


Increased Actin Binding Is a Shared Molecular Consequence of Numerous SCA5 Mutations in β-III-Spectrin
Abstract
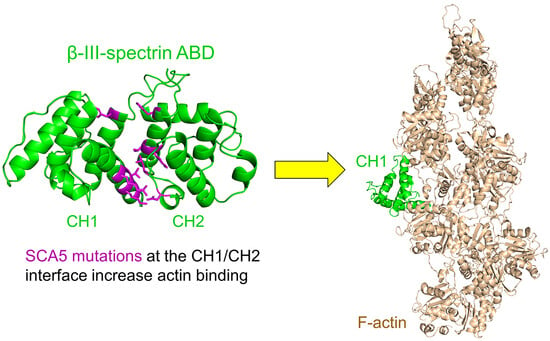
1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Circular Dichroism Measurements
2.3. F-Actin Co-Sedimentation Assays
2.4. Structural Modeling
2.5. Statistical Analysis
3. Results
3.1. Clinical Summary of ABD-Localized Mutations
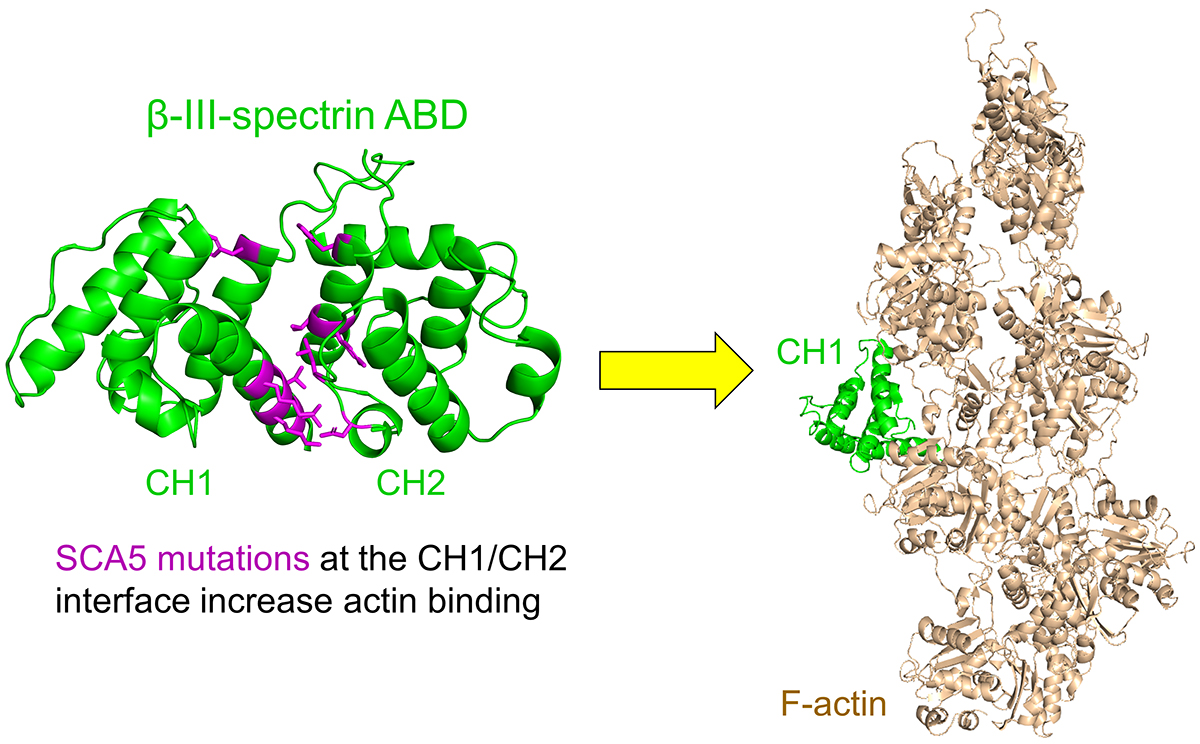
3.2. ABD-Localized Mutations Cluster to the CH1-CH2 Interface
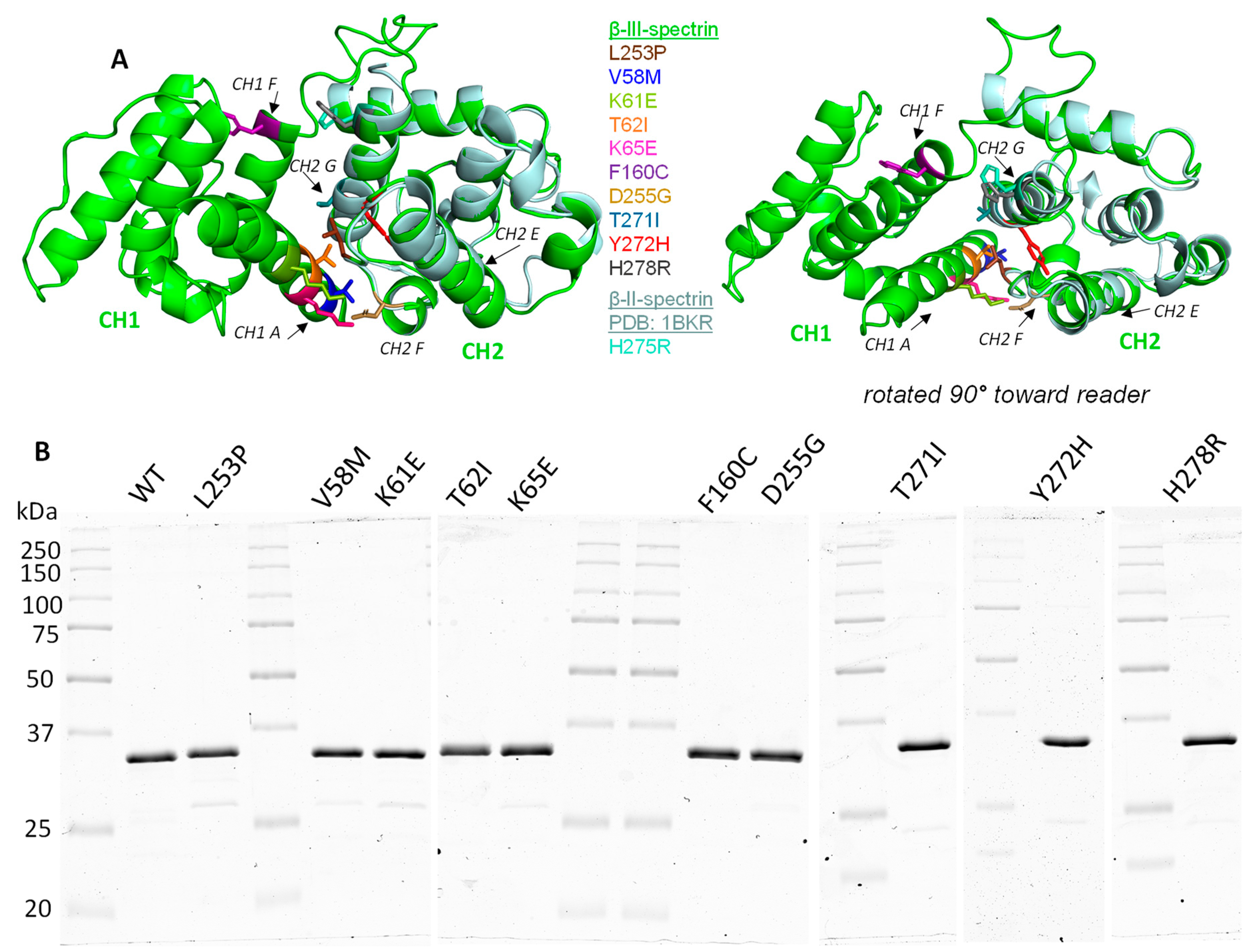
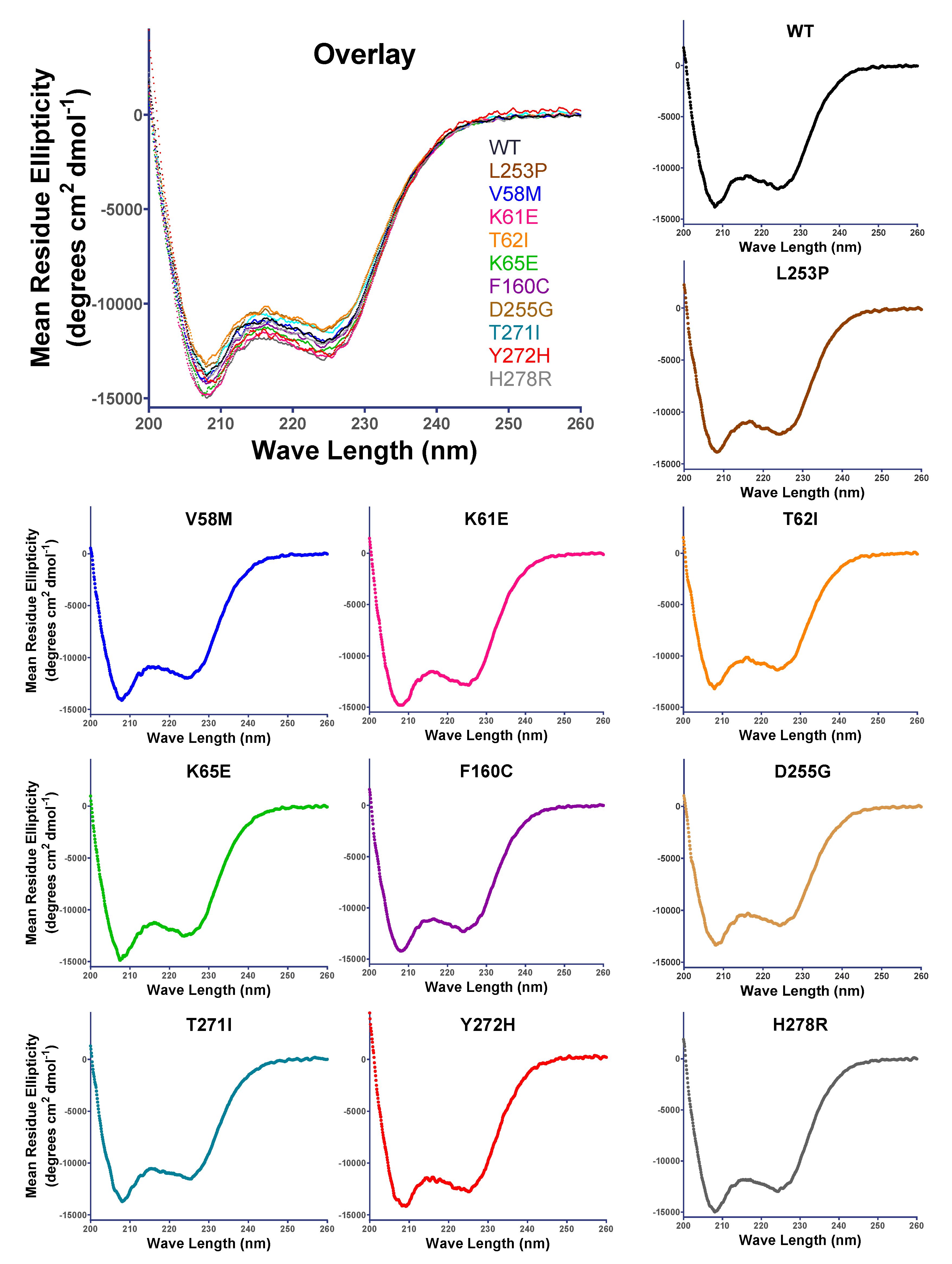
3.3. SCA5 Mutant ABDs Can Attain a Well-Folded State, but Are Destabilized
3.4. All of the SCA5 Mutations Increase Actin-Binding Affinity
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ohara, O.; Ohara, R.; Yamakawa, H.; Nakajima, D.; Nakayama, M. Characterization of a new beta-spectrin gene which is predominantly expressed in brain. Brain Res. Mol. Brain Res. 1998, 57, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Dick, K.A.; Weatherspoon, M.R.; Gincel, D.; Armbrust, K.R.; Dalton, J.C.; Stevanin, G.; Durr, A.; Zuhlke, C.; Burk, K.; et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat. Genet. 2006, 38, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Perkins, E.M.; Clarkson, Y.L.; Tobia, S.; Lyndon, A.R.; Jackson, M.; Rothstein, J.D. beta-III spectrin is critical for development of purkinje cell dendritic tree and spine morphogenesis. J. Neurosci. 2011, 31, 16581–16590. [Google Scholar] [CrossRef]
- Perkins, E.M.; Clarkson, Y.L.; Sabatier, N.; Longhurst, D.M.; Millward, C.P.; Jack, J.; Toraiwa, J.; Watanabe, M.; Rothstein, J.D.; Lyndon, A.R.; et al. Loss of beta-III spectrin leads to Purkinje cell dysfunction recapitulating the behavior and neuropathology of spinocerebellar ataxia type 5 in humans. J. Neurosci. 2010, 30, 4857–4867. [Google Scholar] [CrossRef]
- Stankewich, M.C.; Gwynn, B.; Ardito, T.; Ji, L.; Kim, J.; Robledo, R.F.; Lux, S.E.; Peters, L.L.; Morrow, J.S. Targeted deletion of betaIII spectrin impairs synaptogenesis and generates ataxic and seizure phenotypes. Proc. Natl. Acad. Sci. USA 2010, 107, 6022–6027. [Google Scholar] [CrossRef]
- Efimova, N.; Korobova, F.; Stankewich, M.C.; Moberly, A.H.; Stolz, D.B.; Wang, J.; Kashina, A.; Ma, M.; Svitkina, T. betaIII Spectrin Is Necessary for Formation of the Constricted Neck of Dendritic Spines and Regulation of Synaptic Activity in Neurons. J. Neurosci. 2017, 37, 6442–6459. [Google Scholar] [CrossRef]
- Fujishima, K.; Kurisu, J.; Yamada, M.; Kengaku, M. betaIII spectrin controls the planarity of Purkinje cell dendrites by modulating perpendicular axon-dendrite interactions. Development 2020, 147, dev194530. [Google Scholar] [CrossRef]
- Han, B.; Zhou, R.; Xia, C.; Zhuang, X. Structural organization of the actin-spectrin-based membrane skeleton in dendrites and soma of neurons. Proc. Natl. Acad. Sci. USA 2017, 114, E6678–E6685. [Google Scholar] [CrossRef]
- Clarkson, Y.L.; Perkins, E.M.; Cairncross, C.J.; Lyndon, A.R.; Skehel, P.A.; Jackson, M. beta-III spectrin underpins ankyrin R function in Purkinje cell dendritic trees: Protein complex critical for sodium channel activity is impaired by SCA5-associated mutations. Hum. Mol. Genet. 2014, 23, 3875–3882. [Google Scholar] [CrossRef]
- Stevens, S.R.; van der Heijden, M.E.; Ogawa, Y.; Lin, T.; Sillitoe, R.V.; Rasband, M.N. Ankyrin-R Links Kv3.3 to the Spectrin Cytoskeleton and Is Required for Purkinje Neuron Survival. J. Neurosci. 2022, 42, 2–15. [Google Scholar] [CrossRef]
- Sancho, P.; Andres-Borderia, A.; Gorria-Redondo, N.; Llano, K.; Martinez-Rubio, D.; Yoldi-Petri, M.E.; Blumkin, L.; Rodriguez de la Fuente, P.; Gil-Ortiz, F.; Fernandez-Murga, L.; et al. Expanding the beta-III Spectrin-Associated Phenotypes toward Non-Progressive Congenital Ataxias with Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 2505. [Google Scholar] [CrossRef] [PubMed]
- Burk, K.; Zuhlke, C.; Konig, I.R.; Ziegler, A.; Schwinger, E.; Globas, C.; Dichgans, J.; Hellenbroich, Y. Spinocerebellar ataxia type 5: Clinical and molecular genetic features of a German kindred. Neurology 2004, 62, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Ranum, L.P.; Schut, L.J.; Lundgren, J.K.; Orr, H.T.; Livingston, D.M. Spinocerebellar ataxia type 5 in a family descended from the grandparents of President Lincoln maps to chromosome 11. Nat. Genet. 1994, 8, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Stevanin, G.; Herman, A.; Brice, A.; Durr, A. Clinical and MRI findings in spinocerebellar ataxia type 5. Neurology 1999, 53, 1355–1357. [Google Scholar] [CrossRef]
- Rea, G.; Tirupathi, S.; Williams, J.; Clouston, P.; Morrison, P.J. Infantile Onset of Spinocerebellar Ataxia Type 5 (SCA-5) in a 6 Month Old with Ataxic Cerebral Palsy. Cerebellum 2020, 19, 161–163. [Google Scholar] [CrossRef]
- Nicita, F.; Nardella, M.; Bellacchio, E.; Alfieri, P.; Terrone, G.; Piccini, G.; Graziola, F.; Pignata, C.; Capuano, A.; Bertini, E.; et al. Heterozygous missense variants of SPTBN2 are a frequent cause of congenital cerebellar ataxia. Clin. Genet. 2019, 96, 169–175. [Google Scholar] [CrossRef]
- Nuovo, S.; Micalizzi, A.; D’Arrigo, S.; Ginevrino, M.; Biagini, T.; Mazza, T.; Valente, E.M. Between SCA5 and SCAR14: Delineation of the SPTBN2 p.R480W-associated phenotype. Eur. J. Hum. Genet. 2018, 26, 928–929. [Google Scholar] [CrossRef]
- Lise, S.; Clarkson, Y.; Perkins, E.; Kwasniewska, A.; Sadighi Akha, E.; Schnekenberg, R.P.; Suminaite, D.; Hope, J.; Baker, I.; Gregory, L.; et al. Recessive mutations in SPTBN2 implicate beta-III spectrin in both cognitive and motor development. PLoS Genet. 2012, 8, e1003074. [Google Scholar] [CrossRef]
- Yildiz Bolukbasi, E.; Afzal, M.; Mumtaz, S.; Ahmad, N.; Malik, S.; Tolun, A. Progressive SCAR14 with unclear speech, developmental delay, tremor, and behavioral problems caused by a homozygous deletion of the SPTBN2 pleckstrin homology domain. Am. J. Med. Genet. A 2017, 173, 2494–2499. [Google Scholar] [CrossRef]
- Avery, A.W.; Crain, J.; Thomas, D.D.; Hays, T.S. A human beta-III-spectrin spinocerebellar ataxia type 5 mutation causes high-affinity F-actin binding. Sci. Rep. 2016, 6, 21375. [Google Scholar] [CrossRef]
- Avery, A.W.; Fealey, M.E.; Wang, F.; Orlova, A.; Thompson, A.R.; Thomas, D.D.; Hays, T.S.; Egelman, E.H. Structural basis for high-affinity actin binding revealed by a beta-III-spectrin SCA5 missense mutation. Nat. Commun. 2017, 8, 1350. [Google Scholar] [CrossRef] [PubMed]
- Denha, S.A.; Atang, A.E.; Hays, T.S.; Avery, A.W. beta-III-spectrin N-terminus is required for high-affinity actin binding and SCA5 neurotoxicity. Sci. Rep. 2022, 12, 1726. [Google Scholar] [CrossRef] [PubMed]
- Avery, A.W.; Thomas, D.D.; Hays, T.S. b-III-spectrin spinocerebellar ataxia type 5 mutation reveals a dominant cytoskeletal mechanism that underlies dendritic arborization. Proc. Natl. Acad. Sci. USA 2017, 114, E9376–E9385. [Google Scholar] [CrossRef] [PubMed]
- Legardinier, S.; Legrand, B.; Raguenes-Nicol, C.; Bondon, A.; Hardy, S.; Tascon, C.; Le Rumeur, E.; Hubert, J.F. A Two-amino Acid Mutation Encountered in Duchenne Muscular Dystrophy Decreases Stability of the Rod Domain 23 (R23) Spectrin-like Repeat of Dystrophin. J. Biol. Chem. 2009, 284, 8822–8832. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D. A guide to simple and informative binding assays. Mol. Biol. Cell 2010, 21, 4061–4067. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Sun, M.; Johnson, A.K.; Nelakuditi, V.; Guidugli, L.; Fischer, D.; Arndt, K.; Ma, L.; Sandford, E.; Shakkottai, V.; Boycott, K.; et al. Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet. Med. 2019, 21, 195–206. [Google Scholar] [CrossRef]
- Liu, L.Z.; Ren, M.; Li, M.; Ren, Y.T.; Sun, B.; Sun, X.S.; Chen, S.Y.; Li, S.Y.; Huang, X.S. A Novel Missense Mutation in the Spectrin Beta Nonerythrocytic 2 Gene Likely Associated with Spinocerebellar Ataxia Type 5. Chin. Med. J. 2016, 129, 2516–2517. [Google Scholar] [CrossRef]
- Vissers, L.; van Nimwegen, K.J.M.; Schieving, J.H.; Kamsteeg, E.J.; Kleefstra, T.; Yntema, H.G.; Pfundt, R.; van der Wilt, G.J.; Krabbenborg, L.; Brunner, H.G.; et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet. Med. 2017, 19, 1055–1063. [Google Scholar] [CrossRef]
- Romaniello, R.; Citterio, A.; Panzeri, E.; Arrigoni, F.; De Rinaldis, M.; Trabacca, A.; Bassi, M.T. Novel SPTBN2 gene mutation and first intragenic deletion in early onset spinocerebellar ataxia type 5. Ann. Clin. Transl. Neurol. 2021, 8, 956–963. [Google Scholar] [CrossRef]
- Bian, X.; Wang, S.; Jin, S.; Xu, S.; Zhang, H.; Wang, D.; Shang, W.; Wang, P. Two novel missense variants in SPTBN2 likely associated with spinocerebellar ataxia type 5. Neurol. Sci. 2021, 42, 5195–5203. [Google Scholar] [CrossRef] [PubMed]
- Banuelos, S.; Saraste, M.; Djinovic Carugo, K. Structural comparisons of calponin homology domains: Implications for actin binding. Structure 1998, 6, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Galkin, V.E.; Orlova, A.; Salmazo, A.; Djinovic-Carugo, K.; Egelman, E.H. Opening of tandem calponin homology domains regulates their affinity for F-actin. Nat. Struct. Mol. Biol. 2010, 17, 614–616. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, D.V.; Huehn, A.; Simon, B.; Huet-Calderwood, C.; Baldassarre, M.; Sindelar, C.V.; Calderwood, D.A. Structural basis of the filamin A actin-binding domain interaction with F-actin. Nat. Struct. Mol. Biol. 2018, 25, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Kesarwani, S.; Javoor, M.G.; Vinothkumar, K.R.; Sirajuddin, M. Structural insights into actin filament recognition by commonly used cellular actin markers. EMBO J. 2020, 39, e104006. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, Y.L.; Gillespie, T.; Perkins, E.M.; Lyndon, A.R.; Jackson, M. Beta-III spectrin mutation L253P associated with spinocerebellar ataxia type 5 interferes with binding to Arp1 and protein trafficking from the Golgi. Hum. Mol. Genet. 2010, 19, 3634–3641. [Google Scholar] [CrossRef]
- Guhathakurta, P.; Rebbeck, R.T.; Denha, S.A.; Keller, A.R.; Carter, A.L.; Atang, A.E.; Svensson, B.; Thomas, D.D.; Hays, T.S.; Avery, A.W. Early phase drug discovery of beta-III-spectrin actin-binding modulators for treatment of spinocerebellar ataxia type 5. J. Biol. Chem. 2023, 299, 102956. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Forward Primer | Reverse Primer |
|---|---|---|
| V58M | CTGGCAGATGAACGAGAAGCTATGCAGAAGAAAACCTTCACCAAG | CTTGGTGAAGGTTTTCTTCTGCATAGCTTCTCGTTCATCTGCCAG |
| K61E | GAACGAGAAGCTGTGCAGAAGGAAACCTTCACCAAGTGGGTAAAC | GTTTACCCACTTGGTGAAGGTTTCCTTCTGCACAGCTTCTCGTTC |
| T62I | GAGAAGCTGTGCAGAAGAAAATCTTCACCAAGTGGGTAAACTC | GAGTTTACCCACTTGGTGAAGATTTTCTTCTGCACAGCTTCTC |
| K65E | GTGCAGAAGAAAACCTTCACCGAGTGGGTAAACTCGCACCTGGCC | GGCCAGGTGCGAGTTTACCCACTCGGTGAAGGTTTTCTTCTGCAC |
| F160C | GTCTGGACCATCATCCTTCGATGCCAGATCCAAGACATCAGTGTG | CACACTGATGTCTTGGATCTGGCATCGAAGGATGATGGTCCAGAC |
| D255G | GGACTTACCAAGCTGCTGGGTCCCGAAGACGTGAATGTGGACC | GGTCCACATTCACGTCTTCGGGACCCAGCAGCTTGGTAAGTCC |
| T271I | GCCAGATGAGAAGTCAATCATTATCTATGTGGCTACTTACTACC | GGTAGTAAGTAGCCACATAGATAATGATTGACTTCTCATCTGGC |
| Y272H | GATGAGAAGTCAATCATTACCCATGTGGCTACTTACTACCATTAC | GTAATGGTAGTAAGTAGCCACATGGGTAATGATTGACTTCTCATC |
| H278R | CTATGTGGCTACTTACTACCGTTACTTCTCCAAGATGAAG | CTTCATCTTGGAGAAGTAACGGTAGTAAGTAGCCACATAG |
| SPTBN2 Gene Mutation | Inheritance | Clinical Onset, Sex | Age at Last Evaluation | Clinical Features | Brain MRI Findings |
|---|---|---|---|---|---|
| c. 172 G > A (p. V58M) [27] | familial | 37 y, M n = 1 | 61 y | Progressive cerebellar ataxia; Memory problems | Cerebellar Atrophy (Mild, stable) |
| heterozygous | |||||
| c. 181 A > G (p. K61E) [27] | de novo | Unknown, F n = 1 | 9 y | Cerebellar ataxia; Hyperreflexia; Hypotonia; Dysarthria; Nystagmus | Cerebellar Atrophy |
| heterozygous | |||||
| c.185 C >T (p. T62I) [16] | not known | 8 mo, F n = 1 | 18 y | Cerebellar ataxia; Cognitive delay; Psychomotor delay; Microcephaly; Nystagmus; Hypotonia; Mild bradykinesia | Cerebellar Atrophy |
| heterozygous | |||||
| c. 185 C > A (p. T62N) [30] | de novo | 5 mo, M n = 1 | 1 y 4 mo | Cerebellar ataxia; Hypotonia; Brisk reflexes; Motor and cognitive developmental delay; Nystagmus; Strabismus | Severe Cerebellar Atrophy |
| heterozygous | |||||
| c. 193 A > G (p. K65E) [11] | de novo | 4 mo, M n = 1 | 11 y | Ataxia; Hypotonia; Ataxic gait; Dysarthria (understandable); Strabismus; Horizontal nystagmus; Dysmetria; Intention tremor (mild) | Progressive Cerebellar Atrophy |
| heterozygous | |||||
| c.479 C > T (p. F160C) [16] | de novo | 5 mo, M n = 1 | 5 y | Cerebellar ataxia; Psychomotor delay; Strabismus; Hypotonia; Cognitive delay; Unable to walk; Nonverbal | Progressive Cerebellar Atrophy |
| heterozygous | |||||
| c. 486 C > G (p. I162M) [31] | familial | 48 y, F n = 1 | 53 y | Cerebellar ataxia; Dysarthria; Ataxic gait | Moderate Cerebellar Atrophy |
| heterozygous | |||||
| c. 758 T > C (p. L253P) [12] | familial | 15–50 y, mean = 32.8 y, n = 15 | 20–62 y | Stance, gait, and limb ataxia (14/15); Dysarthria (13/15); Intention tremor (5/15); Rest tremor (2/15); Nystagmus (13/15) | Cerebellar Atrophy |
| heterozygous | |||||
| c. 764 A > G (p. D255G) [11] | de novo | 1 y, F n = 1 | 8 y | Ataxia; Strabismus; Hypotonia; Bradykinesia; Ataxic gait; Dysarthria (difficult to understand); Intention tremor (moderate); ADHD | Progressive Cerebellar Atrophy |
| heterozygous | |||||
| c.812 C > T (p. T271I) [15] | de novo | 6 mo, M n = 1 | 8 y | Cerebellar ataxia; Motor developmental delay; Brisk reflexes; Ataxic gait and limb movements; Dystonia; Intention tremor | Progressive Cerebellar Atrophy |
| heterozygous | |||||
| c. 812 C > A (p. T271N) [29] | de novo | Unknown n = 1 | 4 y 7 mo | Ataxia; Psychomotor retardation | Cerebellar Hypoplasia |
| heterozygous | |||||
| c.888 T > C; + (p. Y272H/+) [16] | familial | Unknown n = 1 | Adulthood | No symptoms | NA |
| heterozygous | |||||
| c.833 A > G (p. H278R) [28] | de novo | 11 y, F n = 1 | 19 y | Cerebellar ataxia; Ataxic gait; Nystagmus | Cerebellar Atrophy |
| Mutation | Kd (µM) | Age of Onset |
|---|---|---|
| L253P | <1 | 15–50 y, mean = 32.8 y, n = 15 |
| T271I | <1 | 6 mo, n = 1 |
| K65E | <1 | 4 mo, n = 1 |
| F160C | <1 | 5 mo, n = 1 |
| K61E | <1 | Not reported; clinical evaluation at 9 y, n = 1 |
| T62I | 2.4 ± 0.3 | 8 mo, n = 1 |
| H278R | 6.7 ± 1.7 | 11 y, n = 1 |
| D255G | 7.3 ± 0.5 | 12 mo, n = 1 |
| V58M | 10.2 ± 1.4 | 37 y, n = 1 |
| Y272H/+ | 15.3 ± 2.0 | Y272H/+ parent is clinically normal, n = 1 |
| Wild-type | 55.2 ± 6.7 | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atang, A.E.; Keller, A.R.; Denha, S.A.; Avery, A.W. Increased Actin Binding Is a Shared Molecular Consequence of Numerous SCA5 Mutations in β-III-Spectrin. Cells 2023, 12, 2100. https://doi.org/10.3390/cells12162100
Atang AE, Keller AR, Denha SA, Avery AW. Increased Actin Binding Is a Shared Molecular Consequence of Numerous SCA5 Mutations in β-III-Spectrin. Cells. 2023; 12(16):2100. https://doi.org/10.3390/cells12162100
Chicago/Turabian StyleAtang, Alexandra E., Amanda R. Keller, Sarah A. Denha, and Adam W. Avery. 2023. "Increased Actin Binding Is a Shared Molecular Consequence of Numerous SCA5 Mutations in β-III-Spectrin" Cells 12, no. 16: 2100. https://doi.org/10.3390/cells12162100
APA StyleAtang, A. E., Keller, A. R., Denha, S. A., & Avery, A. W. (2023). Increased Actin Binding Is a Shared Molecular Consequence of Numerous SCA5 Mutations in β-III-Spectrin. Cells, 12(16), 2100. https://doi.org/10.3390/cells12162100