
Skin-Grafting and Dendritic Cell “Boosted” Humanized Mouse Models Allow the Pre-Clinical Evaluation of Therapeutic Cancer Vaccines
 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. PBMC Isolation and Cryopreservation
2.3. The Humanized K14E7 Skin Grafting Model
2.4. TNE Preparation
2.5. Dendritic Cell-Boosted Humanized Models
2.6. Humanized Tumor Models
2.7. Flow Cytometry
2.8. Immunohistochemistry and Immunofluorescence
2.9. Polyfunctional Assay
2.10. Ex Vivo Re-Stimulation of Engrafted PBMCs
2.11. Statistical Analysis
3. Results
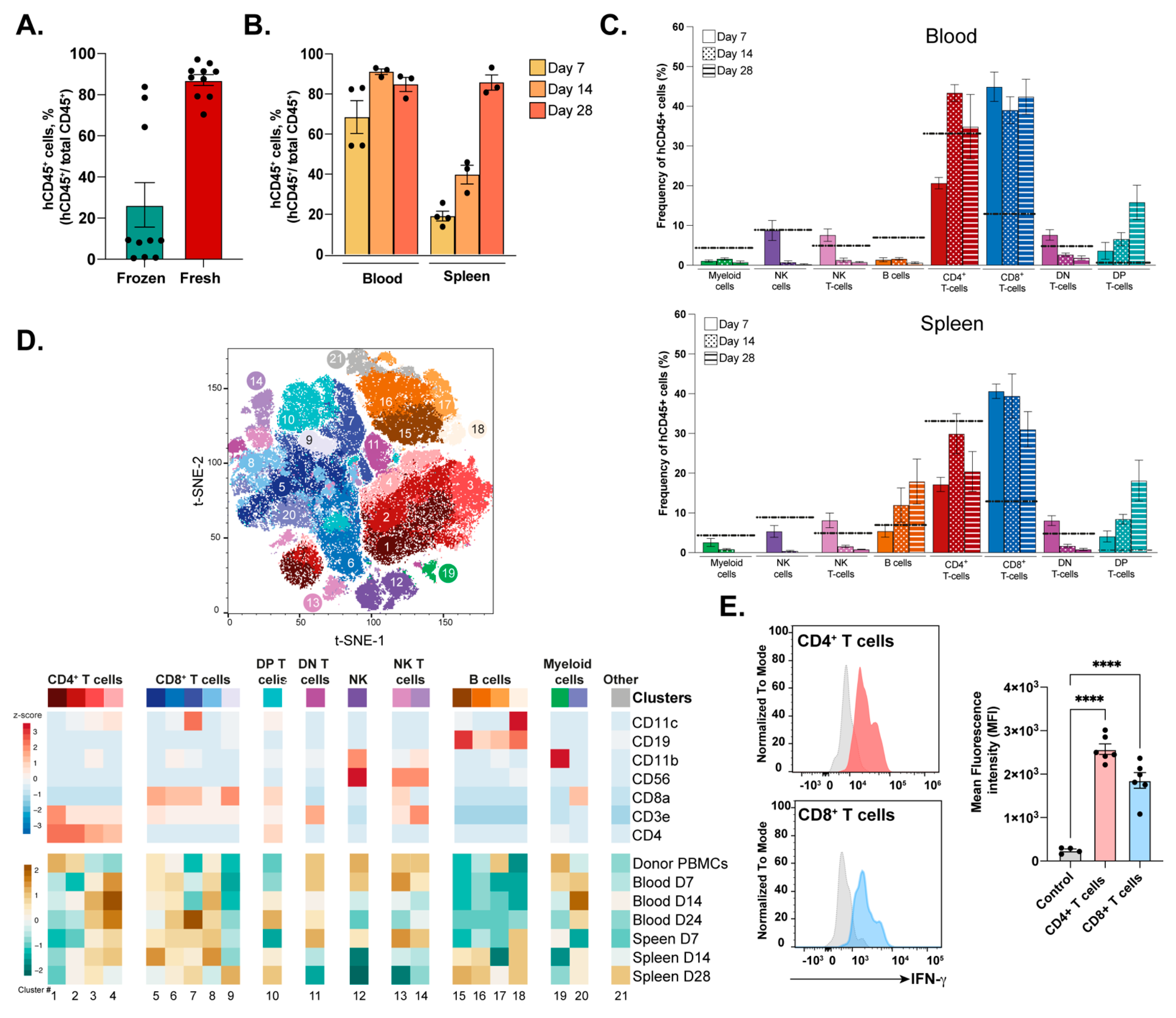
3.1. Engraftment and Immune Profile of Transferred PBMCs into NSG-A2 Mice
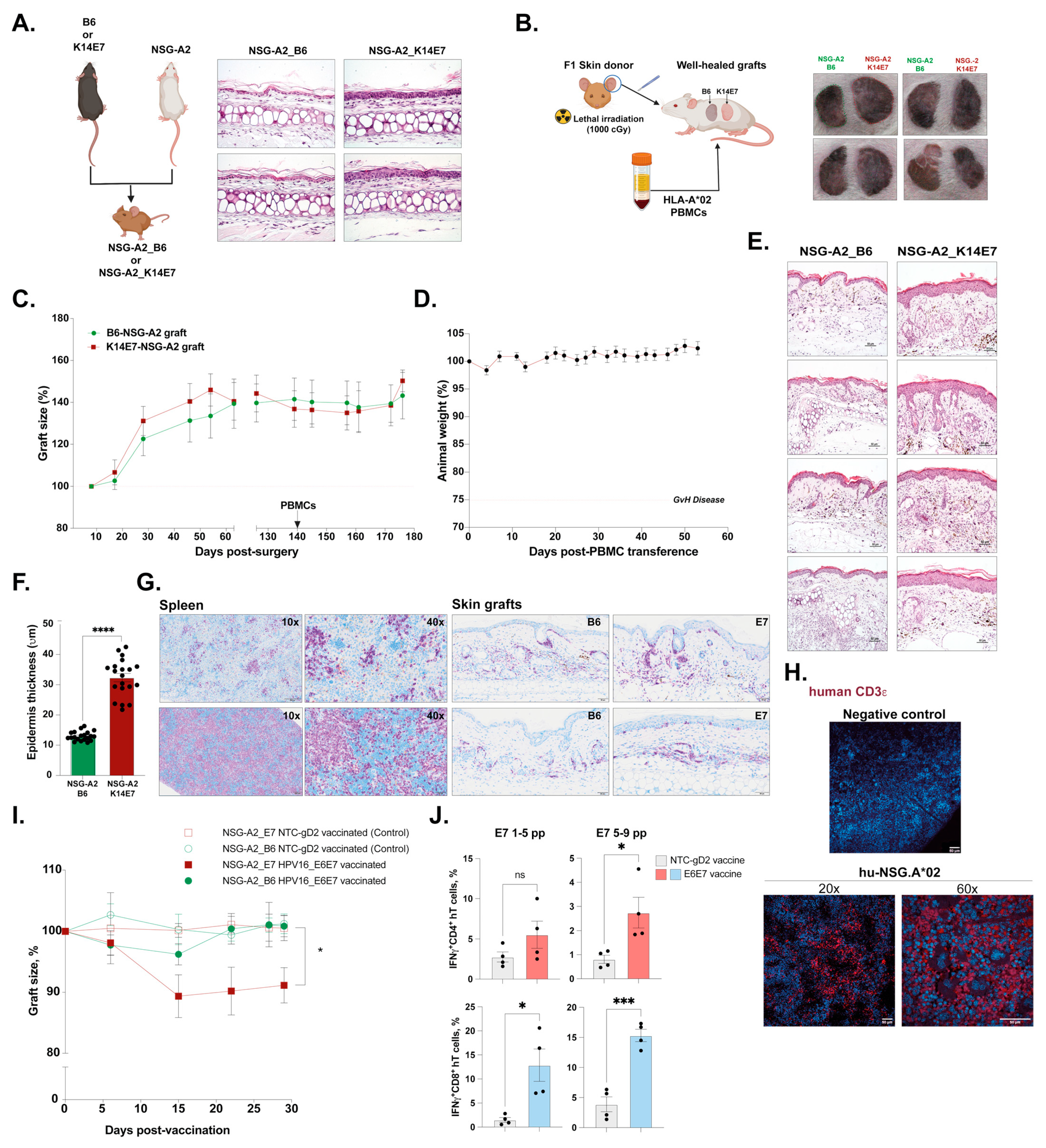
3.2. Dysplastic Skin-Grafting Humanized Mouse Model and Response to DNA-HPV Therapeutic Vaccine
3.3. Boosting PBMC-hu-NSG-A2 Mice with Flt3-L-Treated PBMCs Increases the Number of Human CD141+ cDC1 Cells and Enhances Antigen-Specific T Cell Responses
3.4. Direct Comparison between PBMC-hu-NSG and PBMC-hu-NSG-A2 Models Supports the Use of HLA Matching Systems to Test Cancer Vaccines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tay, B.Q.; Wright, Q.; Ladwa, R.; Perry, C.; Leggatt, G.; Simpson, F.; Wells, J.W.; Panizza, B.J.; Frazer, I.H. Evolution of Cancer Vaccines-Challenges, Achievements, and Future Directions. Vaccines 2021, 9, 535. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Coffman, R.L. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional prop-erties. Annu. Rev. Immunol. 1989, 7, 145–173. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, T.; Kierstead, L.S.; Ranieri, E.; Gesualdo, L.; Schena, F.P.; Finke, J.H.; Bukowski, R.M.; Mueller-Berghaus, J.; Kirkwood, J.M.; Kwok, W.W. Disease-associated bias in T helper type 1 (Th1)/Th2 CD4+ T cell responses against MAGE-6 in HLA-DRB10401+ patients with renal cell carcinoma or melanoma. J. Exp. Med. 2002, 196, 619–628. [Google Scholar] [CrossRef]
- De La Rochere, P.; Guil-Luna, S.; Decaudin, D.; Azar, G.; Sidhu, S.S.; Piaggio, E. Humanized Mice for the Study of Immuno-Oncology. Trends Immunol. 2018, 39, 748–763. [Google Scholar] [CrossRef] [PubMed]
- Chew, H.Y.; De Lima, P.O.; Cruz, J.L.G.; Banushi, B.; Echejoh, G.; Hu, L.; Joseph, S.R.; Lum, B.; Rae, J.; O’donnell, J.S.; et al. Endocytosis Inhibition in Humans to Improve Responses to ADCC-Mediating Antibodies. Cell 2020, 180, 895–914.e27. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Leggatt, G.R.; Zhong, J.; Liu, X.; de Kluyver, R.L.; Peters, T.; Fernando, G.J.P.; Liem, A.; Lambert, P.F.; Frazer, I.H. Impaired antigen presentation and effectiveness of combined active/passive immunotherapy for epithelial tumors. Gynecol. Oncol. 2004, 96, 1611–1619. [Google Scholar] [CrossRef]
- van der Burg, S.; Kwappenberg, K.; O’Neill, T.; Brandt, R.; Melief, C.; Hickling, J.; Offringa, R. Pre-clinical safety and efficacy of TA-CIN, a recombinant HPV16 L2E6E7 fusion protein vaccine, in homologous and heterologous prime-boost regimens. Vaccine 2001, 19, 3652–3660. [Google Scholar] [CrossRef] [PubMed]
- Frazer, I.H.; Quinn, M.; Nicklin, J.L.; Tan, J.; Perrin, L.C.; Ng, P.; Oconnor, V.; White, O.; Wendt, N.; Martin, J.; et al. Phase 1 study of HPV16-specific immunotherapy with E6E7 fusion protein and ISCOMATRIX™ adjuvant in women with cervical intraepithelial neoplasia. Vaccine 2004, 23, 172–181. [Google Scholar] [CrossRef]
- Dunn, L.A.; Evander, M.; Tindle, R.W.; Bulloch, A.L.; de Kluyver, R.L.; Fernando, G.J.; Lambert, P.F.; Frazer, I.H. Presentation of the HPV16E7 protein by skin grafts is insufficient to allow graft rejection in an E7-primed animal. Virology 1997, 235, 94–103. [Google Scholar] [CrossRef]
- Tuong, Z.K.; Noske, K.; Kuo, P.; Bashaw, A.A.; Teoh, S.M.; Frazer, I.H. Murine HPV16 E7-expressing transgenic skin effectively emu-lates the cellular and molecular features of human high-grade squamous intraepithelial lesions. Papillomavirus Res. 2018, 5, 6–20. [Google Scholar] [CrossRef]
- Frazer, I.H.; Leggatt, G.R.; Mattarollo, S.R. Prevention and treatment of papillomavirus-related cancers through immunization. Annu. Rev. Immunol. 2011, 29, 111–138. [Google Scholar] [CrossRef] [PubMed]
- Zschaler, J.; Schlorke, D.; Arnhold, J. Differences in innate immune response between man and mouse. Crit. Rev. Immunol. 2014, 34, 433–454. [Google Scholar] [CrossRef] [PubMed]
- Tullett, K.M.; Leal Rojas, I.M.; Minoda, Y.; Tan, P.S.; Zhang, J.-G.; Smith, C.; Khanna, R.; Shortman, K.; Caminschi, I.; Lahoud, M.H.; et al. Targeting CLEC9A delivers antigen to human CD141+ DC for CD4+ and CD8+ T cell recognition. JCI Insight 2016, 1, e87102. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Middelberg, A.P.; Gemiarto, A.; MacDonald, K.; Baxter, A.G.; Talekar, M.; Moi, D.; Tullett, K.M.; Caminschi, I.; Lahoud, M.H.; et al. Self-adjuvanting nanoemulsion targeting dendritic cell receptor Clec9A enables antigen-specific immunotherapy. J. Clin. Investig. 2018, 128, 1971–1984. [Google Scholar] [CrossRef]
- Frazer, I.H.; De Kluyver, R.; Leggatt, G.R.; Guo, H.Y.; Dunn, L.; White, O.; Harris, C.; Liem, A.; Lambert, P. Tolerance or immunity to a tumor antigen expressed in somatic cells can be determined by systemic proinflammatory signals at the time of first antigen exposure. J. Immunol. 2001, 167, 6180–6187. [Google Scholar] [CrossRef]
- Cruz, J.L.G.; Becares, M.; Sola, I.; Oliveros, J.C.; Enjuanes, L.; Zúñiga, S.; Du, L.; Zhao, G.; Kou, Z.; Ma, C.; et al. Alphacoronavirus protein 7 modulates host innate immune response. J. Virol. 2013, 87, 9754–9767. [Google Scholar] [CrossRef]
- Cruz, J.L.G.; Sola, I.; Becares, M.; Alberca, B.; Plana, J.; Enjuanes, L.; Zuñiga, S. Coronavirus gene 7 counteracts host defenses and modulates virus virulence. PLoS Pathog. 2011, 7, e1002090. [Google Scholar] [CrossRef]
- Eljaafari, A.; Yuruker, O.; Ferrand, C.; Farre, A.; Addey, C.; Tartelin, M.L.; Thomas, X.; Tiberghien, P.; Simpson, E.; Rigal, D. Isolation of human CD4/CD8 double-positive, graft-versus-host disease-protective, minor histocompatibility antigen-specific regulatory T cells and of a novel HLA-DR7-restricted HY-specific CD4 clone. J. Immunol. 2013, 190, 184–194. [Google Scholar] [CrossRef]
- Beyer, M.; Wang, H.; Peters, N.; Doths, S.; Koerner-Rettberg, C.; Openshaw, P.J.; Schwarze, J. The beta2 integrin CD11c distinguishes a subset of cytotoxic pulmonary T cells with potent antiviral effects in vitro and in vivo. Respir. Res. 2005, 6, 70. [Google Scholar] [CrossRef]
- Kim, Y.H.; Seo, S.K.; Choi, B.K.; Kang, W.J.; Kim, C.H.; Lee, S.K.; Kwon, B.S. 4-1BB costimulation enhances HSV-1-specific CD8+ T cell responses by the induction of CD11c+ CD8+ T cells. Cell. Immunol. 2005, 238, 76–86. [Google Scholar] [CrossRef]
- Fujiwara, D.; Chen, L.; Wei, B.; Braun, J. Small intestine CD11c+ CD8+ T cells suppress CD4+ T cell-induced immune colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G939–G947. [Google Scholar] [CrossRef] [PubMed]
- Kubota, K.; Kadoya, Y. Innate IFN-gamma-producing cells in the spleen of mice early after Listeria monocytogenes infection: Importance of microenvironment of the cells involved in the production of innate IFN-gamma. Front. Immunol. 2011, 2, 26. [Google Scholar] [CrossRef] [PubMed]
- Cooney, L.A.; Gupta, M.; Thomas, S.; Mikolajczak, S.; Choi, K.Y.; Gibson, C.; Jang, I.K.; Danziger, S.; Aitchison, J.; Gardner, M.J.; et al. Short-lived effector CD8 T cells induced by genetically attenuated malaria parasite vaccination express CD11c. Infect. Immun. 2013, 81, 4171–4181. [Google Scholar] [CrossRef] [PubMed]
- Qualai, J.; Li, L.-X.; Cantero, J.; Tarrats, A.; Fernández, M.A.; Sumoy, L.; Rodolosse, A.; McSorley, S.J.; Genescà, M. Expression of CD11c Is Associated with Unconventional Activated T Cell Subsets with High Migratory Potential. PLoS ONE 2016, 11, e0154253. [Google Scholar] [CrossRef]
- Rizzo, K.; Stetler-Stevenson, M.; Wilson, W.; Yuan, C.M. Novel CD19 expression in a peripheral T cell lymphoma: A flow cytometry case report with morphologic correlation. Cytom. Part B Clin. Cytom. 2009, 76, 142–149. [Google Scholar] [CrossRef]
- Chandra, J.; Dutton, J.L.; Li, B.; Woo, W.P.; Xu, Y.; Tolley, L.K.; Yong, M.; Wells, J.W.; Leggatt, G.R.; Finlayson, N. DNA Vaccine Encoding HPV16 Oncogenes E6 and E7 Induces Potent Cell-mediated and Humoral Immunity Which Protects in Tumor Challenge and Drives E7-expressing Skin Graft Rejection. J. Immunother. 2017, 40, 62–70. [Google Scholar] [CrossRef]
- Chandra, J.; Woo, W.P.; Finlayson, N.; Liu, H.Y.; McGrath, M.; Ladwa, R.; Brauer, M.; Xu, Y.; Hanson, S.; Panizza, B.; et al. A phase 1, single centre, open label, escalating dose study to assess the safety, tolerability and immunogenicity of a therapeutic human papillomavirus (HPV) DNA vaccine (AMV002) for HPV-associated head and neck cancer (HNC). Cancer Immunol. Immunother. 2021, 70, 743–753. [Google Scholar] [CrossRef]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.-J.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef]
- Maraskovsky, E.; Daro, E.; Roux, E.; Teepe, M.; Maliszewski, C.R.; Hoek, J.; Caron, D.; Lebsack, M.E.; McKenna, H.J. In vivo generation of human dendritic cell subsets by Flt3 ligand. Blood 2000, 96, 878–884. [Google Scholar] [CrossRef]
- Ding, Y.; Wilkinson, A.; Idris, A.; Fancke, B.; O’keeffe, M.; Khalil, D.; Ju, X.; Lahoud, M.H.; Caminschi, I.; Shortman, K.; et al. FLT3-ligand treatment of humanized mice results in the generation of large numbers of CD141+ and CD1c+ dendritic cells in vivo. J. Immunol. 2014, 192, 1982–1989. [Google Scholar] [CrossRef]
- Zarour, H.M.; Maillere, B.; Brusic, V.; Coval, K.; Williams, E.; Pouvelle-Moratille, S.; Castelli, F.; Land, S.; Bennouna, J.; Logan, T.; et al. NY-ESO-1 119-143 Is A Promiscuous Major Histocompatibility Complex Class II T-Helper Epitope Recognized by Th1- and Th2-Type Tumor-reactive CD4+ T Cells. Cancer Res. 2002, 62, 213–218. [Google Scholar] [PubMed]
- Chen, J.-L.; Dunbar, P.R.; Gileadi, U.; Jäger, E.; Gnjatic, S.; Nagata, Y.; Stockert, E.; Panicali, D.L.; Chen, Y.-T.; Knuth, A.; et al. Identification of NY-ESO-1 peptide analogues capable of improved stimulation of tumor-reactive CTL. J. Immunol. 2000, 165, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Guan, S.; Qiao, Y.; Li, X.; Xu, Y.; Yang, L.; Kuai, Z.; Zhang, H.; Shi, Y.; Kong, W.; et al. Effects of poly(I:C) and MF59 co-adjuvants on immunogenicity and efficacy of survivin polypeptide immunogen against melanoma. J. Cell. Physiol. 2018, 233, 4926–4934. [Google Scholar] [CrossRef] [PubMed]
- Muraro, E.; Comaro, E.; Talamini, R.; Turchet, E.; Miolo, G.; Scalone, S.; Militello, L.; Lombardi, D.; Spazzapan, S.; Perin, T. Improved Natural Killer cell activity and retained an-ti-tumor CD8+ T cell responses contribute to the induction of a pathological complete response in HER2-positive breast cancer patients undergoing neoadjuvant chemotherapy. J. Transl. Med. 2015, 13, 204. [Google Scholar] [CrossRef]
- Schag, K.; Schmidt, S.M.; Müller, M.R.; Weinschenk, T.; Appel, S.; Weck, M.M.; Grünebach, F.; Stevanovic, S.; Rammensee, H.-G.; Brossart, P. Identification of C-Met oncogene as a broadly expressed tumor-associated antigen recognized by cytotoxic T-lymphocytes. Clin. Cancer Res. 2004, 10, 3658–3666. [Google Scholar] [CrossRef]
- Wenandy, L.; Sørensen, R.B.; Sengeløv, L.; Svane, I.M.; Straten, P.T.; Andersen, M.H. The Immunogenicity of the hTERT540-548 Peptide in Cancer. Clin. Cancer Res. 2008, 14, 4–7. [Google Scholar] [CrossRef]
- Widenmeyer, M.; Griesemann, H.; Stevanović, S.; Feyerabend, S.; Klein, R.; Attig, S.; Hennenlotter, J.; Wernet, D.; Kuprash, D.V.; Sazykin, A.Y.; et al. Promiscuous survivin peptide induces robust CD4+ T-cell responses in the majority of vaccinated cancer patients. Int. J. Cancer 2012, 131, 140–149. [Google Scholar] [CrossRef]
- McCune, J.M.; Namikawa, R.; Kaneshima, H.; Shultz, L.D.; Lieberman, M.; Weissman, I.L. The SCID-hu mouse: Murine model for the analysis of human hematolymphoid differentiation and function. Science 1988, 241, 1632–1639. [Google Scholar] [CrossRef]
- Allen, T.M.; Brehm, M.A.; Bridges, S.; Ferguson, S.; Kumar, P.; Mirochnitchenko, O.; Palucka, K.; Pelanda, R.; Sanders-Beer, B.; Shultz, L.D.; et al. Humanized immune system mouse models: Progress, challenges and opportunities. Nat. Immunol. 2019, 20, 770–774. [Google Scholar] [CrossRef]
- Yang, J.; Diaz, N.; Adelsberger, J.; Zhou, X.; Stevens, R.; Rupert, A.; Metcalf, J.A.; Baseler, M.; Barbon, C.; Imamichi, T.; et al. The effects of storage temperature on PBMC gene expression. BMC Immunol. 2016, 17, 6. [Google Scholar] [CrossRef]
- Li, B.; Yang, C.; Jia, G.; Liu, Y.; Wang, N.; Yang, F.; Su, R.; Shang, Y.; Han, Y. Comprehensive evaluation of the effects of long-term cryopreservation on peripheral blood mononuclear cells using flow cytometry. BMC Immunol. 2022, 23, 30. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, S.; Tahara, H.; Shirouzu, T.; Kawai, S.; Tanaka, Y.; Ide, K.; Akimoto, S.; Ohdan, H. Development of a humanized mouse model to analyze antibodies specific for human leukocyte antigen (HLA). PLoS ONE 2021, 16, e0236614. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.E.; Crompton, P.D.; Li, S.; Walsh, L.A.; Moir, S.; Traore, B.; Kayentao, K.; Ongoiba, A.; Doumbo, O.K.; Pierce, S.K. Atypical memory B cells are greatly expanded in individuals living in a malaria-endemic area. J. Immunol. 2014, 183, 2176–2182. [Google Scholar] [CrossRef]
- Rubtsov, A.V.; Rubtsova, K.; Fischer, A.; Meehan, R.T.; Gillis, J.Z.; Kappler, J.W.; Marrack, P. Toll-like receptor 7 (TLR7)–driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood 2011, 118, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Golinski, M.-L.; Demeules, M.; Derambure, C.; Riou, G.; Maho-Vaillant, M.; Boyer, O.; Joly, P.; Calbo, S. CD11c+ B Cells Are Mainly Memory Cells, Precursors of Antibody Secreting Cells in Healthy Donors. Front. Immunol. 2020, 11, 32. [Google Scholar] [CrossRef]
- Morton, J.J.; Alzofon, N.; Jimeno, A. The humanized mouse: Emerging translational potential. Mol. Carcinog. 2020, 59, 830–838. [Google Scholar] [CrossRef]
- Orlik, C.; Deibel, D.; Küblbeck, J.; Balta, E.; Ganskih, S.; Habicht, J.; Niesler, B.; Schröder-Braunstein, J.; Schäkel, K.; Wabnitz, G.; et al. Keratinocytes costimulate naive human T cells via CD2: A potential target to prevent the development of proinflammatory Th1 cells in the skin. Cell. Mol. Immunol. 2020, 17, 380–394. [Google Scholar] [CrossRef]
- Black, A.P.; Ardern-Jones, M.R.; Kasprowicz, V.; Bowness, P.; Jones, L.; Bailey, A.S.; Ogg, G.S. Human keratinocyte induction of rapid ef-fector function in antigen-specific memory CD4+ and CD8+ T cells. Eur. J. Immunol. 2007, 37, 1485–1493. [Google Scholar] [CrossRef]
- Wilson, K.R.; Villadangos, J.A.; Mintern, J.D. Dendritic cell Flt3—Regulation, roles and repercussions for immunotherapy. Immunol. Cell Biol. 2021, 99, 962–971. [Google Scholar] [CrossRef]
- Lai, J.; Mardiana, S.; House, I.G.; Sek, K.; Henderson, M.A.; Giuffrida, L.; Chen, A.X.Y.; Todd, K.L.; Petley, E.V.; Chan, J.D.; et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat. Immunol. 2020, 21, 914–926. [Google Scholar] [CrossRef]
- Macri, C.; Jenika, D.; Ouslinis, C.; Mintern, J.D. Targeting dendritic cells to advance cross-presentation and vaccination outcomes. Semin. Immunol. 2023, 68, 101762. [Google Scholar] [CrossRef] [PubMed]
- Chuprin, J.; Buettner, H.; Seedhom, M.O.; Greiner, D.L.; Keck, J.G.; Ishikawa, F.; Shultz, L.D.; Brehm, M.A. Humanized mouse models for immuno-oncology research. Nat. Rev. Clin. Oncol. 2023, 20, 192–206. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Model | Antigen | Type of Epitope | Aminoacidic Sequence |
|---|---|---|---|
| MDA-MB-231 TNBC | Survivin (97–111) | Promiscuous CD4 epitope | TLGEFLKLDRERAKN |
| Survivin (ELT 95–104) | CD8 epitope | ELTLGEFLKL | |
| Telomerase (hTERT 540–548) | CD8 epitope | SLLDRFLATV | |
| A375 melanoma | NY-ESO-1 (119–143) | Pan-DR epitope | PGVLLKEFTVSGNILTIRLTAADHR |
| Tyrosinase (386–406) | Promiscuous CD4 epitope | FLLHHAFVDSIFEQWLQRHRP | |
| Cyclin-I | CD8 epitope | SLLDRFLATV | |
| Tyrosinase (369–377) | CD8 epitope | YMDGTMSQV | |
| STAT1 | CD8 epitope | VLWDRTFSL | |
| Survivin (ELT 95–104) | CD8 epitope | ELTLGEFLKL | |
| MAGE-A3 (271–279) | CD8 epitope | FLWGPRALV |
| Antibody | Fluorochrome | Clone | Cat # | Company | Dilution |
|---|---|---|---|---|---|
| LIVE/DEAD™ | Aqua | NA | L34957 | ThermoFisher | 1:1000 |
| a-mouse-CD45.2 | PE/Dazzle™ | 104 | 109846 | BioLegend | 1:200 |
| a-human-CD45 | Brilliant Violet 421™ | 2D1 | 368522 | BioLegend | 1:200 |
| a-human-CD4 | Alexa Fluor® 700 | RPA-T4 | 300526 | BioLegend | 1:200 |
| a-human-CD8a | FITC | SK1 | 344704 | BioLegend | 1:200 |
| a-human-CD19 | APC | HIB19 | 302212 | BioLegend | 1:200 |
| a-human-CD11c | Brilliant Violet 711™ | 3.9 | 301630 | BioLegend | 1:200 |
| a-human-CD56 | PE | QA17A16 | 985902 | BioLegend | 1:200 |
| a-human-CD11b | PE/Cyanine7 | LM2 | 393104 | BioLegend | 1:200 |
| a-human-CD3e | APC | OKT3 | 317318 | BioLegend | 1:200 |
| a-HLA.A2 | APC | BB7.2 | 343308 | BioLegend | 1:200 |
| a-human-TNFa | Alexa Fluor® 647 | MAb11 | 502916 | BioLegend | 1:100 |
| a-human-IFNγ | Pacific Blue™ | B27 | 506526 | BioLegend | 1:100 |
| a-human-IL2 | Alexa Fluor® 700 | MQ1-17H12 | 500320 | BioLegend | 1:100 |
| Peptide | Sequence | Length (aa) |
|---|---|---|
| E7 #1 (1–22) | MHGDTPTLHEYMLDLQPETTDL | 22 |
| E7 #2 (11–32) | YMLDLQPETTDLYGYGQLNDSS | 22 |
| E7 #3 (21–42) | DLYCYEQLNDSSEEEDEIDGPA | 22 |
| E7 #4 (31–52) | SSEEEDEIDGPAGQAEPDRAHY | 22 |
| E7 #5 (41–62) | PAGQAEPDRAHYNIVTFCCKCD | 22 |
| E7 #6 (51–72) | HYNIVTFCCKCDSTLRLCVQST | 22 |
| E7 #7 (61–82) | CDSTLRLCVQSTHVDIRTLEDL | 22 |
| E7 #8 (71–92) | STHVDIRTLEDLLMGTLGIVCP | 22 |
| E7 #9 (77–98) | RTLEDLLMGTLGIVCPICSQKP | 22 |
| Antibody | Fluorochrome | Clone | Cat # | Company | Dilution |
|---|---|---|---|---|---|
| LIVE/DEAD™ | Aqua | NA | L34957 | ThermoFisher | 1:1000 |
| a-mouse-CD45.2 | FITC | QA17A26 | 157608 | Biolegend | 1:200 |
| a-human-CD45 | PerCP | 2D1 | 368506 | Biolegend | 1:200 |
| a-human-CD3 | Brilliant Violet 711™ | OKT3 | 317328 | Biolegend | 1:200 |
| a-human-CD4 | PE/Cyanine7 | RPA-T4 | 300512 | Biolegend | 1:200 |
| a-human-CD8a | APC/Cyanine7 | SK1 | 344714 | Biolegend | 1:200 |
| a-human-TNFa | Alexa Fluor® 647 | MAb11 | 502916 | Biolegend | 1:100 |
| a-human-IFNγ | Pacific Blue™ | B27 | 506526 | Biolegend | 1:100 |
| a-human-IL2 | Alexa Fluor® 700 | MQ1-17H12 | 500320 | Biolegend | 1:100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, B.; Moi, D.; Tolley, L.; Molotkov, N.; Frazer, I.H.; Perry, C.; Dolcetti, R.; Mazzieri, R.; Cruz, J.L.G. Skin-Grafting and Dendritic Cell “Boosted” Humanized Mouse Models Allow the Pre-Clinical Evaluation of Therapeutic Cancer Vaccines. Cells 2023, 12, 2094. https://doi.org/10.3390/cells12162094
Zeng B, Moi D, Tolley L, Molotkov N, Frazer IH, Perry C, Dolcetti R, Mazzieri R, Cruz JLG. Skin-Grafting and Dendritic Cell “Boosted” Humanized Mouse Models Allow the Pre-Clinical Evaluation of Therapeutic Cancer Vaccines. Cells. 2023; 12(16):2094. https://doi.org/10.3390/cells12162094
Chicago/Turabian StyleZeng, Bijun, Davide Moi, Lynn Tolley, Natalie Molotkov, Ian Hector Frazer, Christopher Perry, Riccardo Dolcetti, Roberta Mazzieri, and Jazmina L. G. Cruz. 2023. "Skin-Grafting and Dendritic Cell “Boosted” Humanized Mouse Models Allow the Pre-Clinical Evaluation of Therapeutic Cancer Vaccines" Cells 12, no. 16: 2094. https://doi.org/10.3390/cells12162094
APA StyleZeng, B., Moi, D., Tolley, L., Molotkov, N., Frazer, I. H., Perry, C., Dolcetti, R., Mazzieri, R., & Cruz, J. L. G. (2023). Skin-Grafting and Dendritic Cell “Boosted” Humanized Mouse Models Allow the Pre-Clinical Evaluation of Therapeutic Cancer Vaccines. Cells, 12(16), 2094. https://doi.org/10.3390/cells12162094