasome mediated mechanism. Sci. Rep. 2016, 6, 37064. [Google Scholar] [CrossRef]
- Mitroi, D.N.; Karunakaran, I.; Graler, M.; Saba, J.D.; Ehninger, D.; Ledesma, M.D.; van Echten-Deckert, G. SGPL1 (sphingosine phosphate lyase 1) modulates neuronal autophagy via phosphatidylethanolamine production. Autophagy 2017, 13, 885–899. [Google Scholar] [CrossRef]
- Alam, S.; Piazzesi, A.; Abd El Fatah, M.; Raucamp, M.; van Echten-Deckert, G. Neurodegeneration Caused by S1P-Lyase Deficiency Involves Calcium-Dependent Tau Pathology and Abnormal Histone Acetylation. Cells 2020, 9, 2189. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, I.; Alam, S.; Jayagopi, S.; Frohberger, S.J.; Hansen, J.N.; Kuehlwein, J.; Holbling, B.V.; Schumak, B.; Hubner, M.P.; Graler, M.H.; et al. Neural sphingosine 1-phosphate accumulation activates microglia and links impaired autophagy and inflammation. Glia 2019, 67, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Afsar, S.Y.; Van Echten-Deckert, G. S1P Released by SGPL1-Deficient Astrocytes Enhances Astrocytic ATP Production via S1PR(2,4), Thus Keeping Autophagy in Check: Potential Consequences for Brain Health. Int. J. Mol. Sci. 2023, 24, 4581. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, L.F.; Ghirnikar, R.S.; Lee, Y.L. Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000). Neurochem. Res. 2000, 25, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Blankenbach, K.V.; Bruno, G.; Wondra, E.; Spohner, A.K.; Aster, N.J.; Vienken, H.; Trautmann, S.; Ferreiros, N.; Wieland, T.; Bruni, P.; et al. The WD40 repeat protein, WDR36, orchestrates sphingosine kinase-1 recruitment and phospholipase C-beta activation by G(q)-coupled receptors. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158704. [Google Scholar] [CrossRef]
- Vienken, H.; Mabrouki, N.; Grabau, K.; Claas, R.F.; Rudowski, A.; Schomel, N.; Pfeilschifter, J.; Lutjohann, D.; van Echten-Deckert, G.; Meyer Zu Heringdorf, D. Characterization of cholesterol homeostasis in sphingosine-1-phosphate lyase-deficient fibroblasts reveals a Niemann-Pick disease type C-like phenotype with enhanced lysosomal Ca2+ storage. Sci. Rep. 2017, 7, 43575. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Franke, H.; Illes, P. Nucleotide signaling in astrogliosis. Neurosci. Lett. 2014, 565, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Delekate, A.; Fuchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, M.; Richardson, J.R. Epigenetic regulation of astrocyte function in neuroinflammation and neurodegeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A. Astroglial Calcium Signaling in Aging and Alzheimer’s Disease. Cold Spring Harb. Perspect. Biol. 2019, 11, a035188. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, P.B.; Knappenberger, J.; Segal, M.; Bennett, M.V.; Charles, A.C.; Kater, S.B. ATP released from astrocytes mediates glial calcium waves. J. Neurosci. 1999, 19, 520–528. [Google Scholar] [CrossRef] [Green Version]
- Toyoshima, T.; Yamagami, S.; Ahmed, B.Y.; Jin, L.; Miyamoto, O.; Itano, T.; Tokuda, M.; Matsui, H.; Hatase, O. Expression of calbindin-D28K by reactive astrocytes in gerbil hippocampus after ischaemia. Neuroreport 1996, 7, 2087–2091. [Google Scholar] [CrossRef]
- Schwaller, B. Cytosolic Ca2+ Buffers Are Inherently Ca2+ Signal Modulators. Cold Spring Harb Perspect. Biol. 2020, 12, a004051. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2014, 7, a020420. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.Y.X.; Zhang, J.; Tee, W.W. Epigenetic Regulation of Inflammatory Signaling and Inflammation-Induced Cancer. Front. Cell Dev. Biol. 2022, 10, 931493. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019, 10, 1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samir, P.; Kesavardhana, S.; Patmore, D.M.; Gingras, S.; Malireddi, R.K.S.; Karki, R.; Guy, C.S.; Briard, B.; Place, D.E.; Bhattacharya, A.; et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 2019, 573, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Volterra, A.; Meldolesi, J. Astrocytes, from brain glue to communication elements: The revolution continues. Nat. Rev. Neurosci. 2005, 6, 626–640. [Google Scholar] [CrossRef]
- Wu, Y.P.; Mizugishi, K.; Bektas, M.; Sandhoff, R.; Proia, R.L. Sphingosine kinase 1/S1P receptor signaling axis controls glial proliferation in mice with Sandhoff disease. Hum. Mol. Genet. 2008, 17, wu2257–wu2264. [Google Scholar] [CrossRef] [PubMed]
- Kolahdooz, Z.; Nasoohi, S.; Asle-Rousta, M.; Ahmadiani, A.; Dargahi, L. Sphingosin-1-phosphate Receptor 1: A Potential Target to Inhibit Neuroinflammation and Restore the Sphingosin-1-phosphate Metabolism. Can. J. Neurol. Sci. 2015, 42, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rivera, R.; Lee, C.W.; Noguchi, K.; Teo, S.T.; Yung, Y.C.; Lu, M.; Kennedy, G.; et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 751–756. [Google Scholar] [CrossRef]
- Kim, S.; Bielawski, J.; Yang, H.; Kong, Y.; Zhou, B.; Li, J. Functional antagonism of sphingosine-1-phosphate receptor 1 prevents cuprizone-induced demyelination. Glia 2018, 66, 654–669. [Google Scholar] [CrossRef]
- Dusaban, S.S.; Chun, J.; Rosen, H.; Purcell, N.H.; Brown, J.H. Sphingosine 1-phosphate receptor 3 and RhoA signaling mediate inflammatory gene expression in astrocytes. J. Neuroinflamm. 2017, 14, 111. [Google Scholar] [CrossRef] [Green Version]
- Fischer, I.; Alliod, C.; Martinier, N.; Newcombe, J.; Brana, C.; Pouly, S. Sphingosine kinase 1 and sphingosine 1-phosphate receptor 3 are functionally upregulated on astrocytes under pro-inflammatory conditions. PLoS ONE 2011, 6, e23905. [Google Scholar] [CrossRef] [Green Version]
- Van Doorn, R.; Van Horssen, J.; Verzijl, D.; Witte, M.; Ronken, E.; Van Het Hof, B.; Lakeman, K.; Dijkstra, C.D.; Van Der Valk, P.; Reijerkerk, A.; et al. Sphingosine 1-phosphate receptor 1 and 3 are upregulated in multiple sclerosis lesions. Glia 2010, 58, 1465–1476. [Google Scholar] [CrossRef]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Ku, Y.C.; Lai, M.H.; Lo, C.C.; Cheng, Y.C.; Qiu, J.T.; Tarn, W.Y.; Lai, M.C. DDX3 Participates in Translational Control of Inflammation Induced by Infections and Injuries. Mol. Cell Biol. 2019, 39, e00285-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, Q.; Wang, Q.; Liu, H.; Zhou, F.; Zhang, Y.; Yuan, M.; Zhao, C.; Guan, Y.; Wang, X. DDX3 binding with CK1epsilon was closely related to motor neuron degeneration of ALS by affecting neurite outgrowth. Am. J. Transl. Res. 2017, 9, 4627–4639. [Google Scholar] [PubMed]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Gallagher, C.J.; Salter, M.W. Differential properties of astrocyte calcium waves mediated by P2Y1 and P2Y2 receptors. J. Neurosci. 2003, 23, 6728–6739. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Yan, W.; Zhou, Z.; Liu, B.; Wang, Z.; Chen, J.; Wang, H. Elevated Levels of NLRP3 in Cerebrospinal Fluid of Patients With Autoimmune GFAP Astrocytopathy. Front. Neurol. 2019, 10, 1019. [Google Scholar] [CrossRef] [Green Version]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [Green Version]
- Abbracchio, M.P.; Brambilla, R.; Ceruti, S.; Cattabeni, F. Signalling mechanisms involved in P2Y receptor-mediated reactive astrogliosis. Prog. Brain Res. 1999, 120, 333–342. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Verderio, C. Pathophysiological roles of P2 receptors in glial cells. Novartis Found. Symp. 2006, 276, 91–103; discussion 103–112, 275–181. [Google Scholar] [PubMed]
- Neary, J.T. Trophic actions of extracellular ATP on astrocytes, synergistic interactions with fibroblast growth factors and underlying signal transduction mechanisms. Ciba Found. Symp. 1996, 198, 130–139; discussion 139–141. [Google Scholar] [CrossRef] [PubMed]
- Franke, H.; Krugel, U.; Illes, P. P2 receptor-mediated proliferative effects on astrocytes in vivo. Glia 1999, 28, 190–200. [Google Scholar] [CrossRef]
- Brambilla, R.; Abbracchio, M.P. Modulation of cyclooxygenase-2 and brain reactive astrogliosis by purinergic P2 receptors. Ann. N. Y. Acad. Sci. 2001, 939, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.J.; Tome, A.R.; Cunha, R.A. ATP as a multi-target danger signal in the brain. Front. Neurosci. 2015, 9, 148. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, T.K.; Bian, J.; Gill, D.L. Sphingosine 1-phosphate generated in the endoplasmic reticulum membrane activates release of stored calcium. J. Biol. Chem. 1994, 269, 22628–22635. [Google Scholar] [CrossRef]
- Baurand, A.; Gachet, C. The P2Y(1) receptor as a target for new antithrombotic drugs: A review of the P2Y(1) antagonist MRS-2179. Cardiovasc. Drug Rev. 2003, 21, 67–76. [Google Scholar] [CrossRef]
- Yelovitch, S.; Barr, H.M.; Camden, J.; Weisman, G.A.; Shai, E.; Varon, D.; Fischer, B. Identification of a promising drug candidate for the treatment of type 2 diabetes based on a P2Y(1) receptor agonist. J. Med. Chem. 2012, 55, 7623–7635. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, S.; Afsar, S.Y.; Wolter, M.A.; Volk, L.M.; Mitroi, D.N.; Meyer zu Heringdorf, D.; van Echten-Deckert, G. S1P Lyase Deficiency in the Brain Promotes Astrogliosis and NLRP3 Inflammasome Activation via Purinergic Signaling. Cells 2023, 12, 1844. https://doi.org/10.3390/cells12141844
Alam S, Afsar SY, Wolter MA, Volk LM, Mitroi DN, Meyer zu Heringdorf D, van Echten-Deckert G. S1P Lyase Deficiency in the Brain Promotes Astrogliosis and NLRP3 Inflammasome Activation via Purinergic Signaling. Cells. 2023; 12(14):1844. https://doi.org/10.3390/cells12141844
Chicago/Turabian StyleAlam, Shah, Sumaiya Yasmeen Afsar, Maya Anik Wolter, Luisa Michelle Volk, Daniel Nicolae Mitroi, Dagmar Meyer zu Heringdorf, and Gerhild van Echten-Deckert. 2023. "S1P Lyase Deficiency in the Brain Promotes Astrogliosis and NLRP3 Inflammasome Activation via Purinergic Signaling" Cells 12, no. 14: 1844. https://doi.org/10.3390/cells12141844