

Nicotinic Acetylcholine Receptor Dysfunction in Addiction and in Some Neurodegenerative and Neuropsychiatric Diseases


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
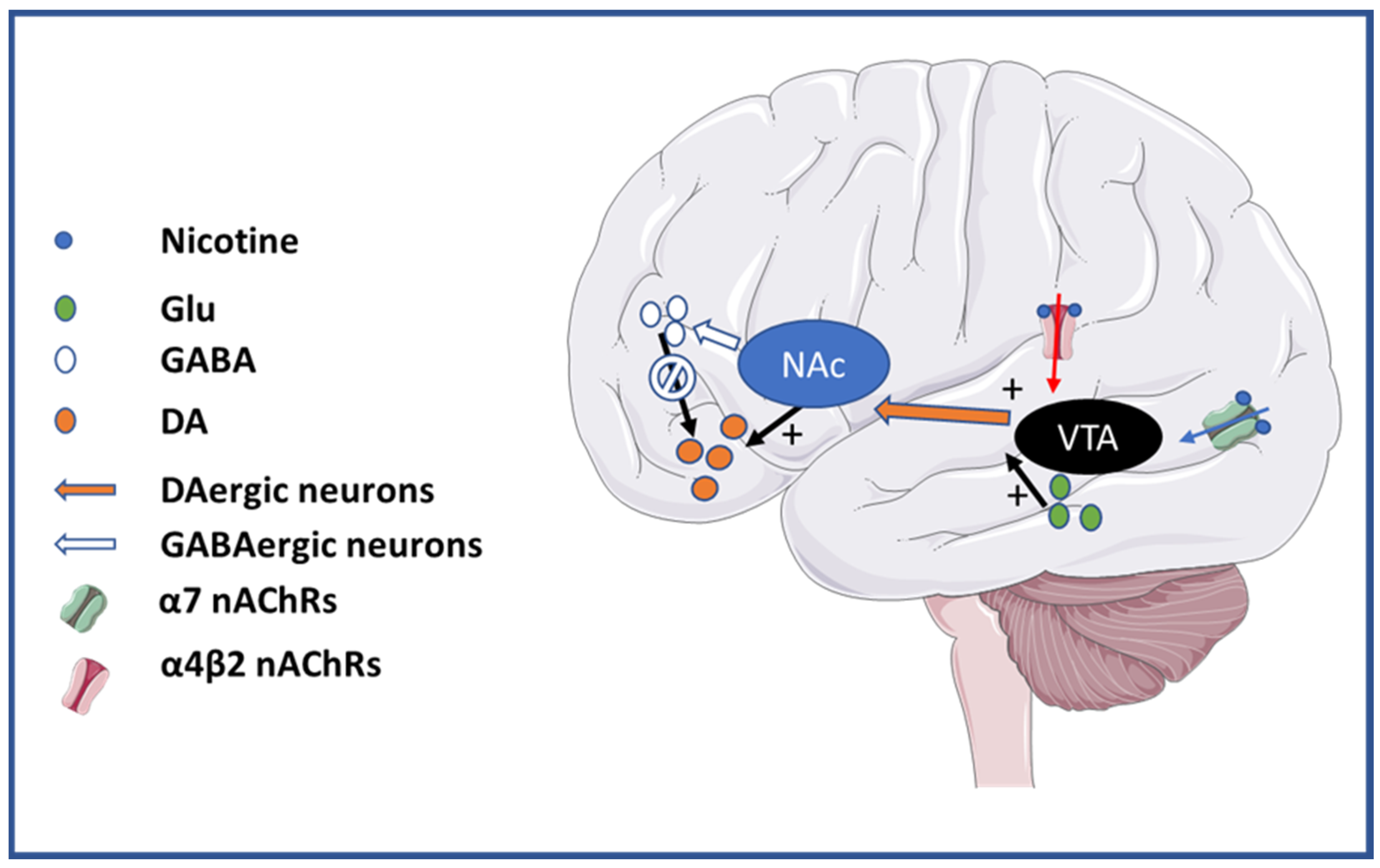
2. Addiction
3. Central and Peripheral Inflammation
4. Alzheimer Disease
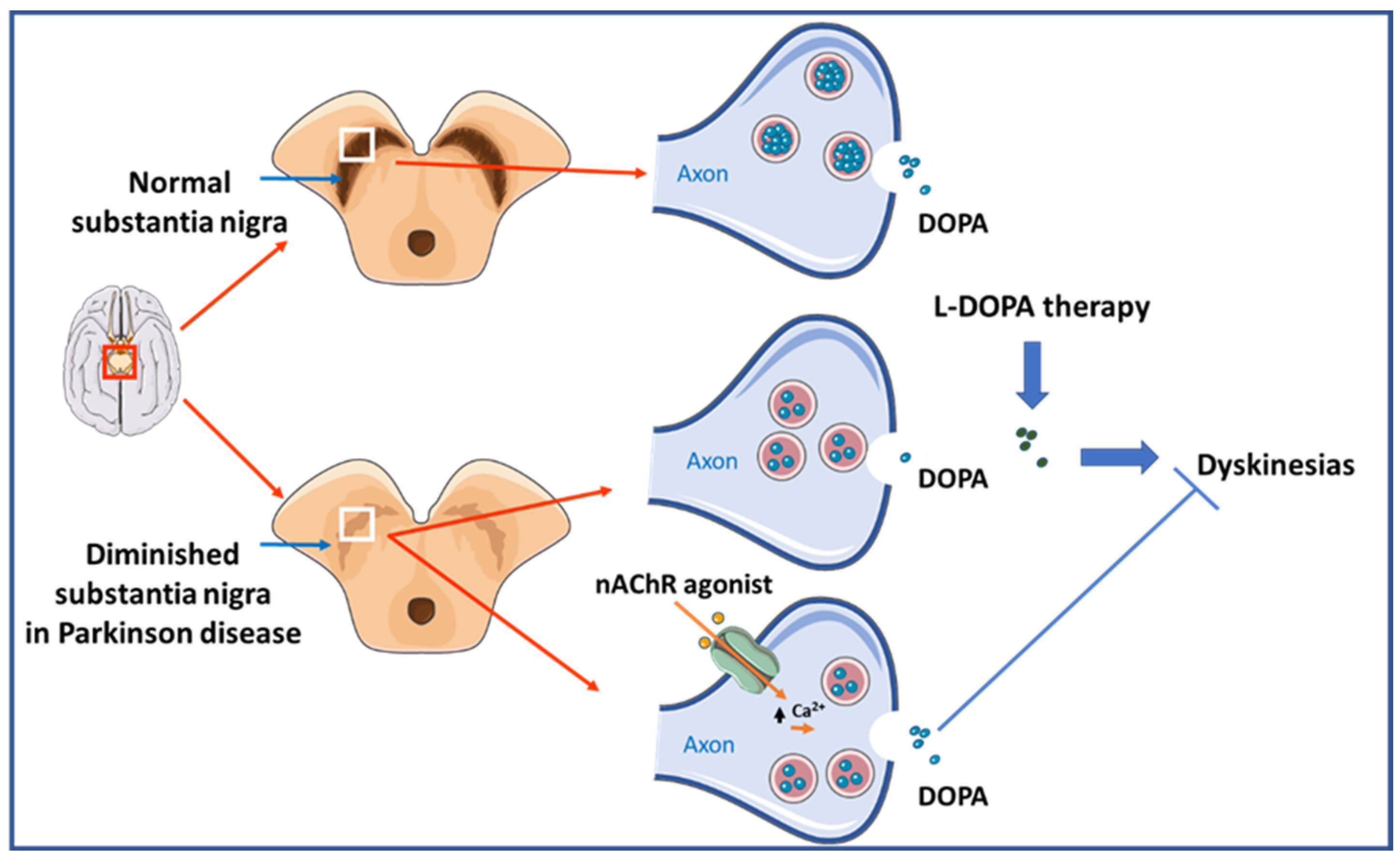
5. Parkinson Disease
6. Schizophrenia Spectrum Disorders
7. Epilepsy
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Jaiteh, M.; Taly, A.; Hénin, J. Evolution of Pentameric Ligand-Gated Ion Channels: Pro-Loop Receptors. PLoS ONE 2016, 11, e0151934. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.K.; Sattelle, D.B. Diversity of insect nicotinic acetylcholine receptor subunits. Adv. Exp. Med. Biol. 2010, 683, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Caton, M.; Ochoa, E.L.M.; Barrantes, F.J. The role of nicotinic cholinergic neurotransmission in delusional thinking. NPJ Schizophr. 2020, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Wessler, I.K.; Kirkpatrick, C.J. The Non-neuronal cholinergic system: An emerging drug target in the airways. Pulm. Pharmacol. Ther. 2001, 14, 423–434. [Google Scholar] [CrossRef]
- Giniatullin, R.; Nistri, A.; Yakel, J.L. Desensitization of nicotinic ACh receptors: Shaping cholinergic signaling. Trends Neurosci. 2005, 28, 371–378. [Google Scholar] [CrossRef]
- Picciotto, M.R.; Addy, N.A.; Mineur, Y.S.; Brunzell, D.H. It is not “either/or”: Activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog. Neurobiol. 2008, 84, 329–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Lindstrom, J. Orthosteric and allosteric potentiation of heteromeric neuronal nicotinic acetylcholine receptors. Br. J. Pharmacol. 2018, 175, 1805–1821. [Google Scholar] [CrossRef] [Green Version]
- Zoli, M.; Pucci, S.; Vilella, A.; Gotti, C. Neuronal and Extraneuronal Nicotinic Acetylcholine Receptors. Curr. Neuropharmacol. 2018, 16, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Veena, J.; Rao, B.S.S.; Srikumar, B.N. Regulation of adult neurogenesis in the hippocampus by stress, acetylcholine and dopamine. J. Nat. Sci. Biol. Med. 2011, 2, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, S.; Adams, C.; Breese, C.R.; Adler, L.E.; Bickford, P.; Byerley, W.; Coon, H.; Griffith, J.M.; Miller, C.; Myles-Worsley, M.; et al. Nicotinic receptor function in schizophrenia. Schizophr. Bull. 1996, 22, 431–445. [Google Scholar] [CrossRef]
- Dineley, K.T.; Pandya, A.A.; Yakel, J.L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 2015, 36, 96–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dani, J.A.; Bertrand, D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 699–729. [Google Scholar] [CrossRef] [PubMed]
- Taly, A.; Corringer, P.-J.; Guedin, D.; Lestage, P.; Changeux, J.-P. Nicotinic receptors: Allosteric transitions and therapeutic targets in the nervous system. Nat. Rev. Drug Discov. 2009, 8, 733–750. [Google Scholar] [CrossRef] [PubMed]
- McQuiston, A.R. Acetylcholine release and inhibitory interneuron activity in hippocampal CA1. Front. Synaptic Neurosci. 2014, 6, 20. [Google Scholar] [CrossRef]
- Oda, A.; Yamagata, K.; Nakagomi, S.; Uejima, H.; Wiriyasermkul, P.; Ohgaki, R.; Nagamori, S.; Kanai, Y.; Tanaka, H. Nicotine induces dendritic spine remodeling in cultured hippocampal neurons. J. Neurochem. 2014, 128, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Yakel, J.L. Timing-Dependent Septal Cholinergic Induction of Dynamic Hippocampal Synaptic Plasticity. Neuron 2011, 71, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pidoplichko, V.I.; Prager, E.M.; Aroniadou-Anderjaska, V.; Braga, M.F.M. α7-Containing nicotinic acetylcholine receptors on interneurons of the basolateral amygdala and their role in the regulation of the network excitability. J. Neurophysiol. 2013, 110, 2358–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udakis, M.; Wright, V.L.; Wonnacott, S.; Bailey, C.P. Integration of inhibitory and excitatory effects of α7 nicotinic acetylcholine receptor activation in the prelimbic cortex regulates network activity and plasticity. Neuropharmacology 2016, 105, 618–629. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Greenberg, M.E. Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu. Rev. Cell Dev. Biol. 2008, 24, 183–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouzat, C.; Lasala, M.; Nielsen, B.E.; Corradi, J.; Esandi, M.D.C. Molecular function of α7 nicotinic receptors as drug targets. J. Physiol. 2018, 596, 1847–1861. [Google Scholar] [CrossRef] [Green Version]
- Wessler, I.; Kirkpatrick, C.J. Acetylcholine beyond neurons: The non-neuronal cholinergic system in humans. Br. J. Pharmacol. 2008, 154, 1558–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanetti, S.R.; Ziblat, A.; Torres, N.I.; Zwirner, N.W.; Bouzat, C. Expression and Functional Role of α7 Nicotinic Receptor in Human Cytokine-stimulated Natural Killer (NK) Cells. J. Biol. Chem. 2016, 291, 16541–16552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, M.R.; Nagele, R.G. Targeting the alpha 7 nicotinic acetylcholine receptor to reduce amyloid accumulation in Alzheimer’s disease pyramidal neurons. Curr. Pharm. Des. 2006, 12, 677–684. [Google Scholar] [CrossRef]
- Freedman, R.; Hall, M.; Adler, L.E.; Leonard, S. Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol. Psychiatry 1995, 38, 22–33. [Google Scholar] [CrossRef]
- Quik, M.; Zhang, D.; McGregor, M.; Bordia, T. Alpha7 nicotinic receptors as therapeutic targets for Parkinson’s disease. Biochem. Pharmacol. 2015, 97, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Steinlein, O.K.; Magnusson, A.; Stoodt, J.; Bertrand, S.; Weiland, S.; Berkovic, S.F.; Nakken, K.O.; Propping, P.; Bertrand, D. An insertion mutation of the CHRNA4 gene in a family with autosomal dominant nocturnal frontal lobe epilepsy. Hum. Mol. Genet. 1997, 6, 943–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallés, A.S.; Barrantes, F.J. Dysregulation of Neuronal Nicotinic Acetylcholine Receptor–Cholesterol Crosstalk in Autism Spectrum Disorder. Front. Mol. Neurosci. 2021, 14, 232. [Google Scholar] [CrossRef]
- Wilens, T.E.; Decker, M.W. Neuronal nicotinic receptor agonists for the treatment of attention-deficit/hyperactivity disorder: Focus on cognition. Biochem. Pharmacol. 2007, 74, 1212–1223. [Google Scholar] [CrossRef] [Green Version]
- Philip, N.S.; Carpenter, L.L.; Tyrka, A.R.; Price, L.H. Nicotinic acetylcholine receptors and depression: A review of the preclinical and clinical literature. Psychopharmacology 2010, 212, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sciamanna, M.A.; Griesmann, G.E.; Williams, C.L.; Lennon, V.A. Nicotinic acetylcholine receptors of muscle and neuronal (alpha7) types coexpressed in a small cell lung carcinoma. J. Neurochem. 1997, 69, 2302–2311. [Google Scholar] [CrossRef] [PubMed]
- Hone, A.J.; McIntosh, J.M. Nicotinic acetylcholine receptors in neuropathic and inflammatory pain. FEBS Lett. 2018, 592, 1045–1062. [Google Scholar] [CrossRef] [PubMed]
- Hone, A.J.; Servent, D.; McIntosh, J.M. α9-containing nicotinic acetylcholine receptors and the modulation of pain. Br. J. Pharmacol. 2018, 175, 1915–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieskopf, J.S.; Mathur, J.; Limapichat, W.; Post, M.R.; Al-Qazzaz, M.; Sorge, R.E.; Martin, L.J.; Zaykin, D.V.; Smith, S.B.; Freitas, K.; et al. The nicotinic α6 subunit gene determines variability in chronic pain sensitivity via cross-inhibition of P2X2/3 receptors. Sci. Transl. Med. 2015, 7, 287ra72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti-Fine, B.M.; Milani, M.; Kaminski, H.J. Myasthenia gravis: Past, present, and future. J. Clin. Investig. 2006, 116, 2843–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, A.G.; Shen, X.-M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet. Neurol. 2015, 14, 420–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, S.; Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015, 96, 255–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz, M.L.; Barrantes, F.J. Autoimmune Attack of the Neuromuscular Junction in Myasthenia Gravis: Nicotinic Acetylcholine Receptors and Other Targets. ACS Chem. Neurosci. 2019, 10, 2186–2194. [Google Scholar] [CrossRef]
- Srinivasan, R.; Henderson, B.J.; Lester, H.A.; Richards, C.I. Pharmacological chaperoning of nAChRs: A therapeutic target for Parkinson’s disease. Pharmacol. Res. 2014, 83, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Lloret, S.; Barrantes, F.J. Deficits in cholinergic neurotransmission and their clinical correlates in Parkinson’s disease. NPJ Park. Dis. 2016, 2, 16001. [Google Scholar] [CrossRef] [Green Version]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders; American Psychiatric Association: Washington, DC, USA, 2013; ISBN 0-89042-555-8. [Google Scholar]
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016, 3, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Wittenberg, R.E.; Wolfman, S.L.; De Biasi, M.; Dani, J.A. Nicotinic acetylcholine receptors and nicotine addiction: A brief introduction. Neuropharmacology 2020, 177, 108256. [Google Scholar] [CrossRef] [PubMed]
- De Biasi, M.; Dani, J.A. Reward, addiction, withdrawal to nicotine. Annu. Rev. Neurosci. 2011, 34, 105–130. [Google Scholar] [CrossRef] [Green Version]
- Dani, J.A.; Radcliffe, K.A.; Pidoplichko, V.I. Variations in desensitization of nicotinic acetylcholine receptors from hippocampus and midbrain dopamine areas. Eur. J. Pharmacol. 2000, 393, 31–38. [Google Scholar] [CrossRef]
- Pidoplichko, V.I.; DeBiasi, M.; Williams, J.T.; Dani, J.A. Nicotine activates and desensitizes midbrain dopamine neurons. Nature 1997, 390, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Wooltorton, J.R.A.; Pidoplichko, V.I.; Broide, R.S.; Dani, J.A. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J. Neurosci. 2003, 23, 3176–3185. [Google Scholar] [CrossRef] [PubMed]
- Mansvelder, H.D.; McGehee, D.S. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 2000, 27, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Mao, D.; Gallagher, K.; McGehee, D.S. Nicotine potentiation of excitatory inputs to ventral tegmental area dopamine neurons. J. Neurosci. 2011, 31, 6710–6720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostroumov, A.; Dani, J.A. Convergent Neuronal Plasticity and Metaplasticity Mechanisms of Stress, Nicotine, and Alcohol. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Saal, D.; Dong, Y.; Bonci, A.; Malenka, R.C. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 2003, 37, 577–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, L.K.; Mao, D.; Chi, H.; Govind, A.P.; Vallejo, Y.F.; Iacoviello, M.; Herrera, S.; Cortright, J.J.; Green, W.N.; McGehee, D.S.; et al. Intermittent nicotine exposure upregulates nAChRs in VTA dopamine neurons and sensitises locomotor responding to the drug. Eur. J. Neurosci. 2013, 37, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Nashmi, R.; Xiao, C.; Deshpande, P.; McKinney, S.; Grady, S.R.; Whiteaker, P.; Huang, Q.; McClure-Begley, T.; Lindstrom, J.M.; Labarca, C.; et al. Chronic Nicotine Cell Specifically Upregulates Functional α4* Nicotinic Receptors: Basis for Both Tolerance in Midbrain and Enhanced Long-Term Potentiation in Perforant Path. J. Neurosci. 2007, 27, 8202–8218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feduccia, A.; Chatterjee, S.; Bartlett, S. Neuronal nicotinic acetylcholine receptors: Neuroplastic changes underlying alcohol and nicotine addictions. Front. Mol. Neurosci. 2012, 5, 83. [Google Scholar] [CrossRef] [Green Version]
- Fenster, C.P.; Hicks, J.H.; Beckman, M.L.; Covernton, P.J.; Quick, M.W.; Lester, R.A. Desensitization of nicotinic receptors in the central nervous system. Ann. N. Y. Acad. Sci. 1999, 868, 620–623. [Google Scholar] [CrossRef]
- Pirhaji, L.; Milani, P.; Dalin, S.; Wassie, B.T.; Dunn, D.E.; Fenster, R.J.; Avila-Pacheco, J.; Greengard, P.; Clish, C.B.; Heiman, M.; et al. Identifying therapeutic targets by combining transcriptional data with ordinal clinical measurements. Nat. Commun. 2017, 8, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, B.J.; Srinivasan, R.; Nichols, W.A.; Dilworth, C.N.; Gutierrez, D.F.; Mackey, E.D.W.; McKinney, S.; Drenan, R.M.; Richards, C.I.; Lester, H.A. Nicotine exploits a COPI-mediated process for chaperone-mediated up-regulation of its receptors. J. Gen. Physiol. 2014, 143, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Matta, S.G.; Balfour, D.J.; Benowitz, N.L.; Boyd, R.T.; Buccafusco, J.J.; Caggiula, A.R.; Craig, C.R.; Collins, A.C.; Damaj, M.I.; Donny, E.C.; et al. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology 2007, 190, 269–319. [Google Scholar] [CrossRef] [PubMed]
- Ikemoto, S. Brain reward circuitry beyond the mesolimbic dopamine system: A neurobiological theory. Neurosci. Biobehav. Rev. 2010, 35, 129–150. [Google Scholar] [CrossRef] [Green Version]
- Lecca, S.; Meye, F.J.; Mameli, M. The lateral habenula in addiction and depression: An anatomical, synaptic and behavioral overview. Eur. J. Neurosci. 2014, 39, 1170–1178. [Google Scholar] [CrossRef]
- Meye, F.J.; Trusel, M.; Soiza-Reilly, M.; Mameli, M. Neural circuit adaptations during drug withdrawal—Spotlight on the lateral habenula. Pharmacol. Biochem. Behav. 2017, 162, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.; Vollmayr, B.; Djodari-Irani, A.; Klein, J.; Sartorius, A. Pharmacological inhibition of the lateral habenula improves depressive-like behavior in an animal model of treatment resistant depression. Behav. Brain Res. 2011, 216, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.J.; Markou, A. Neurobiology of the nicotine withdrawal syndrome. Pharmacol. Biochem. Behav. 2001, 70, 531–549. [Google Scholar] [CrossRef] [PubMed]
- Picciotto, M.R.; Kenny, P.J. Molecular mechanisms underlying behaviors related to nicotine addiction. Cold Spring Harb. Perspect. Med. 2013, 3, a012112. [Google Scholar] [CrossRef] [PubMed]
- Kanasuwan, A.; Deuther-Conrad, W.; Chongruchiroj, S.; Sarasamkan, J.; Chotipanich, C.; Vajragupta, O.; Arunrungvichian, K. Selective α(3)β(4) Nicotinic Acetylcholine Receptor Ligand as a Potential Tracer for Drug Addiction. Int. J. Mol. Sci. 2023, 24, 3614. [Google Scholar] [CrossRef] [PubMed]
- Grady, S.R.; Moretti, M.; Zoli, M.; Marks, M.J.; Zanardi, A.; Pucci, L.; Clementi, F.; Gotti, C. Rodent habenulo-interpeduncular pathway expresses a large variety of uncommon nAChR subtypes, but only the alpha3beta4* and alpha3beta3beta4* subtypes mediate acetylcholine release. J. Neurosci. 2009, 29, 2272–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, C.D.; Lu, Q.; Johnson, P.M.; Marks, M.J.; Kenny, P.J. Habenular α5 nicotinic receptor subunit signalling controls nicotine intake. Nature 2011, 471, 597–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, I.; Dani, J.A.; De Biasi, M. Nicotine withdrawal. Curr. Top. Behav. Neurosci. 2015, 24, 99–123. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, I.; Dani, J.A.; De Biasi, M. The medial habenula and interpeduncular nucleus circuitry is critical in addiction, anxiety, and mood regulation. J. Neurochem. 2017, 142, 130–143. [Google Scholar] [CrossRef] [Green Version]
- Zaveri, N.; Jiang, F.; Olsen, C.; Polgar, W.; Toll, L. Novel α3β4 nicotinic acetylcholine receptor-selective ligands. Discovery, structure-activity studies, and pharmacological evaluation. J. Med. Chem. 2010, 53, 8187–8191. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; Yang, Z.; Cui, W.; Li, M.D. Crucial roles of the CHRNB3-CHRNA6 gene cluster on chromosome 8 in nicotine dependence: Update and subjects for future research. Transl. Psychiatry 2016, 6, e843. [Google Scholar] [CrossRef] [Green Version]
- Cannon, D.S.; Mermelstein, R.J.; Hedeker, D.; Coon, H.; Cook, E.H.; McMahon, W.M.; Hamil, C.; Dunn, D.; Weiss, R.B. Effect of neuronal nicotinic acetylcholine receptor genes (CHRN) on longitudinal cigarettes per day in adolescents and young adults. Nicotine Tob. Res. 2014, 16, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Counotte, D.S.; Smit, A.B.; Pattij, T.; Spijker, S. Development of the motivational system during adolescence, and its sensitivity to disruption by nicotine. Dev. Cogn. Neurosci. 2011, 1, 430–443. [Google Scholar] [CrossRef] [Green Version]
- Day, M.; Pan, J.B.; Buckley, M.J.; Cronin, E.; Hollingsworth, P.R.; Hirst, W.D.; Navarra, R.; Sullivan, J.P.; Decker, M.W.; Fox, G.B. Differential effects of ciproxifan and nicotine on impulsivity and attention measures in the 5-choice serial reaction time test. Biochem. Pharmacol. 2007, 73, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Grottick, A.J.; Higgins, G.A. Effect of subtype selective nicotinic compounds on attention as assessed by the five-choice serial reaction time task. Behav. Brain Res. 2000, 117, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Rycroft, N.; Rusted, J.M.; Hutton, S.B. Acute effects of nicotine on visual search tasks in young adult smokers. Psychopharmacology 2005, 181, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Van Gaalen, M.M.; Brueggeman, R.J.; Bronius, P.F.C.; Schoffelmeer, A.N.M.; Vanderschuren, L.J.M.J. Behavioral disinhibition requires dopamine receptor activation. Psychopharmacology 2006, 187, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Boksa, P. Smoking, psychiatric illness and the brain. J. Psychiatry Neurosci. 2017, 42, 147–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalman, D.; Morissette, S.B.; George, T.P. Co-morbidity of smoking in patients with psychiatric and substance use disorders. Am. J. Addict. 2005, 14, 106–123. [Google Scholar] [CrossRef] [Green Version]
- Breese, C.R.; Lee, M.J.; Adams, C.E.; Sullivan, B.; Logel, J.; Gillen, K.M.; Marks, M.J.; Collins, A.C.; Leonard, S. Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology 2000, 23, 351–364. [Google Scholar] [CrossRef] [Green Version]
- Durany, N.; Zöchling, R.; Boissl, K.W.; Paulus, W.; Ransmayr, G.; Tatschner, T.; Danielczyk, W.; Jellinger, K.; Deckert, J.; Riederer, P. Human post-mortem striatal alpha4beta2 nicotinic acetylcholine receptor density in schizophrenia and Parkinson’s syndrome. Neurosci. Lett. 2000, 287, 109–112. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, D.C.; Esterlis, I.; Carbuto, M.; Krasenics, M.; Seibyl, J.; Bois, F.; Pittman, B.; Ranganathan, M.; Cosgrove, K.; Staley, J. Lower ß2*-nicotinic acetylcholine receptor availability in smokers with schizophrenia. Am. J. Psychiatry 2012, 169, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.N.; Rasmussen, B.A.; Perry, D.C. Binding and functional activity of nicotinic cholinergic receptors in selected rat brain regions are increased following long-term but not short-term nicotine treatment. J. Neurochem. 2004, 90, 40–49. [Google Scholar] [CrossRef]
- Rabin, R.A.; George, T.P. A review of co-morbid tobacco and cannabis use disorders: Possible mechanisms to explain high rates of co-use. Am. J. Addict. 2015, 24, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Amos, A.; Wiltshire, S.; Bostock, Y.; Haw, S.; McNeill, A. ’You can’t go without a fag...you need it for your hash’—A qualitative exploration of smoking, cannabis and young people. Addiction 2004, 99, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Ream, G.L.; Benoit, E.; Johnson, B.D.; Dunlap, E. Smoking tobacco along with marijuana increases symptoms of cannabis dependence. Drug Alcohol Depend. 2008, 95, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallés, A.S.; Barrantes, F.J. Nanoscale Sub-Compartmentalization of the Dendritic Spine Compartment. Biomolecules 2021, 11, 1697. [Google Scholar] [CrossRef] [PubMed]
- Buzzi, B.; Koseli, E.; Moncayo, L.; Shoaib, M.; Damaj, M.I. Role of neuronal nicotinic acetylcholine receptors in cannabinoid dependence. Pharmacol. Res. 2023, 191, 106746. [Google Scholar] [CrossRef] [PubMed]
- Ramo, D.E.; Liu, H.; Prochaska, J.J. Tobacco and marijuana use among adolescents and young adults: A systematic review of their co-use. Clin. Psychol. Rev. 2012, 32, 105–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valjent, E.; Mitchell, J.M.; Besson, M.-J.; Caboche, J.; Maldonado, R. Behavioural and biochemical evidence for interactions between Delta 9-tetrahydrocannabinol and nicotine. Br. J. Pharmacol. 2002, 135, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Lubman, D.I.; Cheetham, A.; Yücel, M. Cannabis and adolescent brain development. Pharmacol. Ther. 2015, 148, 1–16. [Google Scholar] [CrossRef]
- Manwell, L.A.; Miladinovic, T.; Raaphorst, E.; Rana, S.; Malecki, S.; Mallet, P.E. Chronic nicotine exposure attenuates the effects of Δ(9) -tetrahydrocannabinol on anxiety-related behavior and social interaction in adult male and female rats. Brain Behav. 2019, 9, e01375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellazizzo, L.; Potvin, S.; Giguère, S.; Dumais, A. Evidence on the acute and residual neurocognitive effects of cannabis use in adolescents and adults: A systematic meta-review of meta-analyses. Addiction 2022, 117, 1857–1870. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Dar, M.S. Mouse cerebellar nicotinic-cholinergic receptor modulation of Delta9-THC ataxia: Role of the alpha4beta2 subtype. Brain Res. 2006, 1115, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Justinova, Z.; Mascia, P.; Wu, H.-Q.; Secci, M.E.; Redhi, G.H.; Panlilio, L.V.; Scherma, M.; Barnes, C.; Parashos, A.; Zara, T.; et al. Reducing cannabinoid abuse and preventing relapse by enhancing endogenous brain levels of kynurenic acid. Nat. Neurosci. 2013, 16, 1652–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Jiang, Y.; Wedow, R.; Li, Y.; Brazel, D.M.; Chen, F.; Datta, G.; Davila-Velderrain, J.; McGuire, D.; Tian, C.; et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 2019, 51, 237–244. [Google Scholar] [CrossRef]
- Gatta, V.; Mengod, G.; Reale, M.; Tata, A.M. Possible Correlation between Cholinergic System Alterations and Neuro/Inflammation in Multiple Sclerosis. Biomedicines 2020, 8, 153. [Google Scholar] [CrossRef] [PubMed]
- Piovesana, R.; Salazar Intriago, M.S.; Dini, L.; Tata, A.M. Cholinergic Modulation of Neuroinflammation: Focus on α7 Nicotinic Receptor. Int. J. Mol. Sci. 2021, 22, 4912. [Google Scholar] [CrossRef] [PubMed]
- Shytle, R.D.; Mori, T.; Townsend, K.; Vendrame, M.; Sun, N.; Zeng, J.; Ehrhart, J.; Silver, A.A.; Sanberg, P.R.; Tan, J. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J. Neurochem. 2004, 89, 337–343. [Google Scholar] [CrossRef]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain. Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yu, M.; Ochani, M.; Amelia, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, N.; Ulloa, L.; et al. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Lu, X.-X.; Hong, Z.-Q.; Tan, Z.; Sui, M.-H.; Zhuang, Z.-Q.; Liu, H.-H.; Zheng, X.-Y.; Yan, T.-B.; Geng, D.-F.; Jin, D.-M. Nicotinic Acetylcholine Receptor Alpha7 Subunit Mediates Vagus Nerve Stimulation-Induced Neuroprotection in Acute Permanent Cerebral Ischemia by a7nAchR/JAK2 Pathway. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2017, 23, 6072–6081. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Inden, M.; Ueda, T.; Asaka, Y.; Kurita, H.; Hozumi, I. The neuroprotective effects of activated α7 nicotinic acetylcholine receptor against mutant copper-zinc superoxide dismutase 1-mediated toxicity. Sci. Rep. 2020, 10, 22157. [Google Scholar] [CrossRef]
- De Jonge, W.J.; Ulloa, L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br. J. Pharmacol. 2007, 151, 915–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, H.; McIntire, J.; Ryan, S.; Dunah, A.; Loring, R. Anti-inflammatory effects of astroglial α7 nicotinic acetylcholine receptors are mediated by inhibition of the NF-κB pathway and activation of the Nrf2 pathway. J. Neuroinflamm. 2017, 14, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kihara, T.; Sawada, H.; Nakamizo, T.; Kanki, R.; Yamashita, H.; Maelicke, A.; Shimohama, S. Galantamine modulates nicotinic receptor and blocks Aβ-enhanced glutamate toxicity. Biochem. Biophys. Res. Commun. 2004, 325, 976–982. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, J.; Wu, J.; Zhu, C.; Hui, Y.; Han, Y.; Huang, Z.; Ellsworth, K.; Fan, W. α7 nicotinic acetylcholine receptor-mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. J. Neuroinflamm. 2012, 9, 98. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.B.; Schrader, J.W.; Kim, S.U. p38 map kinase regulates TNF-alpha production in human astrocytes and microglia by multiple mechanisms. Cytokine 2000, 12, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Youssef, M.; Ibrahim, A.; Akashi, K.; Hossain, M.S. PUFA-Plasmalogens Attenuate the LPS-Induced Nitric Oxide Production by Inhibiting the NF-kB, p38 MAPK and JNK Pathways in Microglial Cells. Neuroscience 2019, 397, 18–30. [Google Scholar] [CrossRef]
- Hone, A.J.; McIntosh, J.M. Nicotinic acetylcholine receptors: Therapeutic targets for novel ligands to treat pain and inflammation. Pharmacol. Res. 2023, 190, 106715. [Google Scholar] [CrossRef]
- Costantini, E.; Carrarini, C.; Borrelli, P.; De Rosa, M.; Calisi, D.; Consoli, S.; D’Ardes, D.; Cipollone, F.; Di Nicola, M.; Onofrj, M.; et al. Different peripheral expression patterns of the nicotinic acetylcholine receptor in dementia with Lewy bodies and Alzheimer’s disease. Immun. Ageing 2023, 20, 3. [Google Scholar] [CrossRef]
- Chernyavsky, A.I.; Arredondo, J.; Skok, M.; Grando, S.A. Auto/paracrine control of inflammatory cytokines by acetylcholine in macrophage-like U937 cells through nicotinic receptors. Int. Immunopharmacol. 2010, 10, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Maldifassi, M.C.; Atienza, G.; Arnalich, F.; López-Collazo, E.; Cedillo, J.L.; Martín-Sánchez, C.; Bordas, A.; Renart, J.; Montiel, C. A new IRAK-M-mediated mechanism implicated in the anti-inflammatory effect of nicotine via α7 nicotinic receptors in human macrophages. PLoS ONE 2014, 9, e108397. [Google Scholar] [CrossRef] [Green Version]
- Alen, N. V The cholinergic anti-inflammatory pathway in humans: State-of-the-art review and future directions. Neurosci. Biobehav. Rev. 2022, 136, 104622. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Wang, H.; Czura, C.J.; Friedman, S.G.; Tracey, K.J. The cholinergic anti-inflammatory pathway: A missing link in neuroimmunomodulation. Mol. Med. 2003, 9, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef]
- Richter, K.; Papke, R.L.; Stokes, C.; Roy, D.C.; Espinosa, E.S.; Wolf, P.M.K.; Hecker, A.; Liese, J.; Singh, V.K.; Padberg, W.; et al. Comparison of the Anti-inflammatory Properties of Two Nicotinic Acetylcholine Receptor Ligands, Phosphocholine and pCF3-diEPP. Front. Cell. Neurosci. 2022, 16, 779081. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Mathes, V.; Fronius, M.; Althaus, M.; Hecker, A.; Krasteva-Christ, G.; Padberg, W.; Hone, A.J.; McIntosh, J.M.; Zakrzewicz, A.; et al. Phosphocholine – an agonist of metabotropic but not of ionotropic functions of α9-containing nicotinic acetylcholine receptors. Sci. Rep. 2016, 6, 28660. [Google Scholar] [CrossRef] [Green Version]
- Hecker, A.; Küllmar, M.; Wilker, S.; Richter, K.; Zakrzewicz, A.; Atanasova, S.; Mathes, V.; Timm, T.; Lerner, S.; Klein, J.; et al. Phosphocholine-Modified Macromolecules and Canonical Nicotinic Agonists Inhibit ATP-Induced IL-1β Release. J. Immunol. 2015, 195, 2325–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courties, A.; Boussier, J.; Hadjadj, J.; Yatim, N.; Barnabei, L.; Péré, H.; Veyer, D.; Kernéis, S.; Carlier, N.; Pène, F.; et al. Regulation of the acetylcholine/α7nAChR anti-inflammatory pathway in COVID-19 patients. Sci. Rep. 2021, 11, 11886. [Google Scholar] [CrossRef]
- Simon, T.; Kirk, J.; Dolezalova, N.; Guyot, M.; Panzolini, C.; Bondue, A.; Lavergne, J.; Hugues, S.; Hypolite, N.; Saeb-Parsy, K.; et al. The cholinergic anti-inflammatory pathway inhibits inflammation without lymphocyte relay. Front. Neurosci. 2023, 17, 1125492. [Google Scholar] [CrossRef]
- Vonck, K.; Raedt, R.; Naulaerts, J.; De Vogelaere, F.; Thiery, E.; Van Roost, D.; Aldenkamp, B.; Miatton, M.; Boon, P. Vagus nerve stimulation…25 years later! What do we know about the effects on cognition? Neurosci. Biobehav. Rev. 2014, 45, 63–71. [Google Scholar] [CrossRef]
- Vargas-Caballero, M.; Warming, H.; Walker, R.; Holmes, C.; Cruickshank, G.; Patel, B. Vagus Nerve Stimulation as a Potential Therapy in Early Alzheimer’s Disease: A Review. Front. Hum. Neurosci. 2022, 16, 866434. [Google Scholar] [CrossRef]
- Sigurdsson, H.P.; Raw, R.; Hunter, H.; Baker, M.R.; Taylor, J.-P.; Rochester, L.; Yarnall, A.J. Noninvasive vagus nerve stimulation in Parkinson’s disease: Current status and future prospects. Expert Rev. Med. Devices 2021, 18, 971–984. [Google Scholar] [CrossRef]
- Smucny, J.; Visani, A.; Tregellas, J.R. Could vagus nerve stimulation target hippocampal hyperactivity to improve cognition in schizophrenia? Front. Psychiatry 2015, 6, 43. [Google Scholar] [CrossRef] [Green Version]
- Vallés, A.S.; Borroni, M.V.; Barrantes, F.J. Targeting brain α7 nicotinic acetylcholine receptors in alzheimer’s disease: Rationale and current status. CNS Drugs 2014, 28, 975–987. [Google Scholar] [CrossRef]
- Price, J.L.; Davis, P.B.; Morris, J.C.; White, D.L. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer’s disease. Neurobiol. Aging 1991, 12, 295–312. [Google Scholar] [CrossRef]
- Cummings, J.L. Alzheimer’s disease. N. Engl. J. Med. 2004, 351, 56–67. [Google Scholar] [CrossRef]
- Hoskin, J.L.; Al-Hasan, Y.; Sabbagh, M.N. Nicotinic Acetylcholine Receptor Agonists for the Treatment of Alzheimer’s Dementia: An Update. Nicotine Tob. Res. 2019, 21, 370–376. [Google Scholar] [CrossRef]
- Wu, J.; Ishikawa, M.; Zhang, J.; Hashimoto, K. Brain imaging of nicotinic receptors in Alzheimer’s disease. Int. J. Alzheimers. Dis. 2010, 2010, 548913. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.-G.; Qian, Y.-H. Alpha 7 nicotinic acetylcholine receptor and its effects on Alzheimer’s disease. Neuropeptides 2019, 73, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Schliebs, R.; Arendt, T. The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 2011, 221, 555–563. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Green, K.N.; Liang, K.; Tran, L.; Chen, Y.; Leslie, F.M.; LaFerla, F.M. Chronic nicotine administration exacerbates tau pathology in a transgenic model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 3046–3051. [Google Scholar] [CrossRef] [PubMed]
- Barrantes, F.J.; Borroni, V.; Vallés, S. Neuronal nicotinic acetylcholine receptor-cholesterol crosstalk in Alzheimer’s disease. FEBS Lett. 2010, 584, 1856–1863. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, Y.; Liu, T.; Mao, Y.; Peng, B. Novel Microglia-based Therapeutic Approaches to Neurodegenerative Disorders. Neurosci. Bull. 2023, 39, 491–502. [Google Scholar] [CrossRef]
- Xie, J.; Van Hoecke, L.; Vandenbroucke, R.E. The Impact of Systemic Inflammation on Alzheimer’s Disease Pathology. Front. Immunol. 2021, 12, 796867. [Google Scholar] [CrossRef]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [Green Version]
- Leung, R.; Proitsi, P.; Simmons, A.; Lunnon, K.; Güntert, A.; Kronenberg, D.; Pritchard, M.; Tsolaki, M.; Mecocci, P.; Kloszewska, I.; et al. Inflammatory proteins in plasma are associated with severity of Alzheimer’s disease. PLoS ONE 2013, 8, e64971. [Google Scholar] [CrossRef] [Green Version]
- Motta, M.; Imbesi, R.; Di Rosa, M.; Stivala, F.; Malaguarnera, L. Altered plasma cytokine levels in Alzheimer’s disease: Correlation with the disease progression. Immunol. Lett. 2007, 114, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, N.; Wang, K.; Zhang, Z.; Wang, Y. Identification of α7 nicotinic acetylcholine receptor on hippocampal astrocytes cultured in vitro and its role on inflammatory mediator secretion. Neural Regen. Res. 2012, 7, 1709–1714. [Google Scholar] [CrossRef]
- Ballinger, E.C.; Ananth, M.; Talmage, D.A.; Role, L.W. Basal Forebrain Cholinergic Circuits and Signaling in Cognition and Cognitive Decline. Neuron 2016, 91, 1199–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oz, M.; Lorke, D.E.; Yang, K.-H.S.; Petroianu, G. On the interaction of β-amyloid peptides and α7-nicotinic acetylcholine receptors in Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Di Lascio, S.; Fornasari, D.; Benfante, R. The Human-Restricted Isoform of the α7 nAChR, CHRFAM7A: A Double-Edged Sword in Neurological and Inflammatory Disorders. Int. J. Mol. Sci. 2022, 23, 3463. [Google Scholar] [CrossRef]
- Burns, L.H.; Pei, Z.; Wang, H.-Y. Targeting α7 nicotinic acetylcholine receptors and their protein interactions in Alzheimer’s disease drug development. Drug Dev. Res. 2023. early view. [Google Scholar] [CrossRef]
- Gault, J.; Robinson, M.; Berger, R.; Drebing, C.; Logel, J.; Hopkins, J.; Moore, T.; Jacobs, S.; Meriwether, J.; Choi, M.J.; et al. Genomic Organization and Partial Duplication of the Human α7 Neuronal Nicotinic Acetylcholine Receptor Gene (CHRNA7). Genomics 1998, 52, 173–185. [Google Scholar] [CrossRef]
- Riley, B.; Williamson, M.; Collier, D.; Wilkie, H.; Makoff, A. A 3-Mb map of a large Segmental duplication overlapping the alpha7-nicotinic acetylcholine receptor gene (CHRNA7) at human 15q13-q14. Genomics 2002, 79, 197–209. [Google Scholar] [CrossRef]
- O’Bleness, M.; Searles, V.B.; Varki, A.; Gagneux, P.; Sikela, J.M. Evolution of genetic and genomic features unique to the human lineage. Nat. Rev. Genet. 2012, 13, 853–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flomen, R.H.; Davies, A.F.; Di Forti, M.; La Cascia, C.; Mackie-Ogilvie, C.; Murray, R.; Makoff, A.J. The copy number variant involving part of the alpha7 nicotinic receptor gene contains a polymorphic inversion. Eur. J. Hum. Genet. 2008, 16, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Szafranski, P.; Schaaf, C.P.; Person, R.E.; Gibson, I.B.; Xia, Z.; Mahadevan, S.; Wiszniewska, J.; Bacino, C.A.; Lalani, S.; Potocki, L.; et al. Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA7: Benign or pathological? Hum. Mutat. 2010, 31, 840–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araud, T.; Graw, S.; Berger, R.; Lee, M.; Neveu, E.; Bertrand, D.; Leonard, S. The chimeric gene CHRFAM7A, a partial duplication of the CHRNA7 gene, is a dominant negative regulator of α7*nAChR function. Biochem. Pharmacol. 2011, 82, 904–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihnatovych, I.; Nayak, T.K.; Ouf, A.; Sule, N.; Birkaya, B.; Chaves, L.; Auerbach, A.; Szigeti, K. iPSC model of CHRFAM7A effect on α7 nicotinic acetylcholine receptor function in the human context. Transl. Psychiatry 2019, 9, 59. [Google Scholar] [CrossRef] [Green Version]
- Szigeti, K.; Ihnatovych, I.; Birkaya, B.; Chen, Z.; Ouf, A.; Indurthi, D.C.; Bard, J.E.; Kann, J.; Adams, A.; Chaves, L.; et al. CHRFAM7A: A human specific fusion gene, accounts for the translational gap for cholinergic strategies in Alzheimer’s disease. EBioMedicine 2020, 59, 102892. [Google Scholar] [CrossRef]
- Terry, A.V.; Jones, K.; Bertrand, D. Nicotinic acetylcholine receptors in neurological and psychiatric diseases. Pharmacol. Res. 2023, 191, 106764. [Google Scholar] [CrossRef] [PubMed]
- Hsia, A.Y.; Masliah, E.; McConlogue, L.; Yu, G.Q.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Malenka, R.C.; Nicoll, R.A.; Mucke, L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 1999, 96, 3228–3233. [Google Scholar] [CrossRef] [PubMed]
- Orr-Urtreger, A.; Göldner, F.M.; Saeki, M.; Lorenzo, I.; Goldberg, L.; De Biasi, M.; Dani, J.A.; Patrick, J.W.; Beaudet, A.L. Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J. Neurosci. 1997, 17, 9165–9171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziewczapolski, G.; Glogowski, C.M.; Masliah, E.; Heinemann, S.F. Deletion of the alpha 7 nicotinic acetylcholine receptor gene improves cognitive deficits and synaptic pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 2009, 29, 8805–8815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maroli, A.; Di Lascio, S.; Drufuca, L.; Cardani, S.; Setten, E.; Locati, M.; Fornasari, D.; Benfante, R. Effect of donepezil on the expression and responsiveness to LPS of CHRNA7 and CHRFAM7A in macrophages: A possible link to the cholinergic anti-inflammatory pathway. J. Neuroimmunol. 2019, 332, 155–166. [Google Scholar] [CrossRef]
- Meshul, C.K.; Kamel, D.; Moore, C.; Kay, T.S.; Krentz, L. Nicotine alters striatal glutamate function and decreases the apomorphine-induced contralateral rotations in 6-OHDA-lesioned rats. Exp. Neurol. 2002, 175, 257–274. [Google Scholar] [CrossRef]
- Tizabi, Y.; Getachew, B. Nicotinic Receptor Intervention in Parkinson’s Disease: Future Directions. Clin. Pharmacol. Transl. Med. 2017, 1, 14–19. [Google Scholar] [PubMed]
- Tizabi, Y.; Getachew, B.; Csoka, A.B.; Manaye, K.F.; Copeland, R.L. Novel targets for parkinsonism-depression comorbidity. Prog. Mol. Biol. Transl. Sci. 2019, 167, 1–24. [Google Scholar] [CrossRef]
- Lieberman, A.; Deep, A.; Olson, M.C.; Smith Hussain, V.; Frames, C.W.; McCauley, M.; Lockhart, T.E. Falls When Standing, Falls When Walking: Different Mechanisms, Different Outcomes in Parkinson Disease. Cureus 2019, 11, e5329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tizabi, Y.; Getachew, B.; Aschner, M. Novel Pharmacotherapies in Parkinson’s Disease. Neurotox. Res. 2021, 39, 1381–1390. [Google Scholar] [CrossRef]
- Stuckenholz, V.; Bacher, M.; Balzer-Geldsetzer, M.; Alvarez-Fischer, D.; Oertel, W.H.; Dodel, R.C.; Noelker, C. The α7 nAChR agonist PNU-282987 reduces inflammation and MPTP-induced nigral dopaminergic cell loss in mice. J. Parkinsons. Dis. 2013, 3, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.E.; Ross, S.A.; Drago, J.; Loiacono, R.E. Dose-related neuroprotective effects of chronic nicotine in 6-hydroxydopamine treated rats, and loss of neuroprotection in alpha4 nicotinic receptor subunit knockout mice. Br. J. Pharmacol. 2001, 132, 1650–1656. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Mallela, A.; Sohn, D.; Carroll, F.I.; Bencherif, M.; Letchworth, S.; Quik, M. Nicotinic receptor agonists reduce L-DOPA-induced dyskinesias in a monkey model of Parkinson’s disease. J. Pharmacol. Exp. Ther. 2013, 347, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, T.H.; Huot, P.; Fox, S.H.; Koprich, J.B.; Szeliga, K.T.; James, J.W.; Graef, J.D.; Letchworth, S.R.; Jordan, K.G.; Hill, M.P.; et al. TC-8831, a nicotinic acetylcholine receptor agonist, reduces L-DOPA-induced dyskinesia in the MPTP macaque. Neuropharmacology 2013, 73, 337–347. [Google Scholar] [CrossRef]
- Zhang, D.; Bordia, T.; McGregor, M.; McIntosh, J.M.; Decker, M.W.; Quik, M. ABT-089 and ABT-894 reduce levodopa-induced dyskinesias in a monkey model of Parkinson’s disease. Mov. Disord. 2014, 29, 508–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mather, J.; Burdette, C.; Posener, A.; Leventer, Y.; Poole, D.; Fox, R.; Johnston, H. Potential of AZD1446, a novel nicotinic agonist, for the treatment of L-DOPA-induced dyskinesia in Parkinson’s disease. Soc. Neurosci. Abstr 2014, 43, 137. [Google Scholar]
- Zhang, D.; McGregor, M.; Decker, M.W.; Quik, M. The α7 nicotinic receptor agonist ABT-107 decreases L-Dopa-induced dyskinesias in parkinsonian monkeys. J. Pharmacol. Exp. Ther. 2014, 351, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, T.; Grégoire, L.; Feuerbach, D.; Elbast, W.; Weiss, M.; Gomez-Mancilla, B. AQW051, a novel and selective nicotinic acetylcholine receptor α7 partial agonist, reduces l-Dopa-induced dyskinesias and extends the duration of l-Dopa effects in parkinsonian monkeys. Park. Relat. Disord. 2014, 20, 1119–1123. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Bi, G.; Han, S.; Huang, R. MicroRNAs Play a Role in Parkinson’s Disease by Regulating Microglia Function: From Pathogenetic Involvement to Therapeutic Potential. Front. Mol. Neurosci. 2021, 14, 744942. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Du, L.; Hong, J.S. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J. Pharmacol. Exp. Ther. 2000, 293, 607–617. [Google Scholar] [PubMed]
- Vidović, M.; Rikalovic, M.G. Alpha-Synuclein Aggregation Pathway in Parkinson’s Disease: Current Status and Novel Therapeutic Approaches. Cells 2022, 11, 1732. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef]
- Patel, K.R.; Cherian, J.; Gohil, K.; Atkinson, D. Schizophrenia: Overview and treatment options. Pharm. Ther. 2014, 39, 638–645. [Google Scholar]
- Kalkman, H.O.; Feuerbach, D. Modulatory effects of α7 nAChRs on the immune system and its relevance for CNS disorders. Cell. Mol. Life Sci. 2016, 73, 2511–2530. [Google Scholar] [CrossRef] [Green Version]
- Mueser, K.T.; Jeste, D.V. (Eds.) Clinical Handbook of Schizophrenia; The Guilford Press: New York, NY, USA, 2008; ISBN 978-1-59385-652-6. [Google Scholar]
- Müller, N. Inflammation in Schizophrenia: Pathogenetic Aspects and Therapeutic Considerations. Schizophr. Bull. 2018, 44, 973–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trubetskoy, V.; Pardiñas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.-Y.; Dennison, C.A.; Hall, L.S.; et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef]
- Weiland, S.; Bertrand, D.; Leonard, S. Neuronal nicotinic acetylcholine receptors: From the gene to the disease. Behav. Brain Res. 2000, 113, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.; Freedman, R. Genetics of chromosome 15q13-q14 in schizophrenia. Biol. Psychiatry 2006, 60, 115–122. [Google Scholar] [CrossRef]
- Sinkus, M.L.; Lee, M.J.; Gault, J.; Logel, J.; Short, M.; Freedman, R.; Christian, S.L.; Lyon, J.; Leonard, S. A 2-base pair deletion polymorphism in the partial duplication of the alpha7 nicotinic acetylcholine gene (CHRFAM7A) on chromosome 15q14 is associated with schizophrenia. Brain Res. 2009, 1291, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Leonard, S.; Gault, J.; Hopkins, J.; Logel, J.; Vianzon, R.; Short, M.; Drebing, C.; Berger, R.; Venn, D.; Sirota, P.; et al. Association of promoter variants in the alpha7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Arch. Gen. Psychiatry 2002, 59, 1085–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lucas-Cerrillo, A.M.; Maldifassi, M.C.; Arnalich, F.; Renart, J.; Atienza, G.; Serantes, R.; Cruces, J.; Sánchez-Pacheco, A.; Andrés-Mateos, E.; Montiel, C. Function of partially duplicated human α77 nicotinic receptor subunit CHRFAM7A gene: Potential implications for the cholinergic anti-inflammatory response. J. Biol. Chem. 2011, 286, 594–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villiger, Y.; Szanto, I.; Jaconi, S.; Blanchet, C.; Buisson, B.; Krause, K.-H.; Bertrand, D.; Romand, J.-A. Expression of an alpha7 duplicate nicotinic acetylcholine receptor-related protein in human leukocytes. J. Neuroimmunol. 2002, 126, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.; Adler, L.E.; Benhammou, K.; Berger, R.; Breese, C.R.; Drebing, C.; Gault, J.; Lee, M.J.; Logel, J.; Olincy, A.; et al. Smoking and mental illness. Pharmacol. Biochem. Behav. 2001, 70, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.; Coon, H.; Myles-Worsley, M.; Orr-Urtreger, A.; Olincy, A.; Davis, A.; Polymeropoulos, M.; Holik, J.; Hopkins, J.; Hoff, M.; et al. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc. Natl. Acad. Sci. USA 1997, 94, 587–592. [Google Scholar] [CrossRef]
- Freedman, R.; Olincy, A.; Ross, R.G.; Waldo, M.C.; Stevens, K.E.; Adler, L.E.; Leonard, S. The genetics of sensory gating deficits in schizophrenia. Curr. Psychiatry Rep. 2003, 5, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.R.; Hatsukami, D.K.; Mitchell, J.E.; Dahlgren, L.A. Prevalence of smoking among psychiatric outpatients. Am. J. Psychiatry 1986, 143, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C.; Henderson, D.C.; Amico, E. Cigarette smoking in schizophrenia: Relationship to psychopathology and medication side effects. Am. J. Psychiatry 1992, 149, 1189–1194. [Google Scholar] [CrossRef]
- Koike, K.; Hashimoto, K.; Takai, N.; Shimizu, E.; Komatsu, N.; Watanabe, H.; Nakazato, M.; Okamura, N.; Stevens, K.E.; Freedman, R.; et al. Tropisetron improves deficits in auditory P50 suppression in schizophrenia. Schizophr. Res. 2005, 76, 67–72. [Google Scholar] [CrossRef]
- Toyohara, J.; Hashimoto, K. α7 Nicotinic Receptor Agonists: Potential Therapeutic Drugs for Treatment of Cognitive Impairments in Schizophrenia and Alzheimer’s Disease. Open Med. Chem. J. 2010, 4, 37–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackowick, K.M.; Barr, M.S.; Wing, V.C.; Rabin, R.A.; Ouellet-Plamondon, C.; George, T.P. Neurocognitive endophenotypes in schizophrenia: Modulation by nicotinic receptor systems. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 52, 79–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, V.; Kutlu, M.G.; Gould, T.J. nAChR dysfunction as a common substrate for schizophrenia and comorbid nicotine addiction: Current trends and perspectives. Schizophr. Res. 2016, 171, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunii, Y.; Zhang, W.; Xu, Q.; Hyde, T.M.; McFadden, W.; Shin, J.H.; Deep-Soboslay, A.; Ye, T.; Li, C.; Kleinman, J.E.; et al. CHRNA7 and CHRFAM7A mRNAs: Co-localized and their expression levels altered in the postmortem dorsolateral prefrontal cortex in major psychiatric disorders. Am. J. Psychiatry 2015, 172, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- De Luca, V.; Likhodi, O.; Van Tol, H.H.M.; Kennedy, J.L.; Wong, A.H.C. Regulation of alpha7-nicotinic receptor subunit and alpha7-like gene expression in the prefrontal cortex of patients with bipolar disorder and schizophrenia. Acta Psychiatr. Scand. 2006, 114, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Gault, J.; Hopkins, J.; Berger, R.; Drebing, C.; Logel, J.; Walton, C.; Short, M.; Vianzon, R.; Olincy, A.; Ross, R.G.; et al. Comparison of polymorphisms in the alpha7 nicotinic receptor gene and its partial duplication in schizophrenic and control subjects. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2003, 123, 39–49. [Google Scholar] [CrossRef]
- Becchetti, A.; Grandi, L.C.; Cerina, M.; Amadeo, A. Nicotinic acetylcholine receptors and epilepsy. Pharmacol. Res. 2023, 189, 106698. [Google Scholar] [CrossRef]
- Fisher, R.S.; van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J.J. Epileptic seizures and epilepsy: Definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef]
- Pedley, T.A. Major advances in epilepsy in the last century: A personal perspective. Epilepsia 2009, 50, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Alyu, F.; Dikmen, M. Inflammatory aspects of epileptogenesis: Contribution of molecular inflammatory mechanisms. Acta Neuropsychiatr. 2017, 29, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Curia, G.; Longo, D.; Biagini, G.; Jones, R.S.G.; Avoli, M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods 2008, 172, 143–157. [Google Scholar] [CrossRef]
- Hamilton, S.E.; Loose, M.D.; Qi, M.; Levey, A.I.; Hille, B.; McKnight, G.S.; Idzerda, R.L.; Nathanson, N.M. Disruption of the m1 receptor gene ablates muscarinic receptor-dependent M current regulation and seizure activity in mice. Proc. Natl. Acad. Sci. USA 1997, 94, 13311–13316. [Google Scholar] [CrossRef] [PubMed]
- Perucca, P.; Bahlo, M.; Berkovic, S.F. The Genetics of Epilepsy. Annu. Rev. Genomics Hum. Genet. 2020, 21, 205–230. [Google Scholar] [CrossRef] [PubMed]
- Steinlein, O.K.; Mulley, J.C.; Propping, P.; Wallace, R.H.; Phillips, H.A.; Sutherland, G.R.; Scheffer, I.E.; Berkovic, S.F. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 1995, 11, 201–203. [Google Scholar] [CrossRef]
- Becchetti, A.; Grandi, L.C.; Colombo, G.; Meneghini, S.; Amadeo, A. Nicotinic Receptors in Sleep-Related Hypermotor Epilepsy: Pathophysiology and Pharmacology. Brain Sci. 2020, 10, 907. [Google Scholar] [CrossRef] [PubMed]
- Becchetti, A.; Aracri, P.; Meneghini, S.; Brusco, S.; Amadeo, A. The role of nicotinic acetylcholine receptors in autosomal dominant nocturnal frontal lobe epilepsy. Front. Physiol. 2015, 6, 22. [Google Scholar] [CrossRef] [Green Version]
- Lévesque, M.; Biagini, G.; de Curtis, M.; Gnatkovsky, V.; Pitsch, J.; Wang, S.; Avoli, M. The pilocarpine model of mesial temporal lobe epilepsy: Over one decade later, with more rodent species and new investigative approaches. Neurosci. Biobehav. Rev. 2021, 130, 274–291. [Google Scholar] [CrossRef] [PubMed]
- Beleslin, D.B.; Krstić, S.K. Nicotine-induced convulsions in cats and central nicotinic receptors. Pharmacol. Biochem. Behav. 1986, 24, 1509–1511. [Google Scholar] [CrossRef] [PubMed]
- Damaj, M.I.; Glassco, W.; Dukat, M.; Martin, B.R. Pharmacological characterization of nicotine-induced seizures in mice. J. Pharmacol. Exp. Ther. 1999, 291, 1284–1291. [Google Scholar] [PubMed]
- Woolf, A.; Burkhart, K.; Caraccio, T.; Litovitz, T. Self-poisoning among adults using multiple transdermal nicotine patches. J. Toxicol. Clin. Toxicol. 1996, 34, 691–698. [Google Scholar] [CrossRef]
- Bastlund, J.F.; Berry, D.; Watson, W.P. Pharmacological and histological characterisation of nicotine-kindled seizures in mice. Neuropharmacology 2005, 48, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.X.L.; de Oliveira, G.V.; de Araújo, F.Y.R.; de Barros Viana, G.S.; de Sousa, F.C.F.; Hyphantis, T.N.; Grunberg, N.E.; Carvalho, A.F.; Macêdo, D.S. Differences in vulnerability to nicotine-induced kindling between female and male periadolescent rats. Psychopharmacology 2013, 225, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Dobelis, P.; Hutton, S.; Lu, Y.; Collins, A.C. GABAergic systems modulate nicotinic receptor-mediated seizures in mice. J. Pharmacol. Exp. Ther. 2003, 306, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.B.; Manresa, J.J.; Sanberg, P.R.; Shytle, R.D. Nicotine induced seizures blocked by mecamylamine and its stereoisomers. Life Sci. 2001, 69, 2583–2591. [Google Scholar] [CrossRef] [PubMed]
- Iha, H.A.; Kunisawa, N.; Shimizu, S.; Tokudome, K.; Mukai, T.; Kinboshi, M.; Ikeda, A.; Ito, H.; Serikawa, T.; Ohno, Y. Nicotine Elicits Convulsive Seizures by Activating Amygdalar Neurons. Front. Pharmacol. 2017, 8, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.L.; Morley, B.J.; Snead, O.C. An EEG analysis of convulsive activity produced by cholinergic agents. Prog. Neuropsychopharmacol. 1981, 5, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Salas, R.; Orr-Urtreger, A.; Broide, R.S.; Beaudet, A.; Paylor, R.; De Biasi, M. The nicotinic acetylcholine receptor subunit alpha 5 mediates short-term effects of nicotine in vivo. Mol. Pharmacol. 2003, 63, 1059–1066. [Google Scholar] [CrossRef]
- Kedmi, M.; Beaudet, A.L.; Orr-Urtreger, A. Mice lacking neuronal nicotinic acetylcholine receptor beta4-subunit and mice lacking both alpha5- and beta4-subunits are highly resistant to nicotine-induced seizures. Physiol. Genom. 2004, 17, 221–229. [Google Scholar] [CrossRef]
- Wonnacott, S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997, 20, 92–98. [Google Scholar] [CrossRef]
- Tinuper, P.; Bisulli, F.; Cross, J.H.; Hesdorffer, D.; Kahane, P.; Nobili, L.; Provini, F.; Scheffer, I.E.; Tassi, L.; Vignatelli, L.; et al. Definition and diagnostic criteria of sleep-related hypermotor epilepsy. Neurology 2016, 86, 1834–1842. [Google Scholar] [CrossRef]
- Hirose, S.; Iwata, H.; Akiyoshi, H.; Kobayashi, K.; Ito, M.; Wada, K.; Kaneko, S.; Mitsudome, A. A novel mutation of CHRNA4 responsible for autosomal dominant nocturnal frontal lobe epilepsy. Neurology 1999, 53, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Aridon, P.; Marini, C.; Di Resta, C.; Brilli, E.; De Fusco, M.; Politi, F.; Parrini, E.; Manfredi, I.; Pisano, T.; Pruna, D.; et al. Increased sensitivity of the neuronal nicotinic receptor alpha 2 subunit causes familial epilepsy with nocturnal wandering and ictal fear. Am. J. Hum. Genet. 2006, 79, 342–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broide, R.S.; Salas, R.; Ji, D.; Paylor, R.; Patrick, J.W.; Dani, J.A.; De Biasi, M. Increased sensitivity to nicotine-induced seizures in mice expressing the L250T alpha 7 nicotinic acetylcholine receptor mutation. Mol. Pharmacol. 2002, 61, 695–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschini, D.; Paylor, R.; Broide, R.; Salas, R.; Bassetto, L.; Gotti, C.; De Biasi, M. Absence of alpha7-containing neuronal nicotinic acetylcholine receptors does not prevent nicotine-induced seizures. Brain Res. Mol. Brain Res. 2002, 98, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, C.P. Nicotinic acetylcholine receptors in human genetic disease. Genet. Med. 2014, 16, 649–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helbig, I.; Mefford, H.C.; Sharp, A.J.; Guipponi, M.; Fichera, M.; Franke, A.; Muhle, H.; de Kovel, C.; Baker, C.; von Spiczak, S.; et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat. Genet. 2009, 41, 160–162. [Google Scholar] [CrossRef]
- Shinawi, M.; Schaaf, C.P.; Bhatt, S.S.; Xia, Z.; Patel, A.; Cheung, S.W.; Lanpher, B.; Nagl, S.; Herding, H.S.; Nevinny-Stickel, C.; et al. A small recurrent deletion within 15q13.3 is associated with a range of neurodevelopmental phenotypes. Nat. Genet. 2009, 41, 1269–1271. [Google Scholar] [CrossRef] [Green Version]
- Sharp, A.J.; Mefford, H.C.; Li, K.; Baker, C.; Skinner, C.; Stevenson, R.E.; Schroer, R.J.; Novara, F.; De Gregori, M.; Ciccone, R.; et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat. Genet. 2008, 40, 322–328. [Google Scholar] [CrossRef]
- Fejgin, K.; Nielsen, J.; Birknow, M.R.; Bastlund, J.F.; Nielsen, V.; Lauridsen, J.B.; Stefansson, H.; Steinberg, S.; Sorensen, H.B.D.; Mortensen, T.E.; et al. A mouse model that recapitulates cardinal features of the 15q13.3 microdeletion syndrome including schizophrenia- and epilepsy-related alterations. Biol. Psychiatry 2014, 76, 128–137. [Google Scholar] [CrossRef] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallés, A.S.; Barrantes, F.J. Nicotinic Acetylcholine Receptor Dysfunction in Addiction and in Some Neurodegenerative and Neuropsychiatric Diseases. Cells 2023, 12, 2051. https://doi.org/10.3390/cells12162051
Vallés AS, Barrantes FJ. Nicotinic Acetylcholine Receptor Dysfunction in Addiction and in Some Neurodegenerative and Neuropsychiatric Diseases. Cells. 2023; 12(16):2051. https://doi.org/10.3390/cells12162051
Chicago/Turabian StyleVallés, Ana Sofía, and Francisco J. Barrantes. 2023. "Nicotinic Acetylcholine Receptor Dysfunction in Addiction and in Some Neurodegenerative and Neuropsychiatric Diseases" Cells 12, no. 16: 2051. https://doi.org/10.3390/cells12162051