
Complement Dysregulation in Obese Versus Nonobese Polycystic Ovary Syndrome Patients

Abstract
:1. Introduction
2. Materials and Methods
2.1. Obese PCOS and Controls Cohort [10]
2.2. Nonobese PCOS and Control Cohort [9]
2.3. Sample Analysis
2.4. Statistics
3. Results
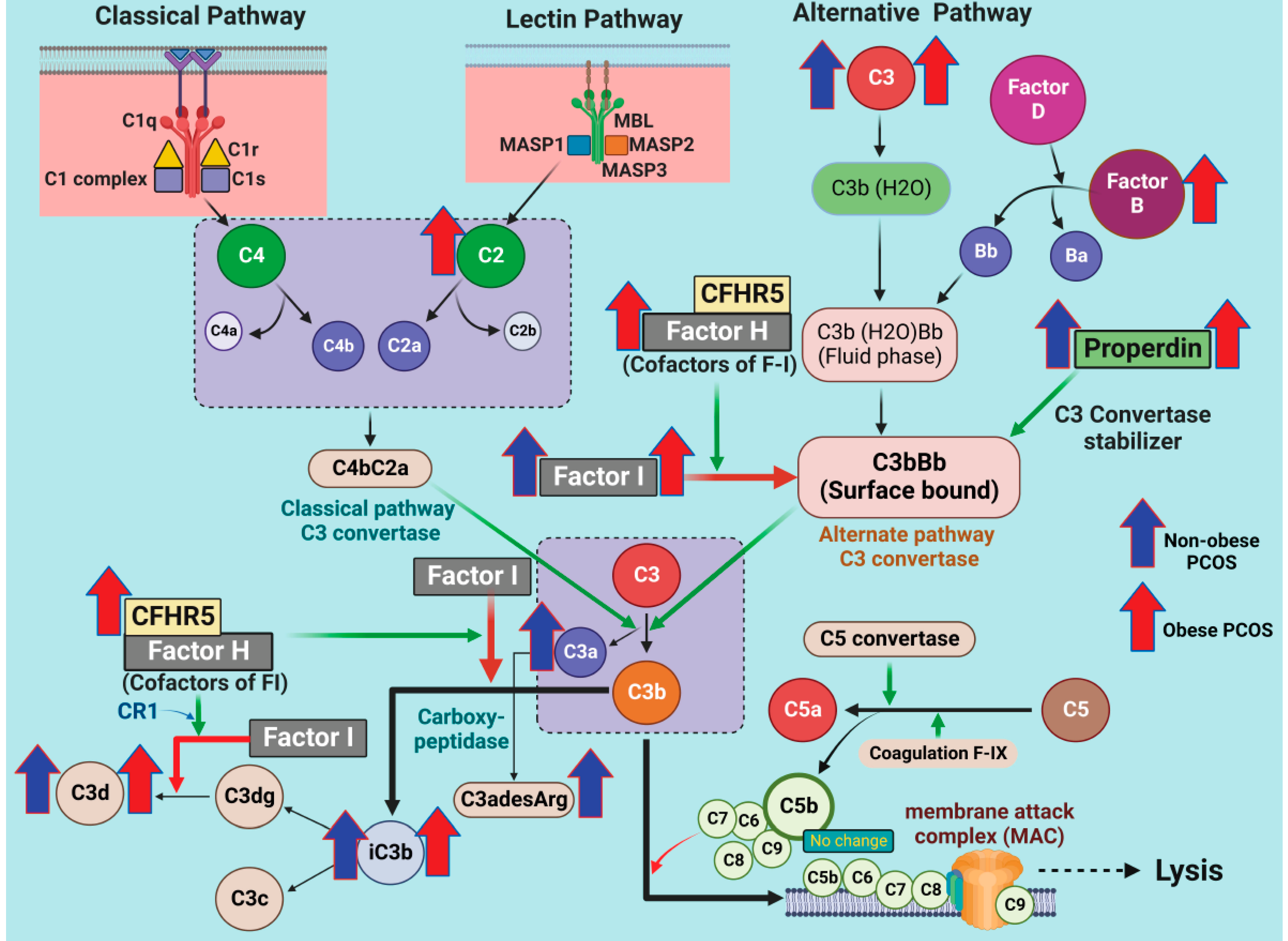
3.1. Alternative Pathway (AP) Proteins of Complement Activation in PCOS
3.2. Lectin Pathway (LP) Proteins of Complement Activation in PCOS
3.3. Classical Pathway (CP) Proteins of Complement Activation in PCOS
3.4. Correlations of Complement Activation-Related Proteins with Hyperandrogenemia and BMI
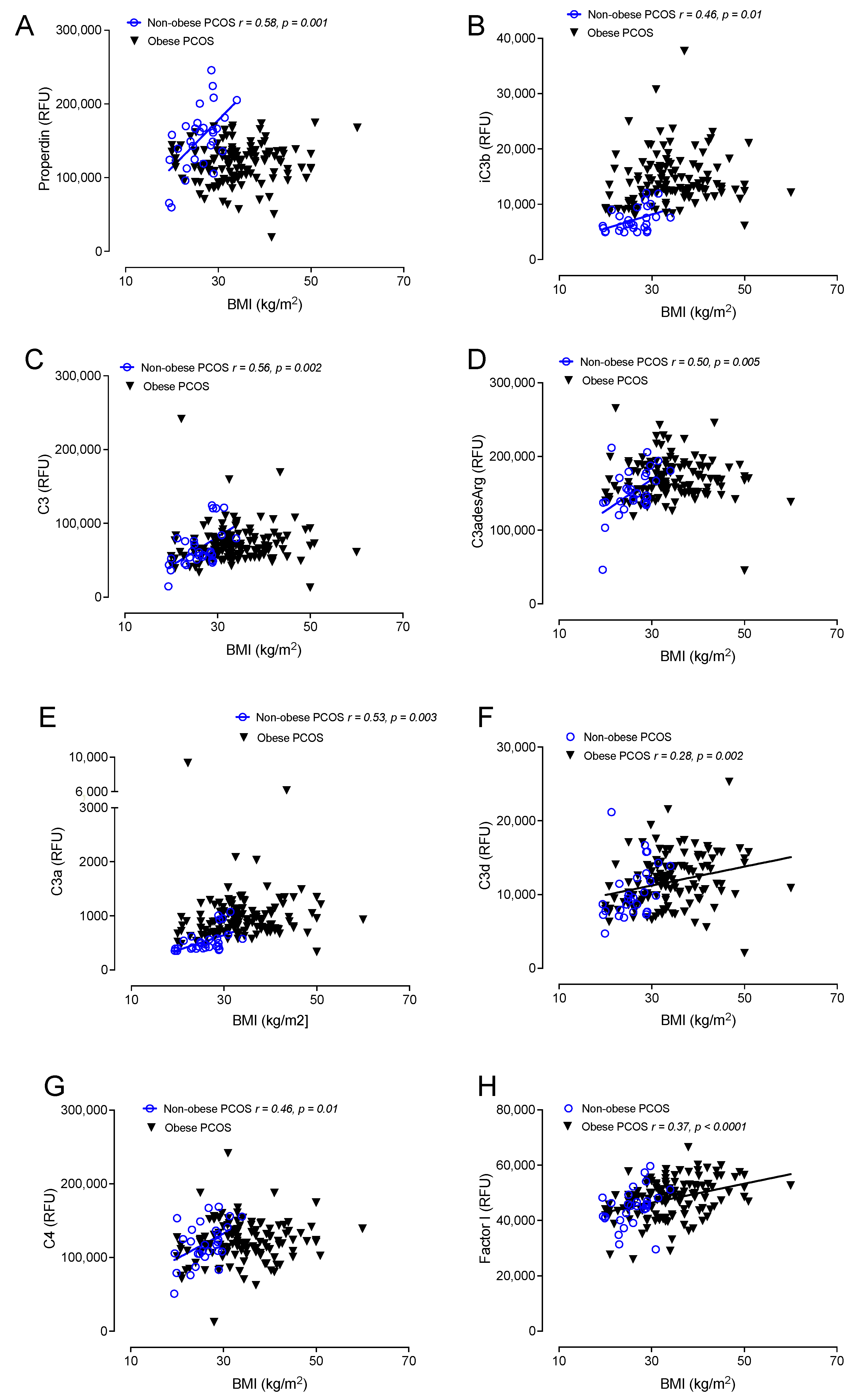
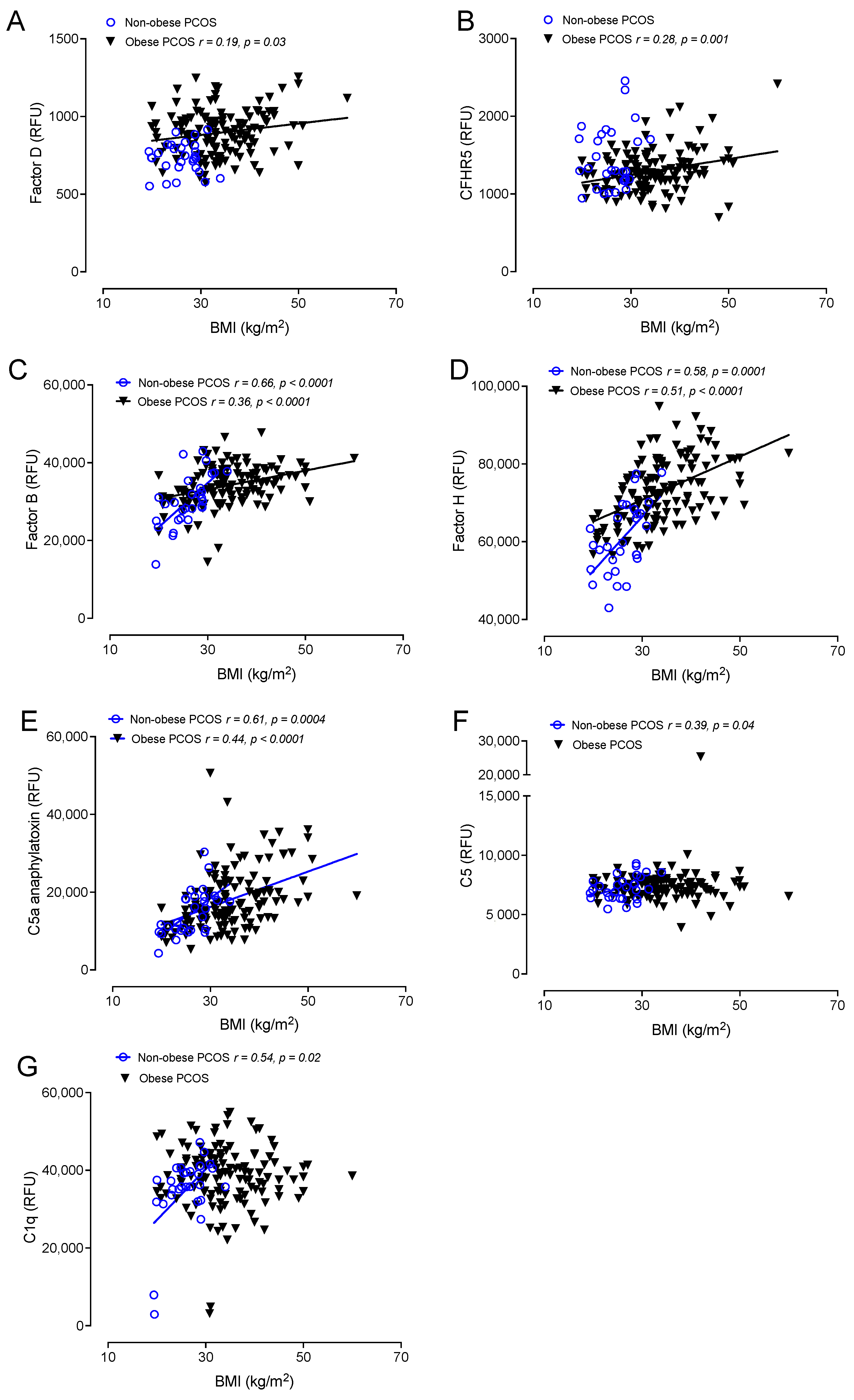
3.5. Correlations of Complement Activation in Obese and Nonobese PCOS with BMI
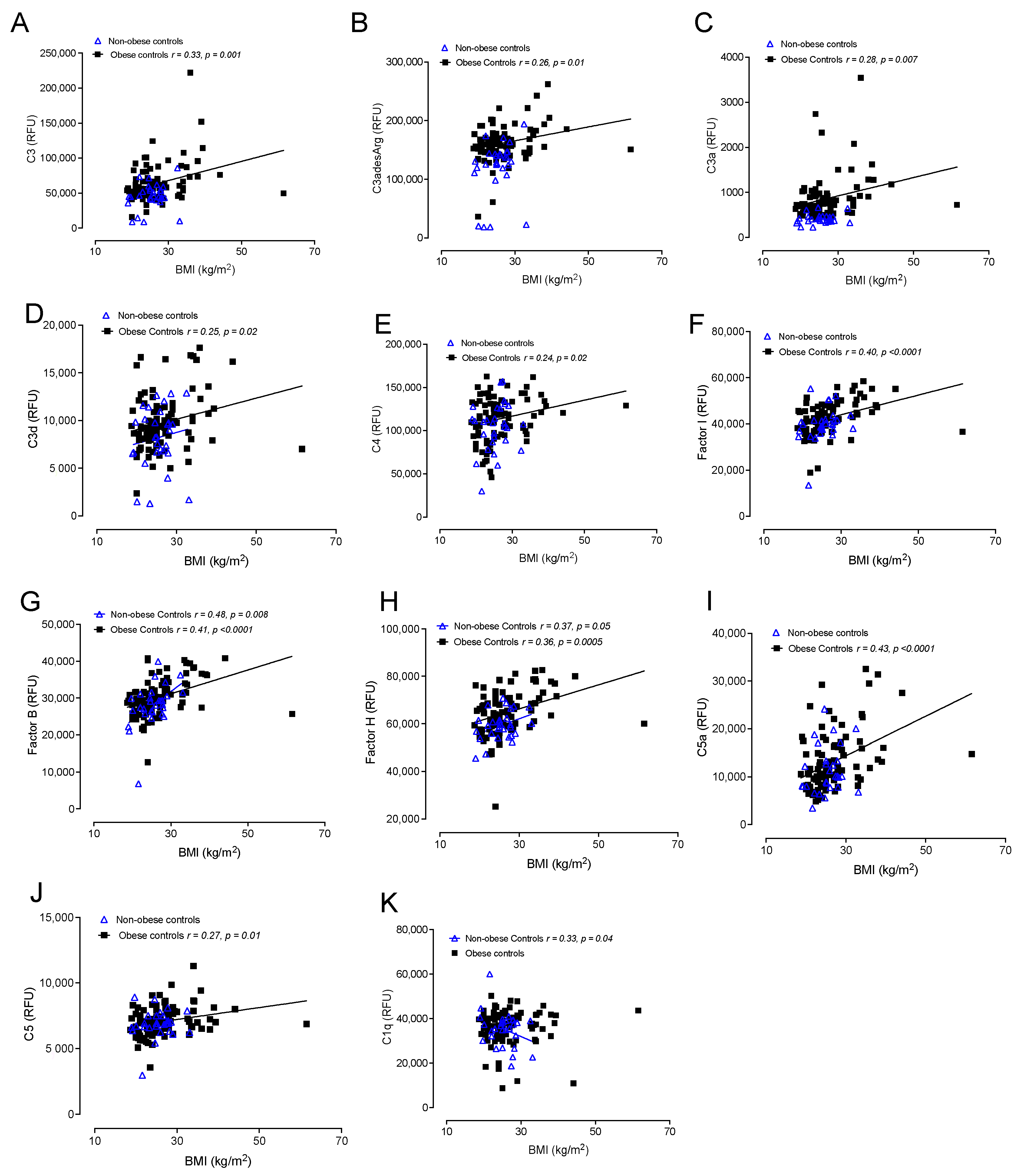
3.6. Correlations of Complement Activation in Obese and Nonobese Control Subjects with BMI
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sathyapalan, T.; Atkin, S.L. Recent advances in cardiovascular aspects of polycystic ovary syndrome. Eur. J. Endocrinol./Eur. Fed. Endocr. Soc. 2012, 166, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Carter, A.M. Complement activation: An emerging player in the pathogenesis of cardiovascular disease. Scientifica 2012, 2012, 402783. [Google Scholar] [PubMed] [Green Version]
- Sathyapalan, T.; Shepherd, J.; Coady, A.M.; Kilpatrick, E.S.; Atkin, S.L. Atorvastatin reduces malondialdehyde concentrations in patients with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2012, 97, 3951–3955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannigida, D.M.; Nayak, B.S.; Vijayaraghavan, R. Insulin resistance and oxidative marker in women with PCOS. Arch. Physiol. Biochem. 2020, 126, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Niinuma, S.A.; Lubbad, L.; Lubbad, W.; Moin, A.S.M.; Butler, A.E. The Role of Heat Shock Proteins in the Pathogenesis of Polycystic Ovarian Syndrome: A Review of the Literature. Int. J. Mol. Sci. 2023, 24, 1838. [Google Scholar] [CrossRef] [PubMed]
- Moin, A.S.M.; Sathyapalan, T.; Butler, A.E.; Atkin, S.L. Coagulation factor dysregulation in polycystic ovary syndrome is an epiphenomenon of obesity. Clin. Endocrinol. 2023, 98, 796–802. [Google Scholar] [CrossRef]
- Lewis, R.D.; Narayanaswamy, A.K.; Farewell, D.; Rees, D.A. Complement activation in polycystic ovary syndrome occurs in the postprandial and fasted state and is influenced by obesity and insulin sensitivity. Clin. Endocrinol. 2021, 94, 74–84. [Google Scholar] [CrossRef]
- Yang, S.; Li, Q.; Song, Y.; Tian, B.; Cheng, Q.; Qing, H.; Zhong, L.; Xia, W. Serum complement C3 has a stronger association with insulin resistance than high-sensitivity C-reactive protein in women with polycystic ovary syndrome. Fertil. Steril. 2011, 95, 1749–1753. [Google Scholar]
- Moin, A.S.M.; Sathyapalan, T.; Butler, A.E.; Atkin, S.L. Classical and alternate complement factor overexpression in non-obese weight matched women with polycystic ovary syndrome does not correlate with vitamin D. Front. Endocrinol. 2022, 13, 935750. [Google Scholar] [CrossRef]
- Butler, A.E.; Moin, A.S.M.; Sathyapalan, T.; Atkin, S.L. Components of the Complement Cascade Differ in Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2022, 23, 12232. [Google Scholar] [CrossRef]
- Dehdashtihaghighat, S.; Mehdizadehkashi, A.; Arbabi, A.; Pishgahroudsari, M.; Chaichian, S. Assessment of C-reactive protein and C3 as inflammatory markers of insulin resistance in women with polycystic ovary syndrome: A case-control study. J. Reprod. Infertil. 2013, 14, 197. [Google Scholar]
- Ramanjaneya, M.; Abdalhakam, I.; Bettahi, I.; Bensila, M.; Jerobin, J.; Aye, M.M.; Alkasem, M.; Sathyapalan, T.; Atkin, S.L.; Abou-Samra, A.-B. Effect of Moderate Aerobic Exercise on Complement Activation Pathways in Polycystic Ovary Syndrome Women. Front. Endocrinol. 2021, 12, 740703. [Google Scholar]
- Carroll, M.C. The complement system in regulation of adaptive immunity. Nat. Immunol. 2004, 5, 981–986. [Google Scholar] [CrossRef]
- Engstrom, G.; Hedblad, B.; Eriksson, K.-F.; Janzon, L.; Lindgärde, F. Complement C 3 Is a Risk Factor for the Development of Diabetes: A Population-Based Cohort Study. Diabetes 2005, 54, 570–575. [Google Scholar]
- Bjerre, M.; Hansen, T.; Flyvbjerg, A. Complement activation and cardiovascular disease. Horm. Metab. Res. 2008, 40, 626–634. [Google Scholar] [CrossRef]
- Sathyapalan, T.; Al-Qaissi, A.; Kilpatrick, E.S.; Dargham, S.R.; Atkin, S.L. Anti-Mullerian hormone measurement for the diagnosis of polycystic ovary syndrome. Clin. Endocrinol. 2017, 88, 258–262. [Google Scholar] [CrossRef]
- Sathyapalan, T.; Al-Qaissi, A.; Kilpatrick, E.S.; Dargham, S.R.; Adaway, J.; Keevil, B.; Atkin, S.L. Salivary testosterone measurement in women with and without polycystic ovary syndrome. Sci. Rep. 2017, 7, 3589. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, T.K.; Allgar, V.; Dargham, S.R.; Kilpatrick, E.; Sathyapalan, T.; Maguiness, S.; Mokhtar Rudin, H.R.; Abdul Ghani, N.M.; Latiff, A.; Atkin, S.L. Association of Vitamin D Metabolites With Embryo Development and Fertilization in Women With and Without PCOS Undergoing Subfertility Treatment. Front. Endocrinol. 2019, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Obesity and Overweight: World Health Organisation. Available online: https://www.who.int/health-topics/obesity#tab=tab_1 (accessed on 9 June 2021).
- Kahal, H.; Halama, A.; Aburima, A.; Bhagwat, A.M.; Butler, A.E.; Grauman, J.; Suhre, K.; Sathyapalan, T.; Atkin, S.L. Effect of induced hypoglycemia on inflammation and oxidative stress in type 2 diabetes and control subjects. Sci. Rep. 2020, 10, 4750. [Google Scholar] [CrossRef] [Green Version]
- Kraemer, S.; Vaught, J.D.; Bock, C.; Gold, L.; Katilius, E.; Keeney, T.R.; Kim, N.; Saccomano, N.A.; Wilcox, S.K.; Zichi, D.; et al. From SOMAmer-based biomarker discovery to diagnostic and clinical applications: A SOMAmer-based, streamlined multiplex proteomic assay. PLoS ONE 2011, 6, e26332. [Google Scholar] [CrossRef] [Green Version]
- Cunnion, K.M.; Hair, P.S.; Buescher, E.S. Cleavage of complement C3b to iC3b on the surface of Staphylococcus aureus is mediated by serum complement factor I. Infect. Immun. 2004, 72, 2858–2863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verschoor, A.; Karsten, C.M.; Broadley, S.P.; Laumonnier, Y.; Köhl, J. Old dogs-new tricks: Immunoregulatory properties of C3 and C5 cleavage fragments. Immunol. Rev. 2016, 274, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Fan, X.; Yao, J.; Tomlinson, S.; Yuan, G.; He, S. The role of complement in nonalcoholic fatty liver disease. Front. Immunol. 2022, 13, 1017467. [Google Scholar] [CrossRef] [PubMed]
- Kazatchkine, M.D.; Fearon, D.T.; Austen, K.F. Human alternative complement pathway: Membrane-associated sialic acid regulates the competition between B and beta1 H for cell-bound C3b. J. Immunol. 1979, 122, 75–81. [Google Scholar] [PubMed]
- Lambris, J.D.; Lao, Z.; Oglesby, T.J.; Atkinson, J.P.; Hack, C.E.; Becherer, J.D. Dissection of CR1, factor H, membrane cofactor protein, and factor B binding and functional sites in the third complement component. J. Immunol. 1996, 156, 4821–4832. [Google Scholar]
- Brodbeck, W.G.; Kuttner-Kondo, L.; Mold, C.; Medof, M.E. Structure/function studies of human decay-accelerating factor. Immunology 2000, 101, 104–111. [Google Scholar] [CrossRef]
- McRae, J.L.; Duthy, T.G.; Griggs, K.M.; Ormsby, R.J.; Cowan, P.J.; Cromer, B.A.; McKinstry, W.J.; Parker, M.W.; Murphy, B.F.; Gordon, D.L. Human factor H-related protein 5 has cofactor activity, inhibits C3 convertase activity, binds heparin and C-reactive protein, and associates with lipoprotein. J. Immunol. 2005, 174, 6250–6256. [Google Scholar] [CrossRef]
- Amara, U.; Flierl, M.A.; Rittirsch, D.; Klos, A.; Chen, H.; Acker, B.; Brückner, U.B.; Nilsson, B.; Gebhard, F.; Lambris, J.D.; et al. Molecular intercommunication between the complement and coagulation systems. J. Immunol. 2010, 185, 5628–5636. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Pang, B.; Ma, Z.; Yi, H. Immunophenotypic Profiles in Polycystic Ovary Syndrome. Mediat. Inflamm. 2020, 2020, 5894768. [Google Scholar] [CrossRef] [Green Version]
- Kyrou, I.; Karteris, E.; Robbins, T.; Chatha, K.; Drenos, F.; Randeva, H.S. Polycystic ovary syndrome (PCOS) and COVID-19: An overlooked female patient population at potentially higher risk during the COVID-19 pandemic. BMC Med. 2020, 18, 220. [Google Scholar]
- Moin, A.S.M.; Sathyapalan, T.; Atkin, S.L.; Butler, A.E. Renin-Angiotensin System Overactivation in Polycystic Ovary Syndrome, a Risk for SARS-CoV-2 Infection? Metab. Open 2020, 7, 100052. [Google Scholar] [CrossRef]
- Basile, U.; Bruno, C.; Napodano, C.; Vergani, E.; Gulli, F.; Piunno, G.; Pocino, K.; Stefanile, A.; Mancini, A. Evaluation of immunoglobulins subclasses and free-light chains in non-obese patients with polycystic ovary syndrome and correlations with hormonal and metabolic parameters: Preliminary data. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 4198–4204. [Google Scholar] [CrossRef]
- Neven, A.C.H.; Laven, J.; Teede, H.J.; Boyle, J.A. A Summary on Polycystic Ovary Syndrome: Diagnostic Criteria, Prevalence, Clinical Manifestations, and Management According to the Latest International Guidelines. Semin. Reprod. Med. 2018, 36, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Barber, T.M.; Golding, S.J.; Alvey, C.; Wass, J.A.; Karpe, F.; Franks, S.; McCarthy, M.I. Global adiposity rather than abnormal regional fat distribution characterizes women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 999–1004. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline Demographics | PCOS Obese (n = 137) | Control Lean (n = 97) | p Value | PCOS Lean (n = 24) | Control Lean (n = 24) | p Value |
|---|---|---|---|---|---|---|
| Mean (SD) | Mean (SD) | Mean (SD) | Mean (SD) | |||
| Age (years) | 29.1 ± 6.1 | 29.6 ± 6.5 | 0.09 | 31 ± 6.4 | 32.5 ± 4.1 | 0.09 |
| BMI (kg/m²) | 34.1 ± 7.5 | 26.7 ± 6.6 | <0.0001 | 25.9 ± 1.8 | 24.8 ± 1.1 | 0.44 |
| Insulin (IU/mL) | 10.2 ± 6.1 | 6.2 ± 3.2 | 0.001 | 8.1 ± 4.7 | 7.7 ± 4.0 | 0.66 |
| HOMA-IR | 3.8 ± 0.6 | 1.6 ± 0.2 | <0.005 | 1.9 ± 1.6 | 1.8 ± 1.0 | 0.39 |
| Testosterone (nmol/L) | 1.6 ± 1.0 | 1.05 ± 0.48 | <0.0001 | 1.4 ± 0.8 | 0.7 ± 0.4 | 0.005 |
| SHBG (nmol/L) | 42.5 ± 39.6 | 77.5 ± 78.4 | 0.0003 | 71.7 ± 62.2 | 104 ± 80 | 0.03 |
| Free androgen index (FAI) | 4.5 ± 3.9 | 2.1 ± 1.4 | <0.0001 | 4.1 ± 2.9 ± 4.08 | 1.3 ± 0.5 | <0.0001 |
| CRP (mg/L) | 4.4 ± 4.2 | 2.4 ± 3.9 | 0.0008 | 2.8 ± 2.6 | 2.3 ± 2.34 | 0.38 |
| AMH (ng/mL) | 40 ± 31 | 18 ± 18 | <0.0001 | 57 ± 14 | 24 ± 13 | <0.0001 |
| LEAN | OBESE | ||||||
|---|---|---|---|---|---|---|---|
| PCOS | Control | p Value Nonobese PCOS vs. Controls | PCOS | Control | p Value Obese PCOS vs. Controls | p Value Nonobese PCOS vs. Obese PCOS | |
| Properdin | 152,592 (42,743) | 117,488 (50,041) | 0.006 | 119,125 (26,794) | 102,491 (26,374) | 0.000003 | 0.00000001 * |
| C3b | 50,982 (28,296) | 46,250 (39,450) | 0.6 | 110,730 (68,743) | 105,010 (59972) | 0.5 | 0.000001 * |
| iC3b | 7148 (2127) | 5991 (1425) | 0.02 | 14,476 (4563) | 11,445 (4188) | 0.0000003 | 1 × 10 13 * |
| C3 | 65,878 (26,872) | 45,742 (18,189) | 0.002 | 71,028 (25,536) | 63,896 (26,822) | 0.04 | 0.33 |
| C3adesArg | 152,050 (32,483) | 121,110 (45,753) | 0.004 | 168,929 (28,060) | 163,048 (30,262) | 0.12 | 0.004 |
| C3a | 534 (204) | 415 (1010 | 0.007 | 1045 (870) | 863 (465) | 0.06 | 0.002 |
| C3d | 10,427 (3675) | 8207 (3261) | 0.02 | 11,905 (3645) | 9816 (2890) | 0.000004 | 0.048 |
| C4 | 119,057 (28,429) | 104,245 (28,069) | 0.05 | 121,193 (26,829) | 114,603 (25,411) | 0.06 | 0.7 |
| C4A | 71,549 (9802) | 73,037 (2258) | 0.43 | 116,060 (4320) | 116,497 (3708) | 0.42 | 1 × 10 14 * |
| Factor I | 44,861 (6786) | 39,960 (7356) | 0.01 | 47,519 (6883) | 42304 (6958) | 0.00000002 | 0.06 |
| Factor D | 733 (99) | 693 (150) | 0.24 | 898 (146) | 933 (314) | 0.24 | 0.00000001 * |
| C2 | 2878 (319) | 2874 (228) | 0.96 | 2694 (386) | 2540 (378) | 0.002 | 0.02 |
| Complement factor H-related 5 | 1454 (399) | 1677 (1416) | 0.42 | 1298 (264) | 1206 (303) | 0.01 | 0.01 |
| Factor B | 30,257 (6541) | 28,172 (5791) | 0.2 | 34,162 (5281) | 30,078 (5054) | 0.00000001 | 0.006 |
| Factor H | 60,898 (9191) | 59,289 (6016) | 0.43 | 72,843 (8954) | 64,750 (9119) | 1 × 10 10 | 1 × 10 10 * |
| C5a | 14,729 (5811) | 11,343 (4953) | 0.02 | 17,983 (7875) | 13,106 (6113) | 0.000001 | 0.04 |
| C5b, 6 Complex | 521 (62) | 502 (62) | 0.27 | 490 (51) | 493 (194) | 0.87 | 0.005 |
| C5 | 7267 (1007) | 6881 (1063) | 0.16 | 7432 (1692) | 7071 (1088) | 0.06 | 0.61 |
| C1q | 35,146 (9269) | 35,294 (7886) | 0.95 | 38,519 (7784) | 37,027 (7612) | 0.14 | 0.04 |
| C1r | 3203 (886) | 3740 (3992) | 0.48 | 3414 (1185) | 3497 (967) | 0.57 | 0.36 |
| C4b | 349 (186) | 335 (202) | 0.78 | 846 (913) | 889 (632) | 0.69 | 0.004 |
| MBL | 12,058 (5835) | 13,322 (7385) | 0.47 | 16,835 (7427) | 16,298 (79,120) | 0.59 | 0.001 * |
| DAF (CD55) | 15,182 (3220) | 13,886 (3339) | 0.14 | 13,958 (2717) | 14,424 (2323) | 0.17 | 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butler, A.E.; Moin, A.S.M.; Sathyapalan, T.; Atkin, S.L. Complement Dysregulation in Obese Versus Nonobese Polycystic Ovary Syndrome Patients. Cells 2023, 12, 2002. https://doi.org/10.3390/cells12152002
Butler AE, Moin ASM, Sathyapalan T, Atkin SL. Complement Dysregulation in Obese Versus Nonobese Polycystic Ovary Syndrome Patients. Cells. 2023; 12(15):2002. https://doi.org/10.3390/cells12152002
Chicago/Turabian StyleButler, Alexandra E., Abu Saleh Md Moin, Thozhukat Sathyapalan, and Stephen L. Atkin. 2023. "Complement Dysregulation in Obese Versus Nonobese Polycystic Ovary Syndrome Patients" Cells 12, no. 15: 2002. https://doi.org/10.3390/cells12152002