
Mathematical Modeling of Clonal Interference by Density-Dependent Selection in Heterogeneous Cancer Cell Lines

 ,
,
Abstract
:1. Introduction
2. Results
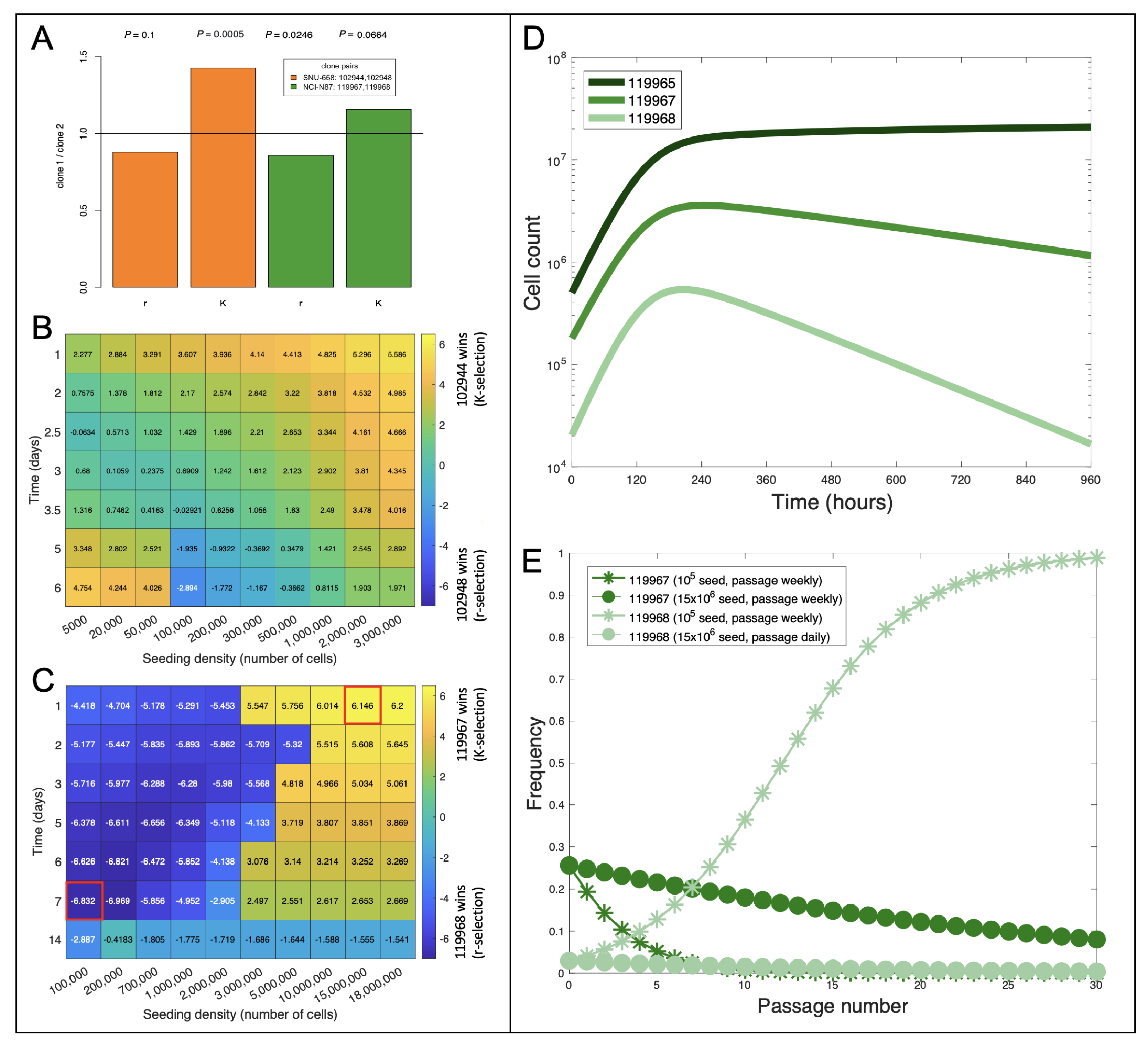
2.1. Identifying Biomarkers of Growth Rate and Carrying Capacity
2.2. Towards Informing Future Experiments: Steering Clonal Evolution
3. Discussion
4. Online Methods
4.1. Cell Culture
4.2. Microscopy
4.3. Determination of Clonal Growth Parameters
4.4. Mathematical Models of In-Vitro Cell Growth
4.5. Identifying r/K Trade-Offs between Co-Existing Clones within a Cell Line
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Identifiability Analysis
Appendix A.2. Analytical Results Optimizing Cell Passaging for Logistic Growth Model (Two Clone Case)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HGC-27 | KATOIII | MKN-45 | NCI-N87 | NUGC-4 | SNU-601 | SNU-638 | SNU-668 | |
|---|---|---|---|---|---|---|---|---|
| Logistic | 1.00 | 0.97 | 0.99 | 0.99 | 0.98 | 0.99 | 0.97 | 0.97 |
| Richards | 1.00 | 0.97 | 0.99 | 0.99 | 0.97 | 0.99 | 0.97 | 0.98 |
| Gompertz | 1.00 | 0.95 | 0.96 | 0.95 | 0.98 | 0.98 | 0.95 | 0.98 |
| r | K | bestFit | adjutedRSquared | doublingTime | cellLine | |
|---|---|---|---|---|---|---|
| HGC-27 | 0.96 | 5,095,556.00 | Logistic | 1.00 | 1.10 | HGC-27 |
| KATOIII | 0.44 | 4,904,316.00 | Logistic | 0.97 | 4.70 | KATOIII |
| MKN-45 | 0.63 | 9,237,232.00 | Logistic | 0.99 | 3.00 | MKN-45 |
| NCI-N87 | 0.45 | 38,306,195.00 | Logistic | 0.99 | 2.50 | NCI-N87 |
| NUGC-4 | 0.92 | 6,899,073.00 | Logistic | 0.98 | 1.10 | NUGC-4 |
| SNU-601 | 0.51 | 7,374,909.00 | Logistic | 0.99 | 2.30 | SNU-601 |
| SNU-638 | 0.71 | 4,791,667.00 | Logistic | 0.97 | 1.20 | SNU-638 |
| SNU-668 | 0.45 | 5,048,178.00 | Logistic | 0.98 | 1.90 | SNU-668 |


| cloneID | K | r | CL |
|---|---|---|---|
| 102944 | 4,690,105 | 0.49 | SNU-668 |
| 102945 | 4,674,250 | 0.38 | SNU-668 |
| 102948 | 3,450,777 | 0.53 | SNU-668 |
| 102950 | 4,331,258 | 0.66 | SNU-668 |
| 102951 | 5,488,043 | 0.53 | SNU-668 |
| 102952 | 4,379,298 | NA | SNU-668 |
| 102954 | 5,046,408 | 0.62 | SNU-668 |
| 102955 | 4,773,230 | 0.52 | SNU-668 |
| 106394 | 3,166,668 | 0.56 | KATOIII |
| 106396 | 3,117,464 | NA | KATOIII |
| 106399 | 3,274,342 | 0.53 | KATOIII |
| 106404 | 3,346,502 | 0.58 | KATOIII |
| 112380 | 7,448,893 | 0.32 | SNU-601 |
| 112382 | 8,148,176 | 0.34 | SNU-601 |
| 112387 | 7,231,439 | NA | SNU-601 |
| 112389 | 6,627,640 | 0.32 | SNU-601 |
| 112392 | 6,798,104 | 0.34 | SNU-601 |
| 112399 | 6,312,316 | NA | SNU-601 |
| 112402 | 8,050,488 | 0.38 | SNU-601 |
| 112404 | 7,081,546 | 0.31 | SNU-601 |
| 112408 | 8,255,791 | 0.37 | SNU-601 |
| 112410 | 7,903,615 | NA | SNU-601 |
| 112413 | 7,196,330 | 0.32 | SNU-601 |
| 114525 | 11,055,814 | 0.65 | MKN-45 |
| 114530 | 10,785,452 | 0.68 | MKN-45 |
| 119963 | 18,156,049 | NA | NCI-N87 |
| 119965 | 22,002,556 | 0.62 | NCI-N87 |
| 119967 | 20,354,070 | 0.57 | NCI-N87 |
| 119968 | 18,548,305 | 0.67 | NCI-N87 |
| 122360 | 5,156,522 | 0.80 | SNU-638 |
| 122361 | 4,331,370 | NA | SNU-638 |
| 122363 | 4,759,109 | 0.80 | SNU-638 |
| 125616 | 9,123,026 | 0.78 | NUGC-4 |
| 125618 | 9,655,395 | 0.69 | NUGC-4 |
| 125619 | 8,845,460 | 0.82 | NUGC-4 |
| 129343 | 9,429,235 | 0.85 | HGC-27 |
| 129344 | 8,996,528 | 0.82 | HGC-27 |
| 129345 | 8,856,699 | 0.76 | HGC-27 |
| 129346 | 8,616,362 | 0.78 | HGC-27 |




References
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef]
- Ben-David, U.; Amon, A. Context is everything: Aneuploidy in cancer. Nat. Rev. Genet. 2020, 21, 44–62. [Google Scholar] [CrossRef]
- Zhu, S.; Qing, T.; Zheng, Y.; Jin, L.; Shi, L. Advances in single-cell RNA sequencing and its applications in cancer research. Oncotarget 2017, 8, 53763–53779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, P.; Lupski, J.R.; Rosenberg, S.M.; Ira, G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009, 10, 551–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Wang, D.D.; Yang, M.; Chen, D.; Pang, L.; Guo, S.; Cai, J.; Wery, J.P.; Li, L.; Li, H.Q.; et al. Comprehensive characterization of chemotherapeutic efficacy on metastases in the established gastric neuroendocrine cancer patient derived xenograft model. Oncotarget 2015, 6, 15639–15651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, X.; Gong, J.; Zhang, Q.; Gao, J.; Cao, Y.; Wang, D.D.; Lin, P.P.; Shen, L. Aneuploidy of chromosome 8 in circulating tumor cells correlates with prognosis in patients with advanced gastric cancer. Chin. J. Cancer Res. 2016, 28, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Fang, J.Y.; Xu, J. Gastric cancer and gene copy number variation: Emerging cancer drivers for targeted therapy. Oncogene 2016, 35, 1475–1482. [Google Scholar] [CrossRef]
- Giam, M.; Rancati, G. Aneuploidy and chromosomal instability in cancer: A jackpot to chaos. Cell Div. 2015, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689. [Google Scholar] [CrossRef] [Green Version]
- Shlien, A.; Malkin, D. Copy number variations and cancer. Genome Med. 2009, 1, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, A.; Nguyen, T.H.M.; Moka, S.B.; Ellis, J.J.; Grady, J.P.; Oey, H.; Cristino, A.S.; Khanna, K.K.; Kroese, D.P.; Krause, L.; et al. Chromosome arm aneuploidies shape tumour evolution and drug response. Nat. Commun. 2020, 11, 449. [Google Scholar] [CrossRef] [Green Version]
- Baslan, T.; Kendall, J.; Volyanskyy, K.; McNamara, K.; Cox, H.; D’Italia, S.; Ambrosio, F.; Riggs, M.; Rodgers, L.; Leotta, A.; et al. Novel insights into breast cancer copy number genetic heterogeneity revealed by single-cell genome sequencing. eLife 2020, 9, e51480. [Google Scholar] [CrossRef]
- Wu, Y.; Grabsch, H.; Ivanova, T.; Tan, I.B.; Murray, J.; Ooi, C.H.; Wright, A.I.; West, N.P.; Hutchins, G.G.A.; Wu, J.; et al. Comprehensive genomic meta-analysis identifies intra-tumoural stroma as a predictor of survival in patients with gastric cancer. Gut 2013, 62, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Jiang, Y.; Li, G.; Fisher, G.A.; Li, R. Natural killer cell and stroma abundance are independently prognostic and predict gastric cancer chemotherapy benefit. JCI Insight 2020, 5, e136570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamlouk, S.; Childs, L.H.; Aust, D.; Heim, D.; Melching, F.; Oliveira, C.; Wolf, T.; Durek, P.; Schumacher, D.; Bläker, H.; et al. DNA copy number changes define spatial patterns of heterogeneity in colorectal cancer. Nat. Commun. 2017, 8, 14093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Bradford, W.D.; Seidel, C.W.; Li, R. Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature 2012, 482, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Lande, R.; Engen, S.; Sæther, B.E. An evolutionary maximum principle for density-dependent population dynamics in a fluctuating environment. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 1511–1518. [Google Scholar] [CrossRef] [Green Version]
- Caswell, H. Life History Theory and the Equilibrium Status of Populations. Am. Nat. 1982, 120, 317–339. [Google Scholar] [CrossRef]
- Li, T.; Liu, J.; Feng, J.; Liu, Z.; Liu, S.; Zhang, M.; Zhang, Y.; Hou, Y.; Wu, D.; Li, C.; et al. Variation in the life history strategy underlies functional diversity of tumors. Natl. Sci. Rev. 2021, 8, nwaa124. [Google Scholar] [CrossRef]
- Aktipis, C.A.; Boddy, A.M.; Gatenby, R.A.; Brown, J.S.; Maley, C.C. Life history trade-offs in cancer evolution. Nat. Rev. Cancer 2013, 13, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andor, N.; Lau, B.T.; Catalanotti, C.; Sathe, A.; Kubit, M.; Chen, J.; Blaj, C.; Cherry, A.; Bangs, C.D.; Grimes, S.M.; et al. Joint single cell DNA-seq and RNA-seq of gastric cancer cell lines reveals rules of in vitro evolution. NAR Genom. Bioinform. 2020, 2, lqaa016. [Google Scholar] [CrossRef] [Green Version]
- Minussi, D.C.; Nicholson, M.D.; Ye, H.; Davis, A.; Wang, K.; Baker, T.; Tarabichi, M.; Sei, E.; Du, H.; Rabbani, M.; et al. Breast tumours maintain a reservoir of subclonal diversity during expansion. Nature 2021, 592, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Tsoularis, A.; Wallace, J. Analysis of logistic growth models. Math. Biosci. 2002, 179, 21–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergman, R.N.; Cobelli, C. Minimal modeling, partition analysis, and the estimation of insulin sensitivity. Fed. Proc. 1980, 39, 110–115. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Andor, N.; Simonds, E.F.; Czerwinski, D.K.; Chen, J.; Grimes, S.M.; Wood-Bouwens, C.; Zheng, G.X.Y.; Kubit, M.A.; Greer, S.; Weiss, W.A.; et al. Single-cell RNA-Seq of lymphoma cancers reveals malignant B cell types and co-expression of T cell immune checkpoints. Blood 2018. [Google Scholar] [CrossRef] [Green Version]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef] [Green Version]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Kinsler, G.; Geiler-Samerotte, K.; Petrov, D.A. Fitness variation across subtle environmental perturbations reveals local modularity and global pleiotropy of adaptation. eLife 2020, 9, e61271. [Google Scholar] [CrossRef]
- Gerlee, P.; Anderson, A.R.A. The evolution of carrying capacity in constrained and expanding tumour cell populations. Phys. Biol. 2015, 12, 056001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubry, M.; de Tayrac, M.; Etcheverry, A.; Clavreul, A.; Saikali, S.; Menei, P.; Mosser, J. ‘From the core to beyond the margin’: A genomic picture of glioblastoma intratumor heterogeneity. Oncotarget 2015, 6, 12094–12109. [Google Scholar] [CrossRef] [Green Version]
- Bastola, S.; Pavlyukov, M.S.; Yamashita, D.; Ghosh, S.; Cho, H.; Kagaya, N.; Zhang, Z.; Minata, M.; Lee, Y.; Sadahiro, H.; et al. Glioma-initiating cells at tumor edge gain signals from tumor core cells to promote their malignancy. Nat. Commun. 2020, 11, 4660. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Silva, A.S.; Gillies, R.J.; Frieden, B.R. Adaptive therapy. Cancer Res. 2009, 69, 4894–4903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, A.; Barkley, D.; França, G.S.; Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 2021, 596, 211–220. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Brown, J.S. The Evolution and Ecology of Resistance in Cancer Therapy. Cold Spring Harb. Perspect. Med. 2020, 10, a040972. [Google Scholar] [CrossRef]
- Liu, Y.; Mi, Y.; Mueller, T.; Kreibich, S.; Williams, E.G.; Van Drogen, A.; Borel, C.; Frank, M.; Germain, P.L.; Bludau, I.; et al. Multi-omic measurements of heterogeneity in HeLa cells across laboratories. Nat. Biotechnol. 2019, 37, 314–322. [Google Scholar] [CrossRef]
- Stringer, C.; Wang, T.; Michaelos, M.; Pachitariu, M. Cellpose: A generalist algorithm for cellular segmentation. Nat. Methods 2021, 18, 100–106. [Google Scholar] [CrossRef]
- Ronneberger, O.; Fischer, P.; Brox, T. U-Net: Convolutional Networks for Biomedical Image Segmentation. arXiv 2015, arXiv:1505.04597. [Google Scholar]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [Green Version]
- Kimmel, G.J.; Beck, R.J.; Yu, X.; Veith, T.; Bakhoum, S.; Altrock, P.M.; Andor, N. Intra-tumor heterogeneity, turnover rate and karyotype space shape susceptibility to missegregation-induced extinction. PLoS Comput. Biol. 2023, 19, e1010815. [Google Scholar] [CrossRef]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [Green Version]
- Gerlee, P. The Model Muddle: In Search of Tumor Growth Laws. Cancer Res. 2013, 73, 2407–2411. [Google Scholar] [CrossRef] [Green Version]
- Murphy, H.; Jaafari, H.; Dobrovolny, H.M. Differences in predictions of ODE models of tumor growth: A cautionary example. BMC Cancer 2016, 16, 163. [Google Scholar] [CrossRef] [Green Version]
- Heuser, L.; Spratt, J.S.; Polk, H.C., Jr. Growth rates of primary breast cancers. Cancer 1979, 43, 1888–1894. [Google Scholar] [CrossRef]
- Voulgarelis, D.; Bulusu, K.C.; Yates, J.W.T. Comparison of classical tumour growth models for patient derived and cell-line derived xenografts using the nonlinear mixed-effects framework. J. Biol. Dyn. 2022, 16, 160–185. [Google Scholar] [CrossRef]
- Hall, B.G.; Acar, H.; Nandipati, A.; Barlow, M. Growth Rates Made Easy. Mol. Biol. Evol. 2014, 31, 232–238. [Google Scholar] [CrossRef] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veith, T.; Schultz, A.; Alahmari, S.; Beck, R.; Johnson, J.; Andor, N. Mathematical Modeling of Clonal Interference by Density-Dependent Selection in Heterogeneous Cancer Cell Lines. Cells 2023, 12, 1849. https://doi.org/10.3390/cells12141849
Veith T, Schultz A, Alahmari S, Beck R, Johnson J, Andor N. Mathematical Modeling of Clonal Interference by Density-Dependent Selection in Heterogeneous Cancer Cell Lines. Cells. 2023; 12(14):1849. https://doi.org/10.3390/cells12141849
Chicago/Turabian StyleVeith, Thomas, Andrew Schultz, Saeed Alahmari, Richard Beck, Joseph Johnson, and Noemi Andor. 2023. "Mathematical Modeling of Clonal Interference by Density-Dependent Selection in Heterogeneous Cancer Cell Lines" Cells 12, no. 14: 1849. https://doi.org/10.3390/cells12141849