Neuronal atg1 Coordinates Autophagy Induction and Physiological Adaptations to Balance mTORC1 Signalling



Abstract
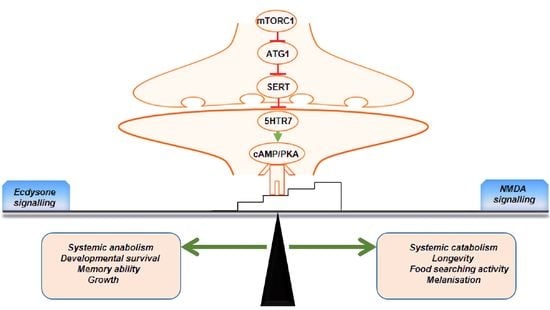
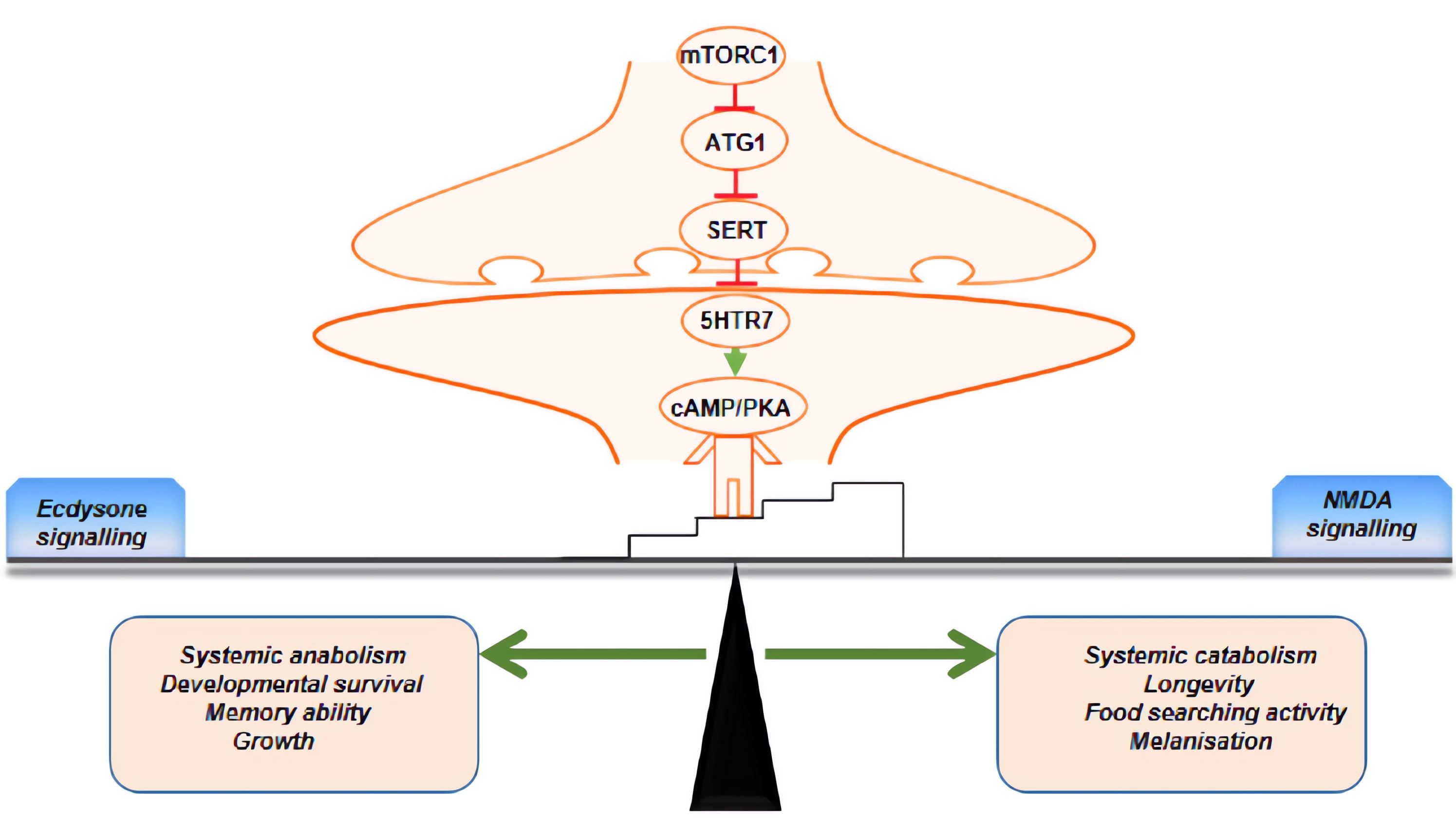
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Flies’ Maintenance, Lifespan Analysis, and Mutants
2.2. Drosophila Drug Treatment
2.3. Drosophila Behavioural Assays
2.4. Zebrafish Maintenance
2.5. Zebrafish Drug Treatment
2.6. Zebrafish Learning and Memory Assays
2.7. Quantitative RT-PCR
2.8. Western Blot Analysis and Plasma Membranes Isolation
2.9. Fecundity Measurements in Drosophila
2.10. Epifluorescence Microscopy
2.11. Measurement of Extracellular Serotonin in the Brain of Drosophila
2.12. cAMP Detection
2.13. Novel Tank Test
2.14. Statistical Analysis
3. Results
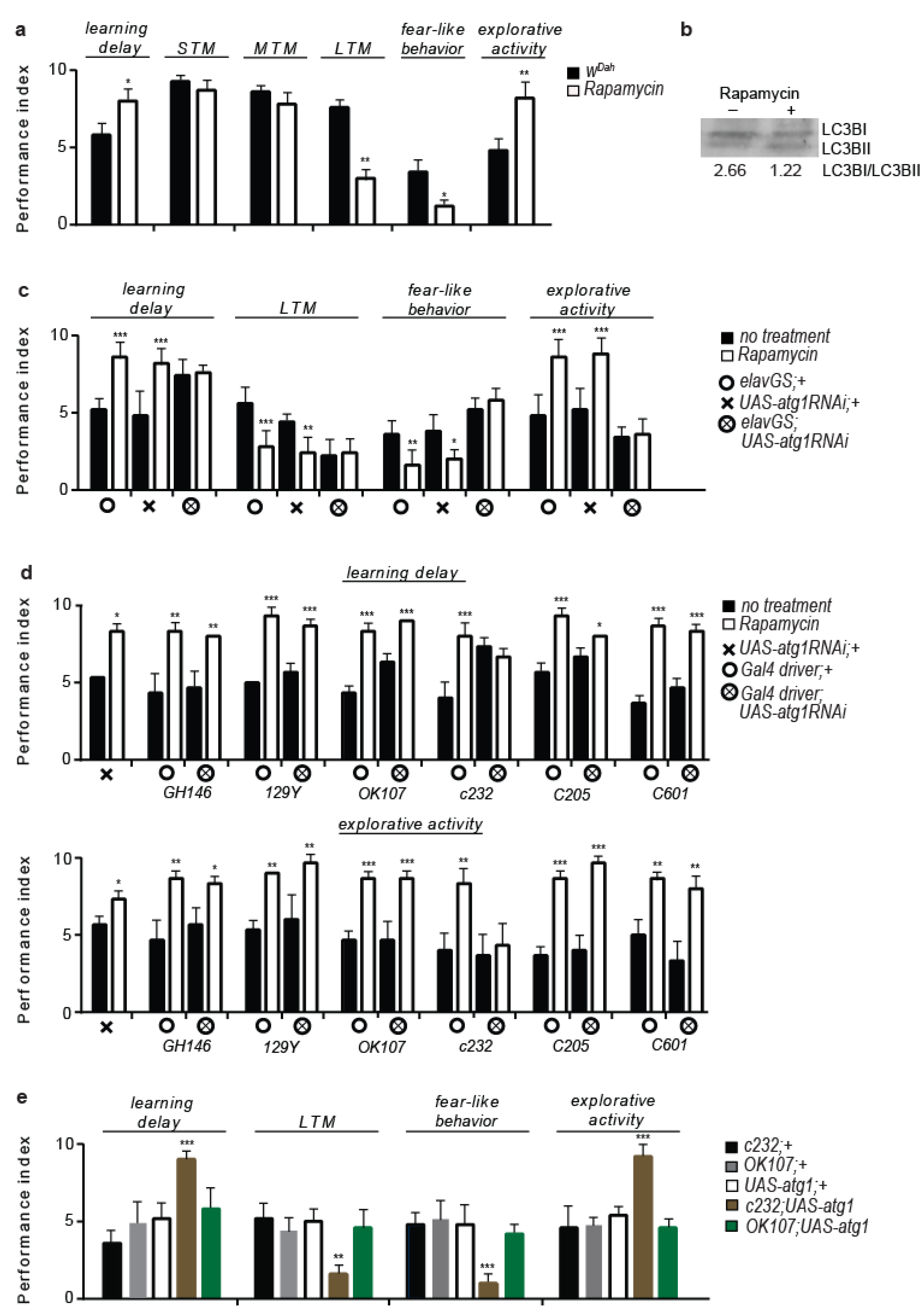
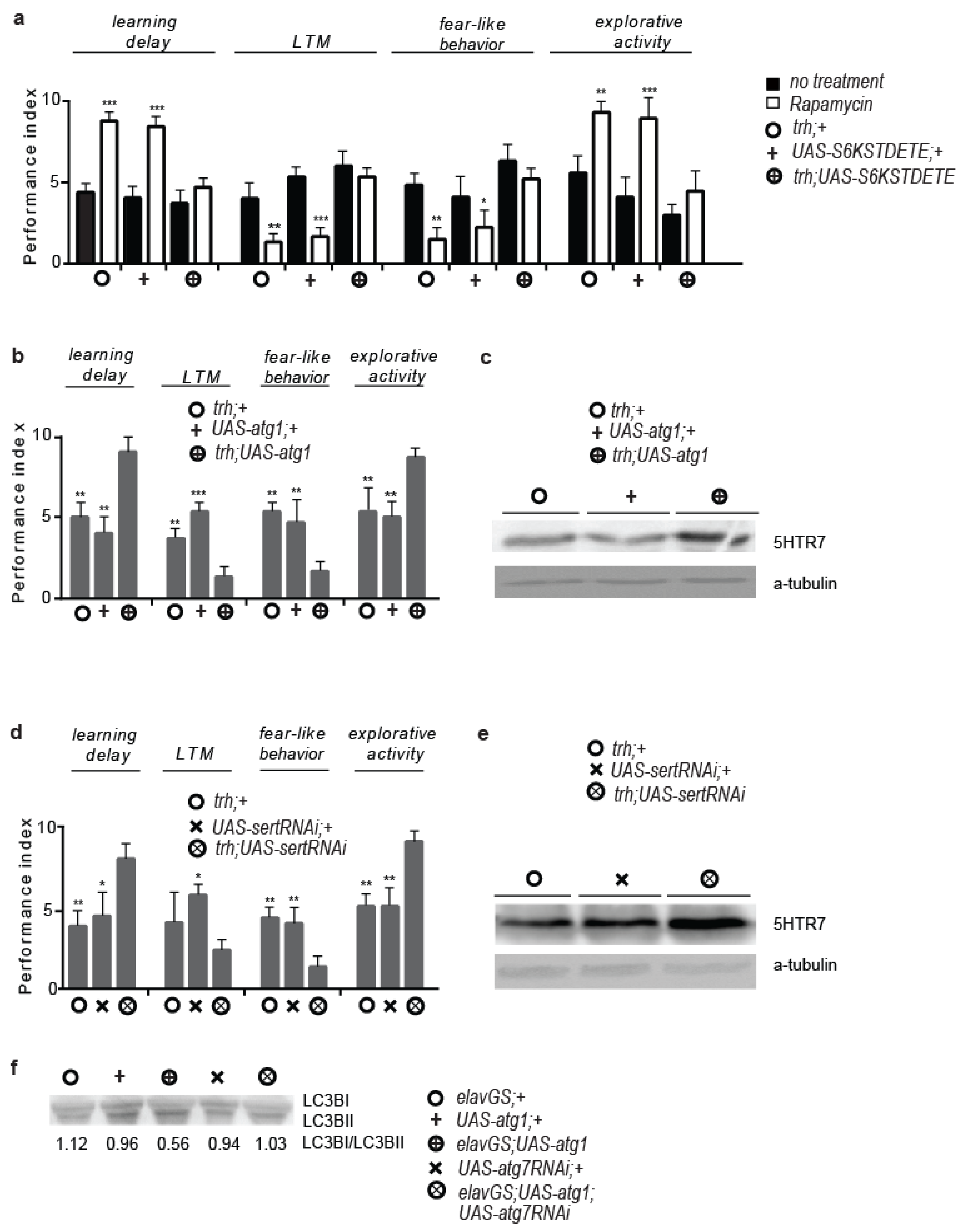
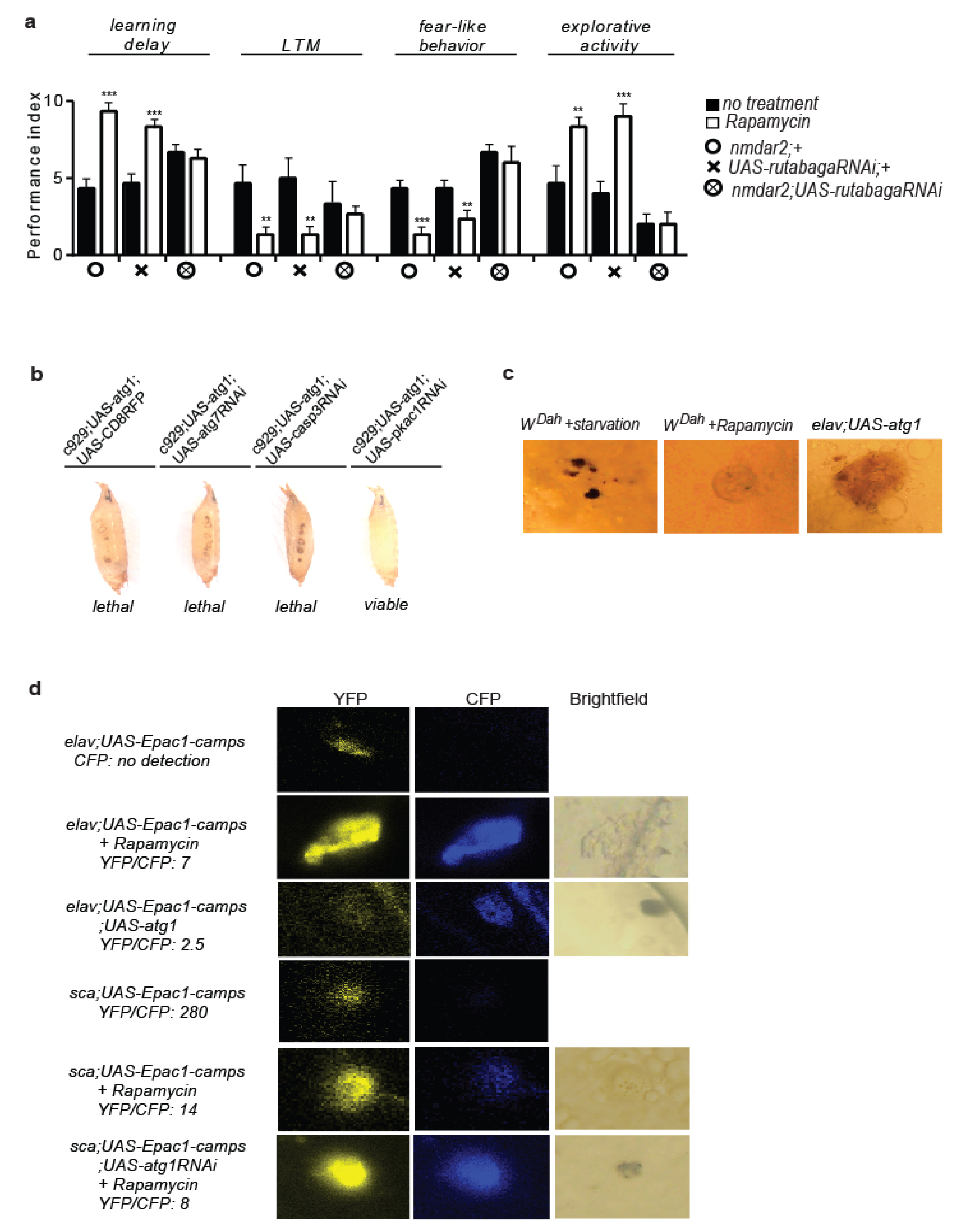
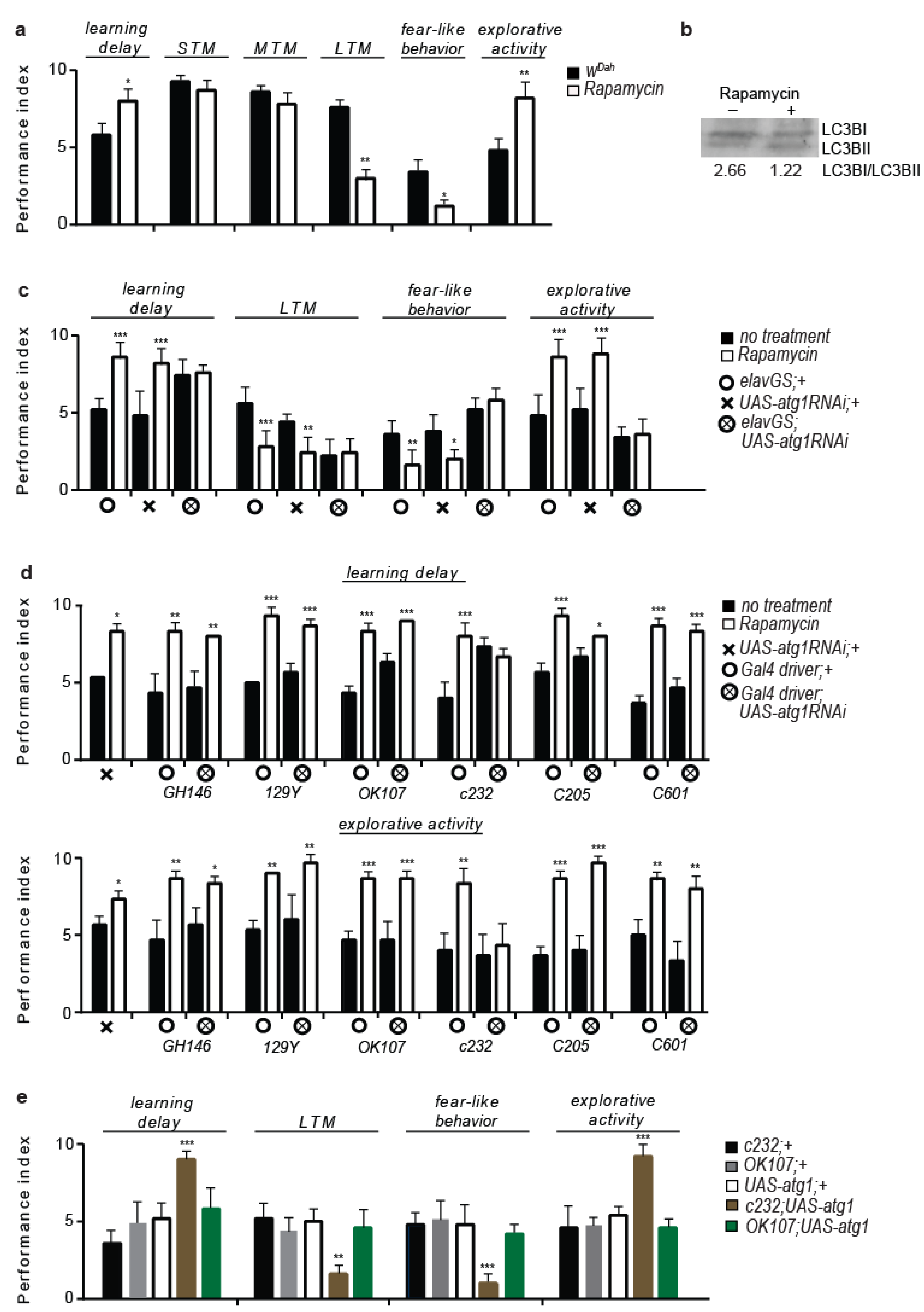
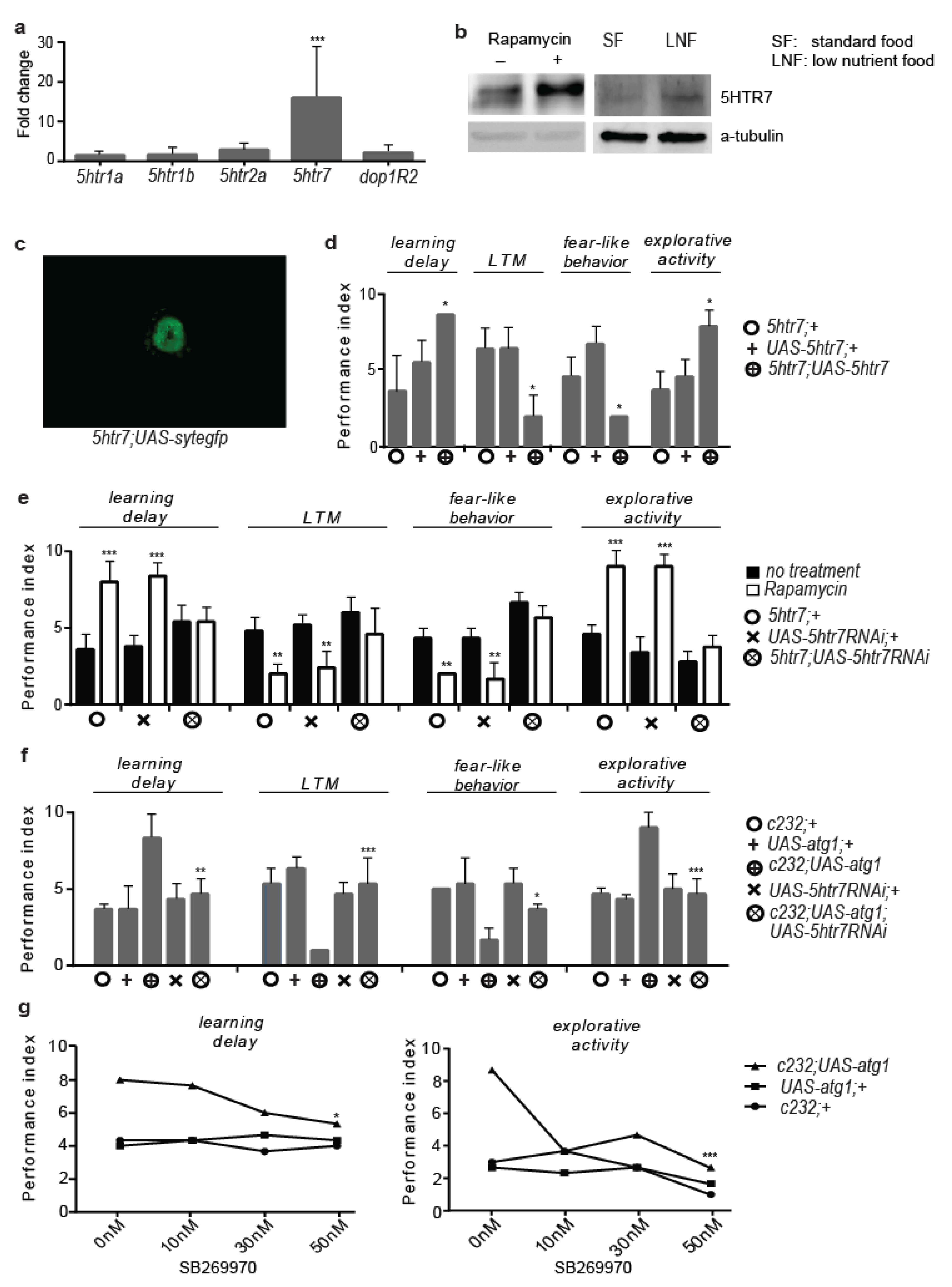
3.1. Rapamycin Treatment Alters Behaviour and Cognitive Performance
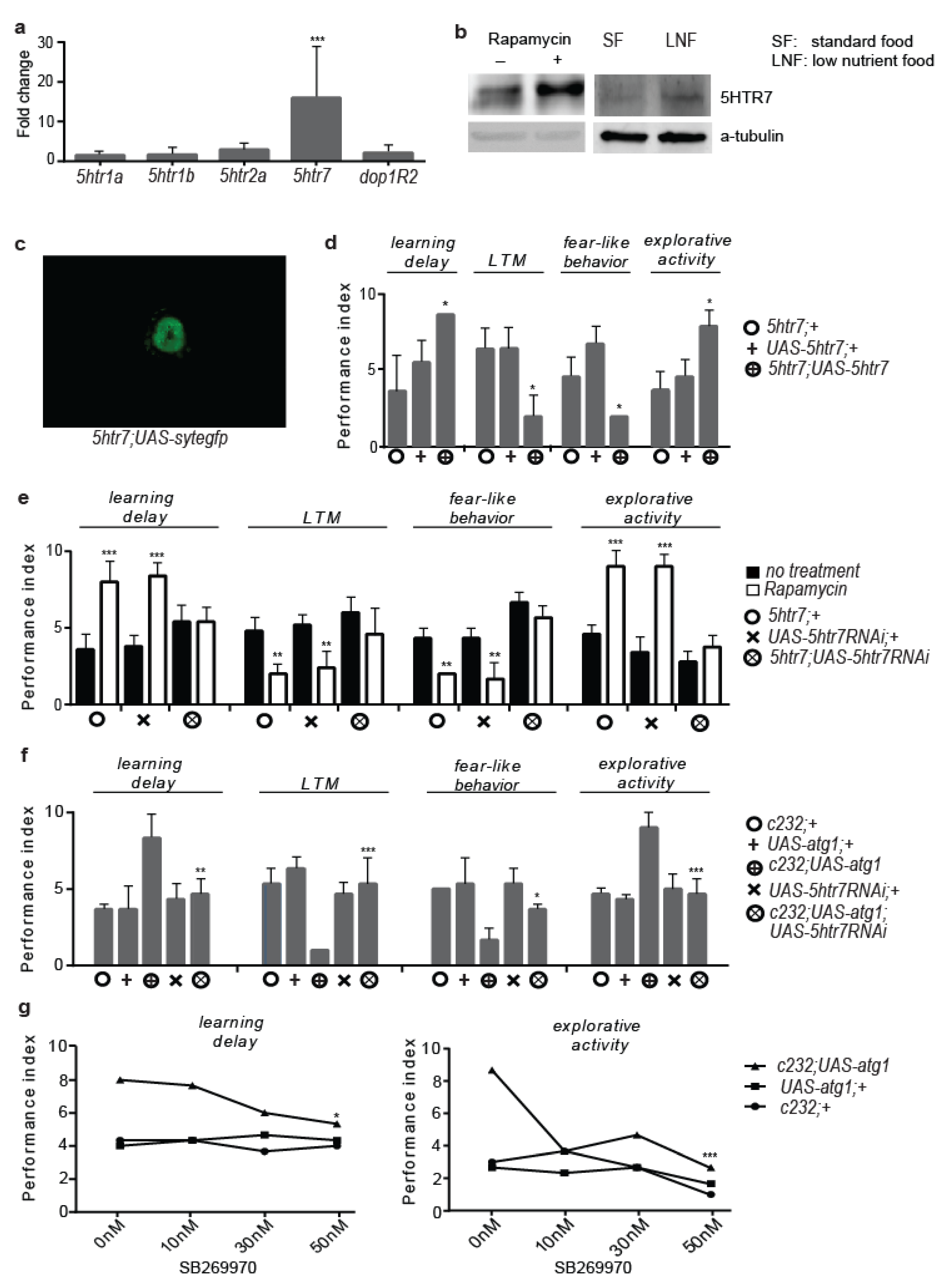
3.2. 5htr7 Inhibition Ameliorates atg1-Evoked Behavioural Patterns
3.3. Serotonergic Cell-Specific atg1 Induction Augments Postsynaptic 5HTR7 Expression and Mimics Rapamycin-Induced Behaviours
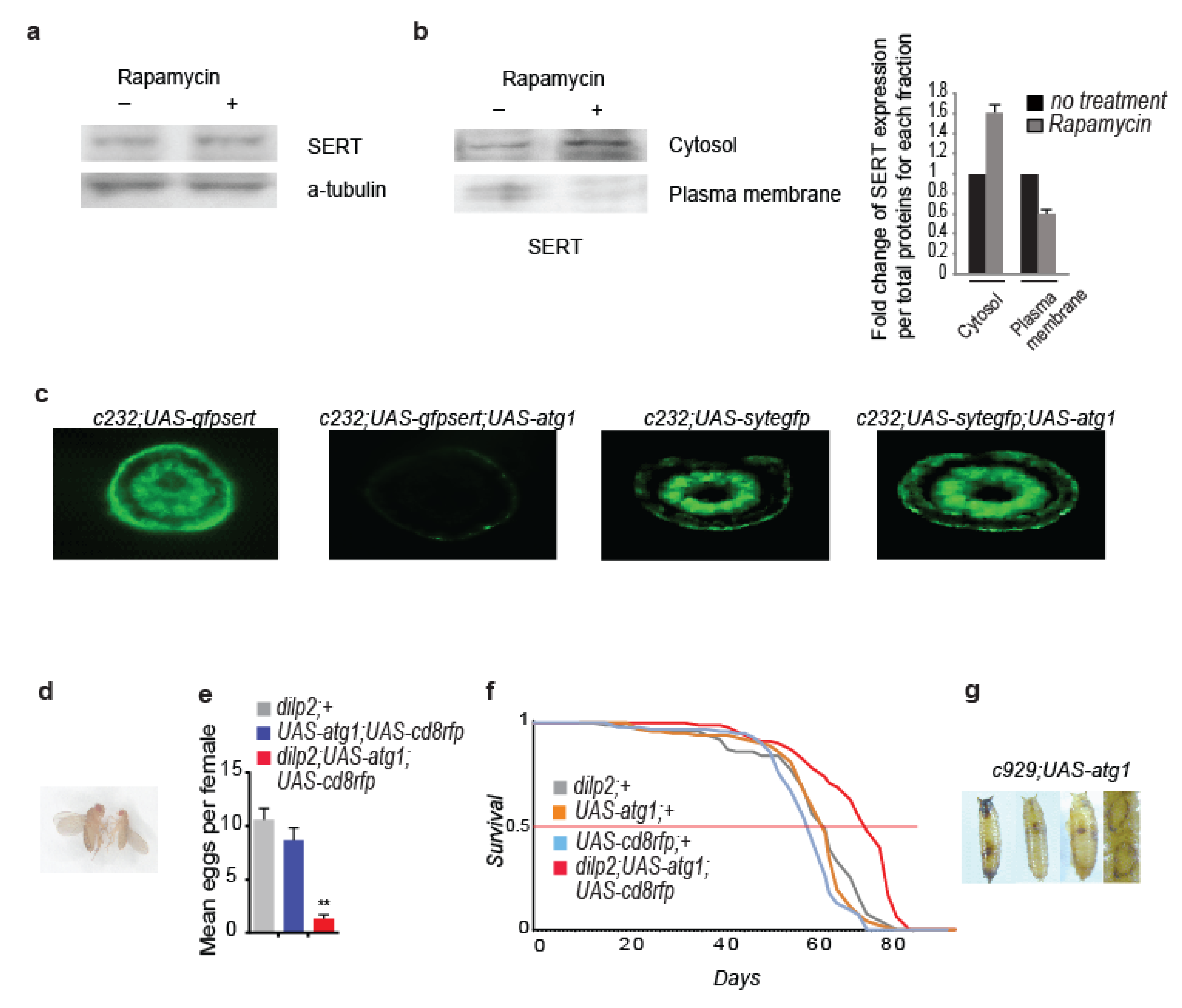
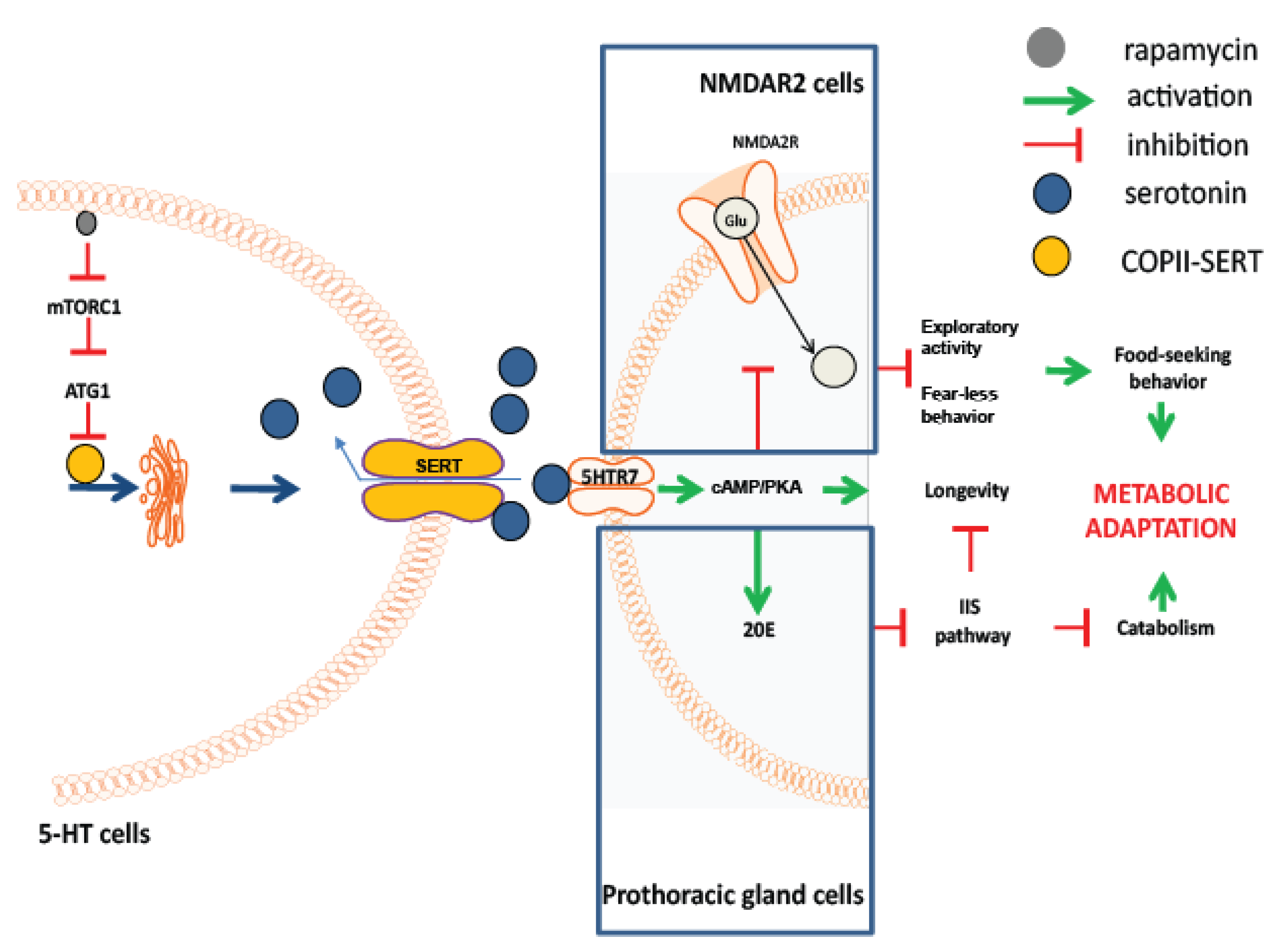
3.4. Rapamycin Alters the Cellular Localization of the Serotonin Transporter
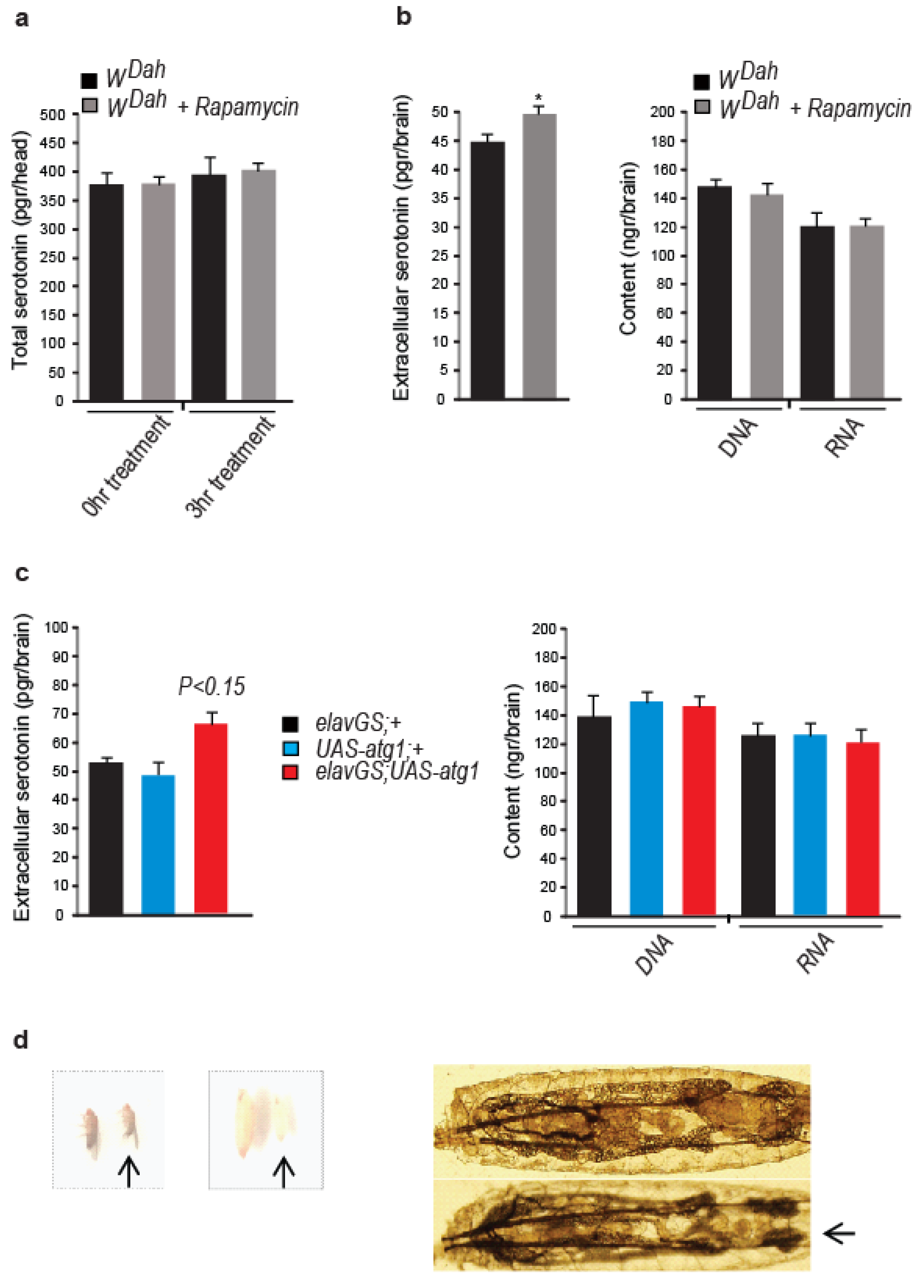
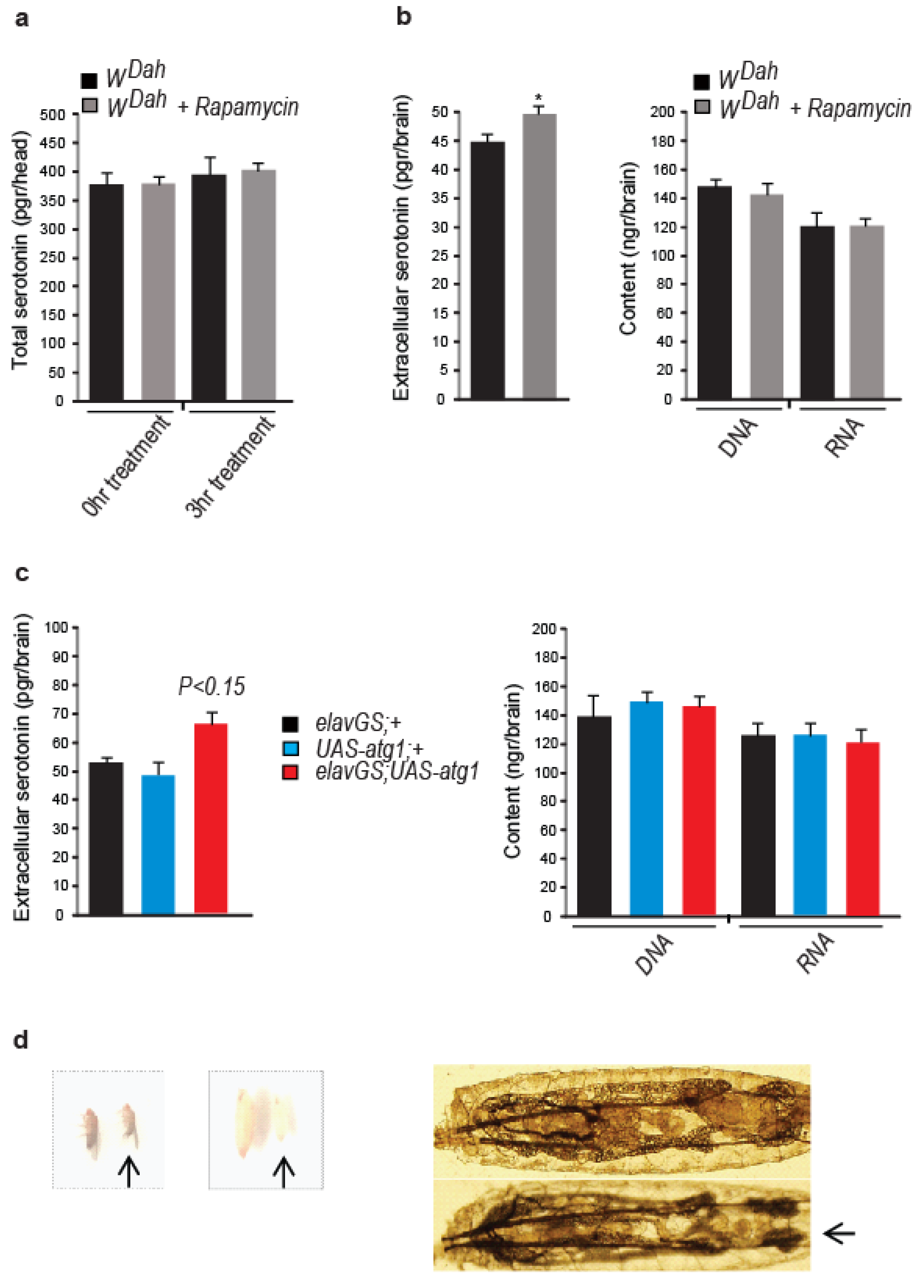
3.5. Neuronal atg1 Increases Extracellular Serotonin in Drosophila Brain and Enhances Serotonergic Neurotransmission
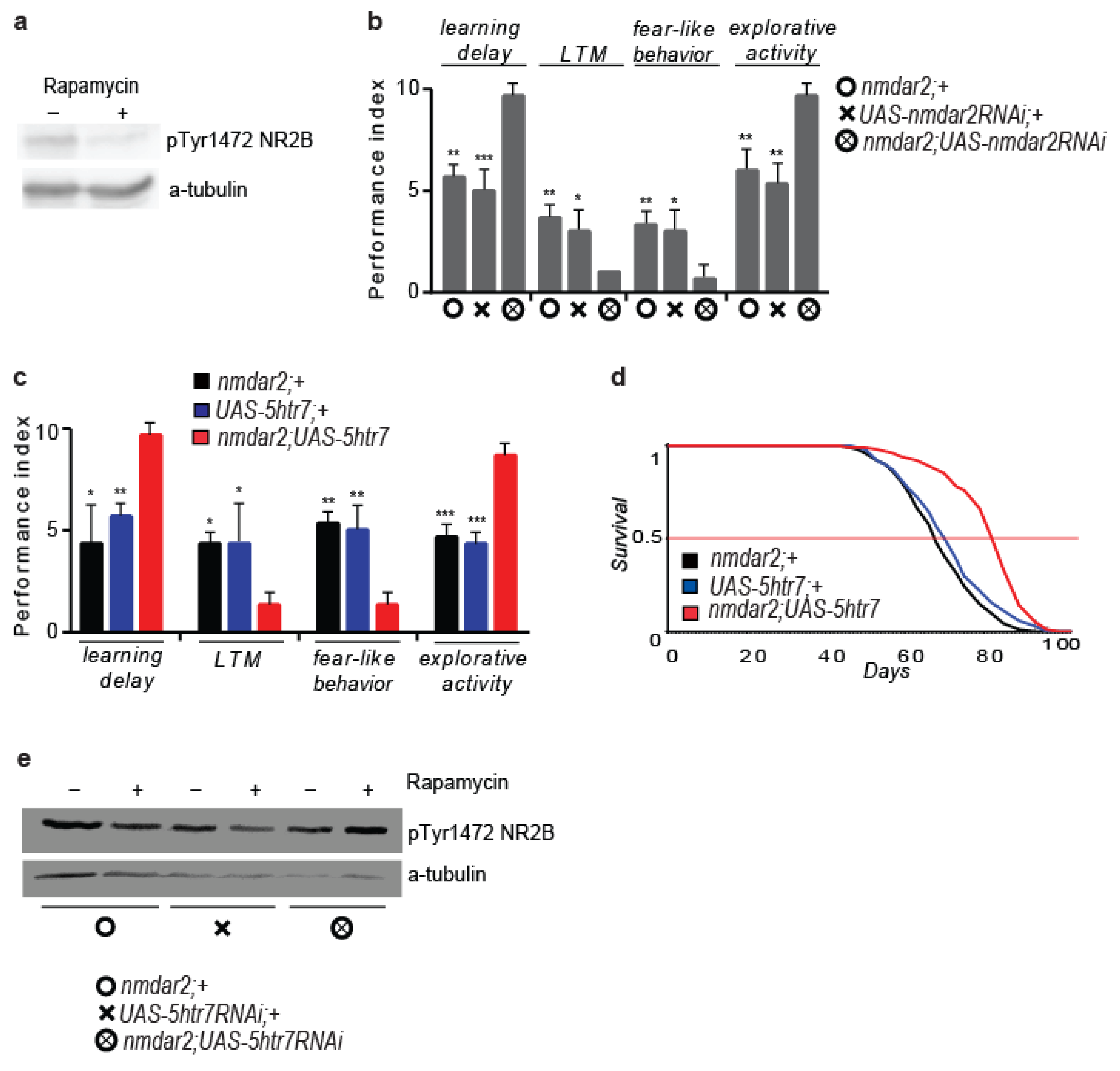
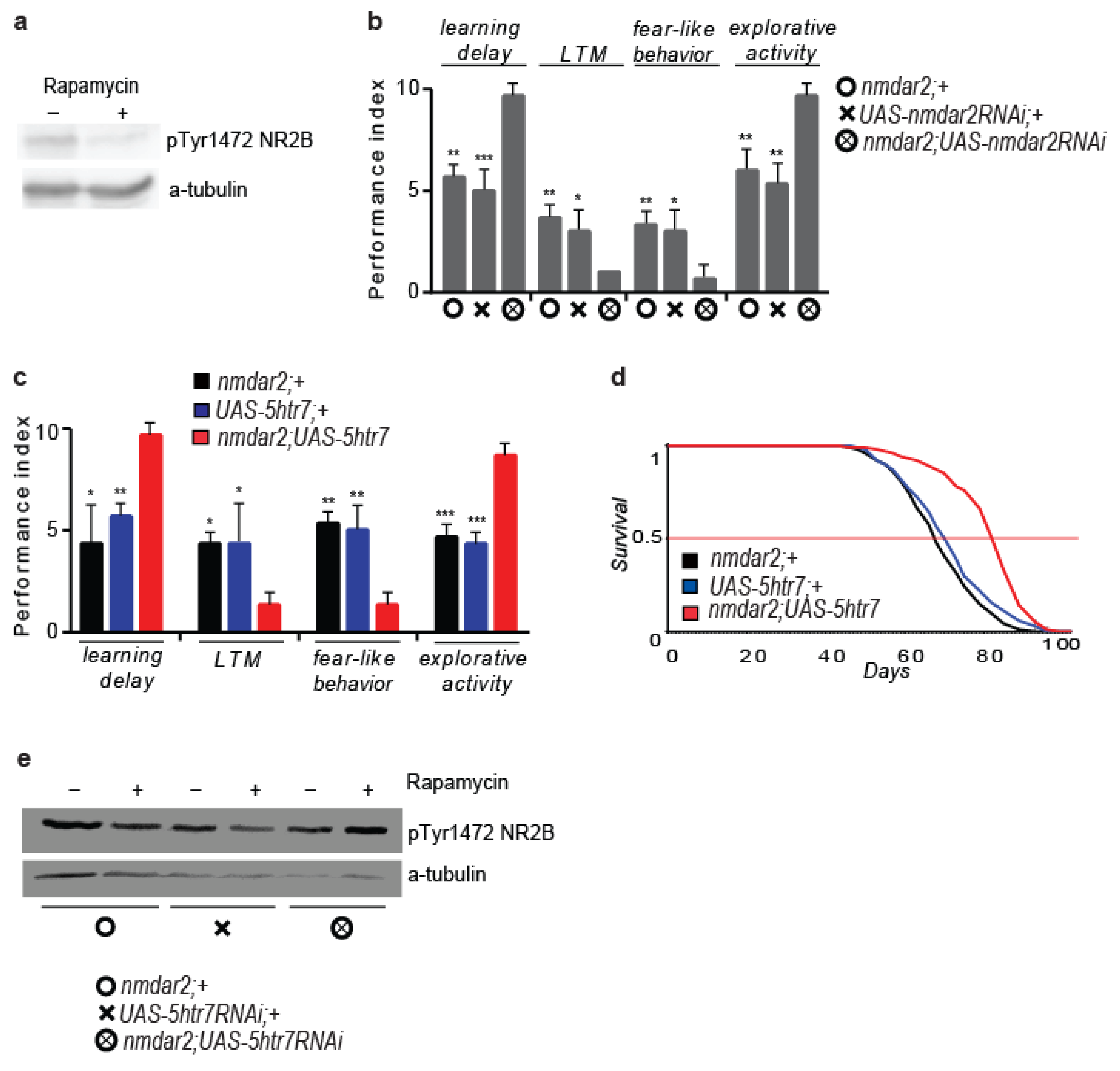
3.6. 5HTR7 Activation Reduces NMDA Signalling in the Drosophila Brain
3.7. Rapamycin Treatment Stimulates Non-Cell Autonomous cAMP/PKA Signalling
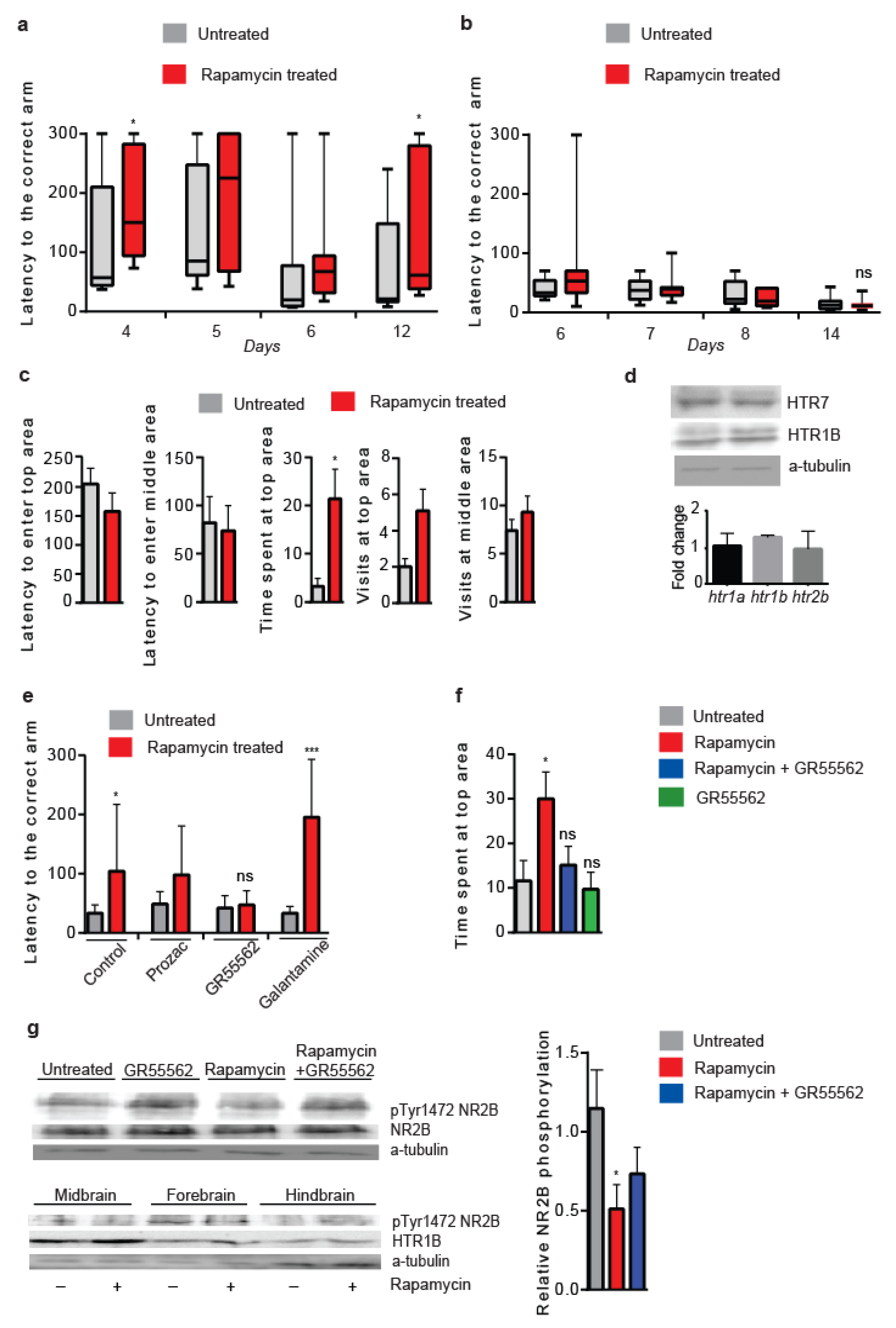
3.8. Lowered Serotonergic Signalling Abrogates Rapamycin-Induced Effects in Vertebrates
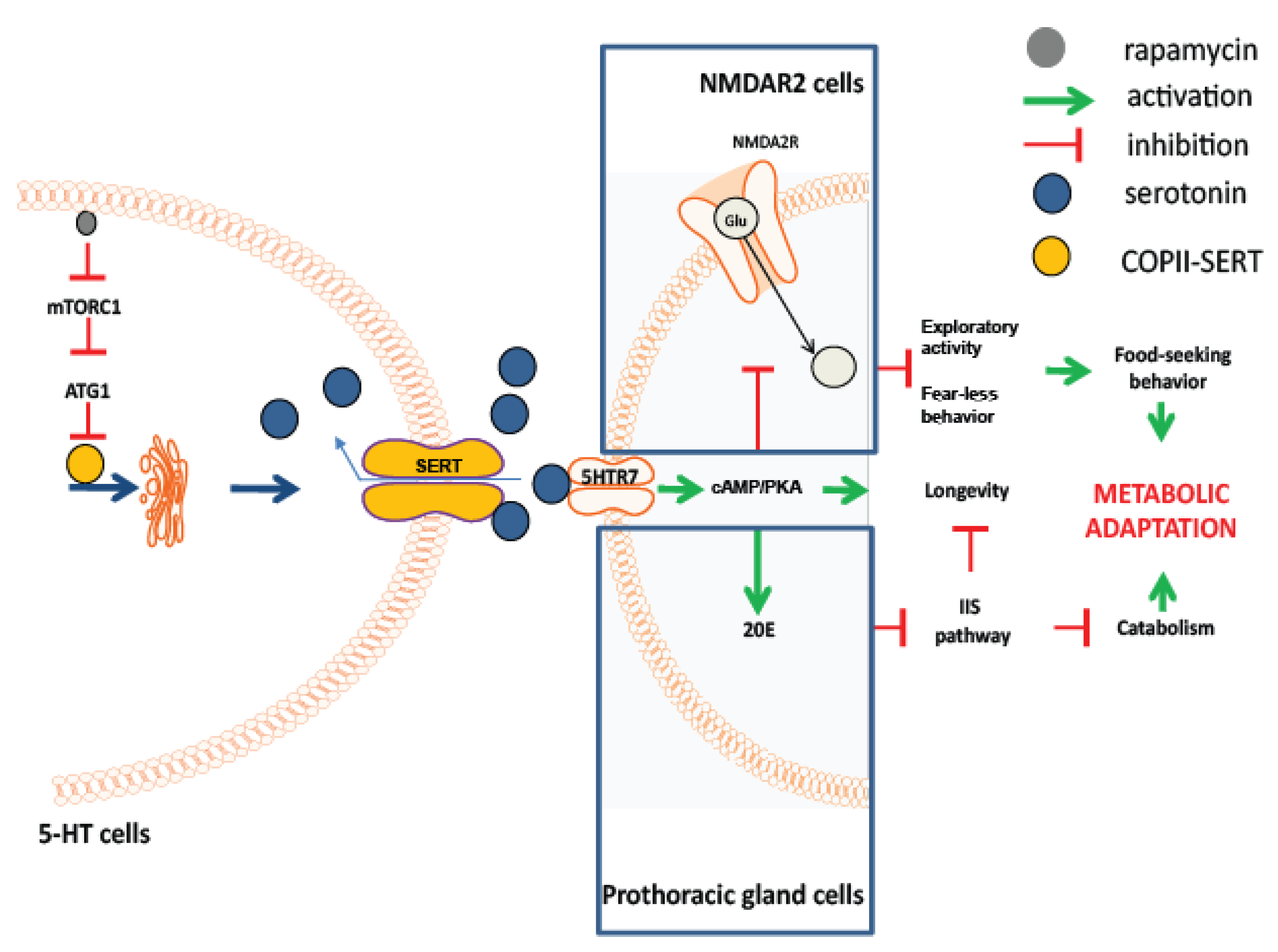
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cota, D.; Proulx, K.; Smith, K.A.; Kozma, S.C.; Thomas, G.; Woods, S.C.; Seeley, R.J. Hypothalamic mTOR signalling regulates food intake. Science 2006, 312, 927–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proulx, K.; Cota, D.; Woods, S.C.; Seeley, R.J. Fatty acid synthase inhibitors modulate energy balance via mammalian target of rapamycin complex 1 signalling in the central nervous system. Diabetes 2008, 57, 3231–3238. [Google Scholar] [CrossRef] [Green Version]
- Blouet, C.; Ono, H.; Schwartz, G.J. Mediobasal hypothalamic p70 S6 kinase 1 modulates the control of energy homeostasis. Cell Metab. 2008, 8, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, E.C.; Munzberg, H.; Cota, D.; Leshan, R.L.; Kopp, K.; Ishida-Takahashi, R.; Jones, J.C.; Fingar, D.C.; Seeley, R.J.; Myers, M.G., Jr. Complex regulation of mammalian target of rapamycin complex 1 in the basomedial hypothalamus by leptin and nutritional status. Endocrinology 2009, 150, 4541–4551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar] [CrossRef]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef] [Green Version]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [Green Version]
- Ye, P.; Liu, Y.; Chen, C.; Tang, F.; Wu, Q.; Wang, X.; Liu, C.G.; Liu, X.; Liu, R.; Liu, Y.; et al. An mTORC1-Mdm2-Drosha axis for miRNA biogenesis in response to glucose- and amino acid-deprivation. Mol. Cell 2015, 57, 708–720. [Google Scholar] [CrossRef] [Green Version]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Qiao, M.; Iglehart, J.D.; Pardee, A.B. Metastatic potential of 21T human breast cancer cells depends on Akt/protein kinase B activation. Cancer Res. 2007, 67, 5293–5299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Feature Article: mTOR complex 2-Akt signalling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signalling, is inhibited by TSC1 and 2. Mol Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes. Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [Green Version]
- Tzatsos, A.; Kandror, K.V. Nutrients suppress phosphatidylinositol 3-kinase/Akt signalling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol. Cell. Biol. 2006, 26, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.N.; Ha, S.H.; Kim, J.; Koh, A.; Lee, C.S.; Kim, J.H.; Jeon, H.; Kim, D.H.; Suh, P.G.; Ryu, S.H. Glycolytic flux signals to mTOR through glyceraldehyde-3-phosphate dehydrogenase-mediated regulation of Rheb. Mol. Cell. Biol. 2009, 29, 3991–4001. [Google Scholar] [CrossRef] [Green Version]
- Dagon, Y.; Hur, E.; Zheng, B.; Wellenstein, K.; Cantley, L.C.; Kahn, B.B. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake. Cell Metab. 2012, 16, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizuka, Y.; Kakiya, N.; Witters, L.A.; Oshiro, N.; Shirao, T.; Nawa, H.; Takei, N. AMP-activated protein kinase counteracts brain-derived neurotrophic factor-induced mammalian target of rapamycin complex 1 signalling in neurons. J. Neurochem. 2013, 127, 66–77. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Sidiropoulou, K.; Kallergi, E.; Dalezios, Y.; Tavernarakis, N. Modulation of Autophagy by BDNF Underlies Synaptic Plasticity. Cell Metab. 2017, 26, 230. [Google Scholar] [CrossRef] [Green Version]
- Muta, K.; Morgan, D.A.; Rahmouni, K. The role of hypothalamic mTORC1 signalling in insulin regulation of food intake, body weight, and sympathetic nerve activity in male mice. Endocrinology 2015, 156, 1398–1407. [Google Scholar] [CrossRef] [Green Version]
- Ono, H.; Pocai, A.; Wang, Y.; Sakoda, H.; Asano, T.; Backer, J.M.; Schwartz, G.J.; Rossetti, L. Activation of hypothalamic S6 kinase mediates diet-induced hepatic insulin resistance in rats. J. Clin. Investig. 2008, 118, 2959–2968. [Google Scholar] [CrossRef] [PubMed]
- Procaccini, C.; De Rosa, V.; Galgani, M.; Carbone, F.; Cassano, S.; Greco, D.; Qian, K.; Auvinen, P.; Calì, G.; Stallone, G.; et al. Leptin-induced mTOR activation defines a specific molecular and transcriptional signature controlling CD4+ effector T cell responses. J. Immunol. 2012, 189, 2941–2953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harlan, S.M.; Guo, D.F.; Morgan, D.A.; Fernandes-Santos, C.; Rahmouni, K. Hypothalamic mTORC1 signalling controls sympathetic nerve activity and arterial pressure and mediates leptin effects. Cell Metab. 2013, 17, 599–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Lehtinen, M.K.; Sessa, A.; Zappaterra, M.W.; Cho, S.H.; Gonzalez, D.; Boggan, B.; Austin, C.A.; Wijnholds, J.; Gambello, M.J.; et al. The apical complex couples cell fate and cell survival to cerebral cortical development. Neuron 2010, 66, 69–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.B.; Tien, A.C.; Boddupalli, G.; Xu, A.W.; Jan, Y.N.; Jan, L.Y. Rapamycin ameliorates age-dependent obesity associated with increased mTOR signalling in hypothalamic POMC neurons. Neuron 2012, 75, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Cloetta, D.; Thomanetz, V.; Baranek, C.; Lustenberger, R.M.; Lin, S.; Oliveri, F.; Atanasoski, S.; Rüegg, M.A. Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J. Neurosci. 2013, 33, 7799–7810. [Google Scholar] [CrossRef] [Green Version]
- Hartman, N.W.; Lin, T.V.; Zhang, L.; Paquelet, G.E.; Feliciano, D.M.; Bordey, A. mTORC1 targets the translational repressor 4E-BP2, but not S6 kinase 1/2, to regulate neural stem cell self-renewal in vivo. Cell Rep. 2013, 5, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Bercury, K.K.; Dai, J.; Sachs, H.H.; Ahrendsen, J.T.; Wood, T.L.; Macklin, W.B. Conditional ablation of raptor or rictor has differential impact on oligodendrocyte differentiation and CNS myelination. J. Neurosci. 2014, 34, 4466–4480. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Ganetzky, B. Autophagy promotes synapse development in Drosophila. J. Cell Biol. 2009, 187, 71–79. [Google Scholar] [CrossRef]
- Binotti, B.; Pavlos, N.J.; Riedel, D.; Wenzel, D.; Vorbrüggen, G.; Schalk, A.M.; Kühnel, K.; Boyken, J.; Erck, C.; Martens, H.; et al. The GTPase Rab26 links synaptic vesicles to the autophagy pathway. Elife 2015, 4, e05597. [Google Scholar] [CrossRef]
- Luningschror, P.; Binotti, B.; Dombert, B.; Heimann, P.; Perez-Lara, A.; Slotta, C.; Thau-Habermann, N.; von Collenberg, C.R.; Karl, F.; Damme, M.; et al. Plekhg5-regulated autophagy of synaptic vesicles reveals a pathogenic mechanism in motoneuron disease. Nat. Commun. 2017, 8, 678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, A.M.; Richmond, J.E.; Olsen, J.G.; Hall, D.H.; Bamber, B.A. Presynaptic terminals independently regulate synaptic clustering and autophagy of GABA(A) receptors in Caenorhabditis elegans. J. Neurosci. 2006, 26, 1711–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehata, M.; Matsumura, H.; Okubo-Suzuki, R.; Ohkawa, N.; Inokuchi, K. Neuronal Stimulation Induces Autophagy in Hippocampal Neurons That Is Involved in AMPA Receptor Degradation after Chemical Long-Term Depression. J. Neurosci. 2012, 32, 10413–10422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.M.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, D.; Torres, C.A.; Setlik, W.; Cebrián, C.; Mosharov, E.V.; Tang, G.; Cheng, H.C.; Kholodilov, N.; Yarygina, O.; Burke, R.E.; et al. Regulation of presynaptic neurotransmission by macroautophagy. Neuron 2012, 74, 277–284. [Google Scholar] [CrossRef] [Green Version]
- Friedman, L.G.; Lachenmayer, M.L.; Wang, J.; He, L.; Poulose, S.M.; Komatsu, M.; Holstein, G.R.; Yue, Z. Disrupted Autophagy Leads to Dopaminergic Axon and Dendrite Degeneration and Promotes Presynaptic Accumulation of alpha-Synuclein and LRRK2 in the Brain. J. Neurosci. 2012, 32, 7585–7593. [Google Scholar] [CrossRef] [Green Version]
- Sheehan, P.; Zhu, M.; Beskow, A.; Vollmer, C.; Waites, C.L. Activity-Dependent Degradation of Synaptic Vesicle Proteins Requires Rab35 and the ESCRT Pathway. J. Neurosci. 2016, 36, 8668–8686. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, S.; Orlando, M.; Andrzejak, E.; Bruns, C.; Trimbuch, T.; Rosenmund, C.; Garner, C.C.; Ackermann, F. Light-Activated ROS Production Induces Synaptic Autophagy. J. Neurosci. 2019, 39, 2163–2183. [Google Scholar] [CrossRef] [Green Version]
- Kiral, F.R.; Linneweber, G.A.; Mathejczyk, T.; Georgiev, S.V.; Wernet, M.F.; Hassan, B.A.; von Kleist, M.; Hiesinger, P.R. Autophagy-dependent filopodial kinetics restrict synaptic partner choice during Drosophila brain wiring. Nat. Commun. 2020, 11, 1325. [Google Scholar] [CrossRef] [Green Version]
- Theodoridi, A.; Tsalafouta, A.; Pavlidis, M. Acute Exposure to Fluoxetine Alters Aggressive Behaviour of Zebrafish and Expression of Genes Involved in Serotonergic System Regulation. Front. Neurosci. 2017, 11, 223. [Google Scholar] [CrossRef]
- Bennett, P.C.; Schmidt, L.; Lawen, A.; Moutsoulas, P.; Ng, K.T. Cyclosporin A, FK506 and rapamycin produce multiple, temporally distinct, effects on memory following single-trial, passive avoidance training in the chick. Brain Res. 2002, 927, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sanchis, S.; Bernal, M.C.; Montagud, J.V.; Candela, G.; Crespo, J.; Sancho, A.; Pallardó, L.M. Effects of immunosuppressive drugs on the cognitive functioning of renal transplant recipients: A pilot study. J. Clin. Exp. Neuropsychol. 2011, 33, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Ries, A.S.; Hermanns, T.; Poeck, B.; Strauss, R. Serotonin modulates a depression-like state in Drosophila responsive to lithium treatment. Nat. Commun. 2017, 8, 15738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jope, R.S. Glycogen synthase kinase-3 in the aetiology and treatment of mood disorders. Front. Mol. Neurosci. 2011, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.J.; Fuchikami, M.; Dwyer, J.M.; Lepack, A.E.; Duman, R.S.; Aghajanian, G.K. GSK-3 Inhibition Potentiates the Synaptogenic and Antidepressant-Like Effects of Subthreshold Doses of Ketamine. Neuropsychopharmacology 2013, 38, 2268–2277. [Google Scholar] [CrossRef] [Green Version]
- Klein, P.S.; Melton, D.A. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA 1996, 93, 8455–8459. [Google Scholar] [CrossRef]
- Holz, M.K.; Blenis, J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J. Biol. Chem. 2005, 280, 26089–26093. [Google Scholar] [CrossRef] [Green Version]
- Casadio, A.; Martin, K.C.; Giustetto, M.; Zhu, H.; Chen, M.; Bartsch, D.; Bailey, C.H.; Kandel, E.R. A transient, neuron-wide form of CREB-mediated long-term facilitation can be stabilized at specific synapses by local protein synthesis. Cell 1999, 99, 221–237. [Google Scholar] [CrossRef] [Green Version]
- Billes, V.; Kovacs, T.; Manzeger, A.; Lőrincz, P.; Szincsák, S.; Regős, Á.; Kulcsár, P.I.; Korcsmáros, T.; Lukácsovich, T.; Hoffmann, G.; et al. Developmentally regulated autophagy is required for eye formation in Drosophila. Autophagy 2018, 14, 1499–1519. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Zhou, Y.; Guo, C.; Gong, H.; Gong, Z.; Liu, L. Differential roles of the fan-shaped body and the ellipsoid body in Drosophila visual pattern memory. Learn. Mem. 2009, 16, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Valles, A.M.; White, K. Serotonin-containing neurons in Drosophila melanogaster: Development and distribution. J. Comp. Neurol. 1988, 268, 414–428. [Google Scholar] [CrossRef]
- Sitaraman, D.; Zars, M.; LaFerriere, H.; Zars, T. Serotonin is necessary for place memory in Drosophila. Proc. Natl. Acad. Sci. USA 2008, 105, 5579–5584. [Google Scholar] [CrossRef]
- Johnson, O.; Becnel, J.; Nichols, C.D. Serotonin Receptor Activity Is Necessary for Olfactory Learning and Memory in Drosophila melanogaster. Neuroscience 2011, 192, 372–381. [Google Scholar] [CrossRef] [Green Version]
- Kahsai, L.; Carlsson, M.A.; Winther, A.M.; Nässel, D.R. Distribution of metabotropic receptors of serotonin, dopamine, GABA, glutamate, and short neuropeptide F in the central complex of Drosophila. Neuroscience 2012, 208, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Azanchi, R.; Kaun, K.R.; Heberlein, U. Competing dopamine neurons drive oviposition choice for ethanol in Drosophila. Proc. Natl. Acad. Sci. USA 2013, 110, 21153–21158. [Google Scholar] [CrossRef] [PubMed]
- Becnel, J.; Johnson, O.; Luo, J.N.; Nässel, D.R.; Nichols, C.D. The Serotonin 5-HT(7)Dro Receptor Is Expressed in the Brain of Drosophila, and Is Essential for Normal Courtship and Mating. PLoS ONE 2011, 6, e20800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saller, C.F.; Stricker, E.M. Hyperphagia and Increased Growth in Rats after Intraventricular-Injection of 5,7-Dihydroxytryptamine. Science 1976, 192, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, P.J. Increased Food-Intake in Satiated Rats Induced by the 5-Ht Antagonists Methysergide, Metergoline and Ritanserin. Psychopharmacology 1988, 96, 237–242. [Google Scholar] [CrossRef]
- Lassen, J.B. Potent and long-lasting potentiation of two 5-hydroxytryptophan- induced effects in mice by three selective 5-ht uptake inhibitors. Eur. J. Pharmacol. 1978, 47, 351–358. [Google Scholar] [CrossRef]
- Stark, P.; Fuller, R.W.; Wong, D.T. The pharmacologic profile of fluoxetine. J. Clin. Psychiatry 1985, 46, 7–13. [Google Scholar]
- Rosario, F.J.; Kanai, Y.; Powell, T.L.; Jansson, T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J. Physiol.-Lond. 2013, 591, 609–625. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, S.; Lee, J.; Park, J.; Lee, G.; Kim, Y.; Kim, J.M.; Chung, J. ATG1, an autophagy regulator, inhibits cell growth by negatively regulating S6 kinase. EMBO Rep. 2007, 8, 360–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, J.H.; Wang, B.; Frankel, E.; Ge, L.; Xu, L.; Iyengar, R.; Li-Harms, X.; Wright, C.; Shaw, T.I.; Lindsten, T. The Noncanonical Role of ULK/ATG1 in ER-to-Golgi Trafficking Is Essential for Cellular Homeostasis. Mol. Cell 2016, 62, 491–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, W.J.; Zhang, C.Y.; Siu, K.Y.; Satoh, A.; Tanner, J.A.; Yu, S. ULK1 phosphorylates Sec23A and mediates autophagy-induced inhibition of ER-to-Golgi traffic. Bmc Cell Biol. 2017, 18, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.K.; George, R.; Cai, Y.; Chang, H.Y.; Krantz, D.E.; Friggi-Grelin, F.; Birman, S.; Hirsh, J. Cell-type-specific limitation on in vivo serotonin storage following ectopic expression of the Drosophila serotonin transporter, dSERT. J. Neurobiol. 2006, 66, 452–462. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Rodesch, C.K.; Broadie, K. Living synaptic vesicle marker: Synaptotagmin-GFP. Genesis 2002, 34, 142–145. [Google Scholar] [CrossRef]
- Fang, J.; Liu, M.; Zhang, X.; Sakamoto, T.; Taatjes, D.J.; Jena, B.P.; Sun, F.; Woods, J.; Bryson, T.; Kowluru, A.; et al. COPII-Dependent ER Export: A Critical Component of Insulin Biogenesis and beta-Cell ER Homeostasis. Mol. Endocrinol. 2015, 29, 1156–1169. [Google Scholar] [CrossRef] [Green Version]
- Gronke, S.; Clarke, D.F.; Broughton, S.; Andrews, T.D.; Partridge, L. Molecular evolution and functional characterization of Drosophila insulin-like peptides. PLoS Genet. 2010, 6, e1000857. [Google Scholar] [CrossRef] [Green Version]
- Park, D.; Veenstra, J.A.; Park, J.H.; Taghert, P.H. Mapping peptidergic cells in Drosophila: Where DIMM fits in. PLoS ONE 2008, 3, e1896. [Google Scholar] [CrossRef] [Green Version]
- Shimada-Niwa, Y.; Niwa, R. Serotonergic neurons respond to nutrients and regulate the timing of steroid hormone biosynthesis in Drosophila. Nat. Commun. 2014, 5, 5778. [Google Scholar] [CrossRef] [Green Version]
- Orme, M.H.; Leevers, S.J. Files on steroids: The interplay between ecdysone and insulin signalling. Cell Metab. 2005, 2, 277–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maura, G.; Marcoli, M.; Pepicelli, O.; Rosu, C.; Viola, C.; Raiteri, M. Serotonin inhibition of the NMDA receptor/nitric oxide/cyclic GMP pathway in human neocortex slices: Involvement of 5-HT(2C) and 5-HT(1A) receptors. Br. J. Pharmacol. 2000, 130, 1853–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.L.; Xia, S.; Fu, T.F.; Wang, H.; Chen, Y.H.; Leong, D.; Chiang, A.S.; Tully, T. Specific requirement of NMDA receptors for long-term memory consolidation in Drosophila ellipsoid body. Nat. Neurosci. 2007, 10, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Caccamo, A.; Medina, D.X.; Benavides, A.D.; Javors, M.A.; Kraig, E.; Strong, R.; Richardson, A.; Oddo, S. Lifelong rapamycin administration ameliorates age-dependent cognitive deficits by reducing IL-1beta and enhancing NMDA signalling. Aging Cell 2012, 11, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Vasefi, M.S.; Yang, K.; Li, J.; Kruk, J.S.; Heikkila, J.J.; Jackson, M.F.; MacDonald, J.F.; Beazely, M.A. Acute 5-HT7 receptor activation increases NMDA-evoked currents and differentially alters NMDA receptor subunit phosphorylation and trafficking in hippocampal neurons. Mol. Brain 2013, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Bjedov, I.; Toivonen, J.M.; Kerr, F.; Slack, C.; Jacobson, J.; Foley, A.; Partridge, L. Mechanisms of Life Span Extension by Rapamycin in the Fruit Fly Drosophila melanogaster. Cell Metab. 2010, 11, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Witz, P.; Amlaiky, N.; Plassat, J.L.; Maroteaux, L.; Borrelli, E.; Hen, R. Cloning and Characterization of a Drosophila Serotonin Receptor That Activates Adenylate-Cyclase. Proc. Natl. Acad. Sci. USA 1990, 87, 8940–8944. [Google Scholar] [CrossRef]
- Davis, M.M.; O’Keefe, S.L.; Primrose, D.A.; Hodgetts, R.B. A neuropeptide hormone cascade controls the precise onset of post-eclosion cuticular tanning in Drosophila melanogaster. Development 2007, 134, 4395–4404. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.W.; Dewey, E.M.; Sudo, S.; Ewer, J.; Hsu, S.Y.; Honegger, H.W.; Hsueh, A.J. Bursicon, the insect cuticle-hardening hormone, is a heterodimeric cystine knot protein that activates G protein-coupled receptor LGR2. Proc. Natl. Acad. Sci. USA 2005, 102, 2820–2825. [Google Scholar] [CrossRef]
- Loveall, B.J.; Deitcher, D.L. The essential role of bursicon during Drosophila development. Bmc Dev. Biol. 2010, 10, 92. [Google Scholar] [CrossRef]
- Minakhina, S.; Steward, R. Melanotic mutants in Drosophila: Pathways and phenotypes. Genetics 2006, 174, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Ponsioen, B.; Zhao, J.; Riedl, J.; Zwartkruis, F.; van der Krogt, G.; Zaccolo, M.; Moolenaar, W.H.; Bos, J.L.; Jalink, K. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. Embo Rep. 2004, 5, 1176–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.C.; Klionsky, D.J. Regulation Mechanisms and Signalling Pathways of Autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Rinkwitz, S.; Mourrain, P.; Becker, T.S. Zebrafish: An integrative system for neurogenomics and neurosciences. Prog. Neurobiol. 2011, 93, 231–243. [Google Scholar] [CrossRef]
- Glover, E.M.; Ressler, K.J.; Davis, M. Differing effects of systemically administered rapamycin on consolidation and reconsolidation of context vs. cued fear memories. Learn. Mem. 2010, 17, 577–581. [Google Scholar]
- Ahlander-Luttgen, M.; Madjid, N.; Schott, P.A.; Sandin, J.; Ogren, S.O. Analysis of the role of the 5-HT1B receptor in spatial and aversive learning in the rat. Neuropsychopharmacology 2003, 28, 1642–1655. [Google Scholar] [CrossRef] [PubMed]
- Chenu, F.; David, D.J.; Leroux-Nicollet, I.; Le Maître, E.; Gardier, A.M.; Bourin, M. Serotonin1B heteroreceptor activation induces an antidepressant-like effect in mice with an alteration of the serotonergic system. J. Psychiatry Neurosci. 2008, 33, 541–550. [Google Scholar] [PubMed]
- Eriksson, T.M.; Madjid, N.; Elvander-Tottie, E.; Stiedl, O.; Svenningsson, P.; Ogren, S.O. Blockade of 5-HT 1B receptors facilitates contextual aversive learning in mice by disinhibition of cholinergic and glutamatergic neurotransmission. Neuropharmacology 2008, 54, 1041–1050. [Google Scholar] [CrossRef] [Green Version]
- Tiger, M.; Varnas, K.; Okubo, Y.; Lundberg, J. The 5-HT1B receptor—A potential target for antidepressant treatment. Psychopharmacology 2018, 235, 1317–1334. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Kallarackal, A.J.; Kvarta, M.D.; Goluskin, S.; Gaylor, K.; Bailey, A.M.; Lee, H.K.; Huganir, R.L.; Thompson, S.M. Local potentiation of excitatory synapses by serotonin and its alteration in rodent models of depression. Nat. Neurosci. 2013, 16, 464–472. [Google Scholar] [CrossRef]
- Daws, L.C.; Gerhardt, G.A.; Frazer, A. 5-HT1B antagonists modulate clearance of extracellular serotonin in rat hippocampus. Neurosci. Lett. 1999, 266, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Albert, V.; Hall, M.N. mTOR signalling in cellular and organismal energetics. Curr. Opin. Cell Biol. 2015, 33, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Xu, Y.; Liu, F. Hypothalamic roles of mTOR complex I: Integration of nutrient and hormone signals to regulate energy homeostasis. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E994–E1002. [Google Scholar] [CrossRef] [Green Version]
- Chintapalli, V.R.; Wang, J.; Dow, J.A. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet. 2007, 39, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Blandino-Rosano, M.; Barbaresso, R.; Jimenez-Palomares, M.; Bozadjieva, N.; Werneck-de-Castro, J.P.; Hatanaka, M.; Mirmira, R.G.; Sonenberg, N.; Liu, M.; Rüegg, M.A.; et al. Loss of mTORC1 signalling impairs beta-cell homeostasis and insulin processing. Nat. Commun. 2017, 8, 16014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C-elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lanjuin, A.; Chowdhury, S.R.; Mistry, M.; Silva-García, C.G.; Weir, H.J.; Lee, C.L.; Escoubas, C.C.; Tabakovic, E.; Mair, W.B. Neuronal TORC1 modulates longevity via AMPK and cell nonautonomous regulation of mitochondrial dynamics in C. elegans. Elife 2019, 8, e49158. [Google Scholar] [CrossRef]
- Bjedov, I.; Cocheme, H.M.; Foley, A.; Wieser, D.; Woodling, N.S.; Castillo-Quan, J.I.; Norvaisas, P.; Lujan, C.; Regan, J.C.; Toivonen, J.M.; et al. Fine-tuning autophagy maximises lifespan and is associated with changes in mitochondrial gene expression in Drosophila. PLoS Genet. 2020, 16, e1009083. [Google Scholar] [CrossRef]
- Van Galen, K.A.; Ter Horst, K.W.; Serlie, M.J. Serotonin, food intake, and obesity. Obes. Rev. 2021, 22, e13210. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Metaxakis, A.; Pavlidis, M.; Tavernarakis, N. Neuronal atg1 Coordinates Autophagy Induction and Physiological Adaptations to Balance mTORC1 Signalling. Cells 2023, 12, 2024. https://doi.org/10.3390/cells12162024
Metaxakis A, Pavlidis M, Tavernarakis N. Neuronal atg1 Coordinates Autophagy Induction and Physiological Adaptations to Balance mTORC1 Signalling. Cells. 2023; 12(16):2024. https://doi.org/10.3390/cells12162024
Chicago/Turabian StyleMetaxakis, Athanasios, Michail Pavlidis, and Nektarios Tavernarakis. 2023. "Neuronal atg1 Coordinates Autophagy Induction and Physiological Adaptations to Balance mTORC1 Signalling" Cells 12, no. 16: 2024. https://doi.org/10.3390/cells12162024
APA StyleMetaxakis, A., Pavlidis, M., & Tavernarakis, N. (2023). Neuronal atg1 Coordinates Autophagy Induction and Physiological Adaptations to Balance mTORC1 Signalling. Cells, 12(16), 2024. https://doi.org/10.3390/cells12162024