
The Microtubule-Targeting Agent Pretubulysin Impairs the Inflammatory Response in Endothelial Cells by a JNK-Dependent Deregulation of the Histone Acetyltransferase Brd4

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Cell Culture
2.3. Animals
2.4. Psoriasiform Dermatitis Model
2.5. Intravital Microscopy of the Mouse Cremaster Muscle
2.6. Cell Adhesion Assay under Flow Conditions
2.7. Flow Cytometric Analysis
2.8. Western Blot Analysis
2.9. Western Blot Analysis of Cell Fractions
2.10. Quantitative Polymerase Chain Reaction (qPCR)
2.11. mRNA Decay
2.12. Reporter Gene Assay
2.13. Chromatin Immunoprecipitation (ChIP) qPCR
2.14. Protein Translation Inhibition Assay
2.15. Metabolic Activity Assay
2.16. Lactate Dehydrogenase (LDH) Release Assay
2.17. Cell Viability Assay
2.18. siRNA Transfection
2.19. Statistical Analysis
3. Results
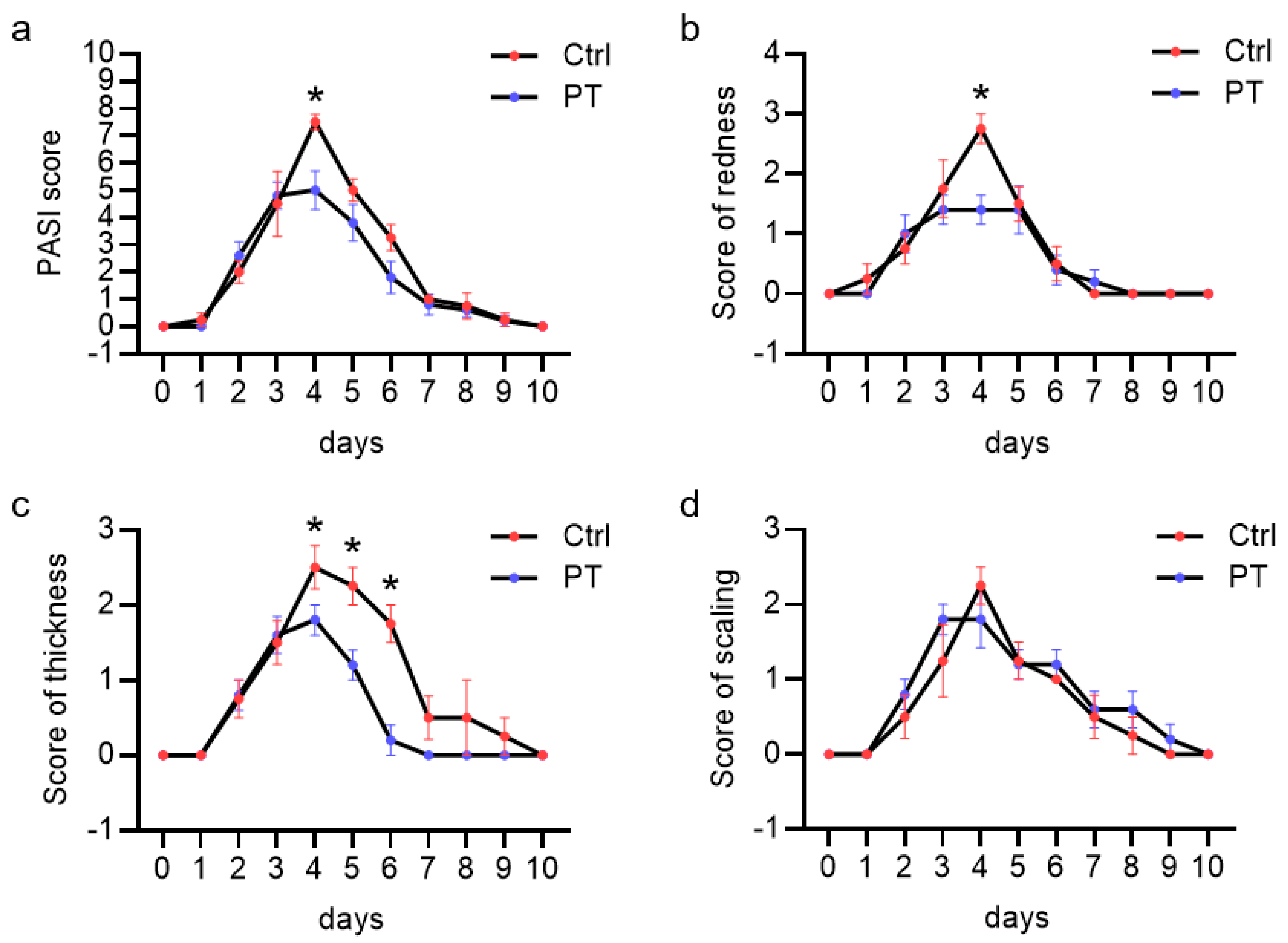
3.1. Pretubulysin Reduces Inflammation In Vivo in a Psoriasiform Dermatitis Mouse Model
3.2. Pretubulysin Reduces TNF-Induced Inflammatory Processes In Vivo and In Vitro by Down-Regulation of Endothelial Components of the Leukocyte Adhesion Cascade
3.3. Pretubulysin Increases the TNF-Induced NFκB and AP-1 Signaling Cascades in HUVECs, but Reduces the Promoter Activity of NFκB and AP-1
3.4. The Reduced Promoter Activity of NFκB-p65 and AP-1-cJun Is Not Due to a Decreased DNA-Binding Activity These Transcription Factors
3.5. Treatment with Pretubulysin Leads to a Time-Dependent Reduction of RNA Polymerase II Enrichment in the icam-1 and vcam-1 Genes Independent on cjun and p65 Enrichment and Promotes Super-Induction of the CAM mRNAs at Mean Pre-Treatment Times
3.6. The Pretubulysin-Evoked Down-Regulation of the Cell Adhesion Molecules ICAM-1 and VCAM-1 after Long-Term but Not Short-Term Treatment Can Be Reversed by Inhibition of JNK
3.7. Pretubulysin Exerts Its Inhibitory Effects on the Cell Adhesion Molecule Synthesis in Part by Differentially Regulating the DNA-Binding Behavior and Protein Levels of the Histone-Acetyl Transferase Bromodomain-Containing Protein 4 via Induction of JNK
4. Discussion
4.1. Pretubulysin and Other Depolymerizing MTAs Reduce Inflammatory Processes in ECs by Deregulation of Brd4 in a JNK-Dependent Mechanism
4.2. Microtubule Stabilizer Do Not Show an Inhibitory Action on the Leukocyte Adhesion Cascade
4.3. Depolymerizing MTAs Induce Pro-Inflammatory Signaling Cascades but Disconnect Promoter Action from Constructive Elongation
4.4. PT Induces Different Mechanisms of Action Depending on the Treatment Time
4.5. The PT-Evoked Down-Regulation of Both CAMs Is Due to Increased JNK Activity but Differs in the Functional Dependency on Brd4
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831. [Google Scholar] [CrossRef]
- Cook-Mills, J.M.; Deem, T.L. Active participation of endothelial cells in inflammation. J. Leukoc. Biol. 2005, 77, 487–495. [Google Scholar] [CrossRef]
- Madge, L.A.; Pober, J.S. TNF Signaling in Vascular Endothelial Cells. Exp. Mol. Pathol. 2001, 70, 317–325. [Google Scholar] [CrossRef]
- Bradley, J. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef]
- Lawrence, T. The Nuclear Factor NF-kappa B Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Ventura, J.-J.; Kennedy, N.J.; Lamb, J.A.; Flavell, R.A.; Davis, R.J. c-Jun NH2-Terminal Kinase Is Essential for the Regulation of AP-1 by Tumor Necrosis Factor. Mol. Cell. Biol. 2003, 23, 2871–2882. [Google Scholar] [CrossRef]
- Reinhard, C.; Shamoon, B.; Shyamala, V.; Williams, L.T. Tumor necrosis factor alpha -induced activation of c-jun N-terminal kinase is mediated by TRAF2. EMBO J. 1997, 16, 1080–1092. [Google Scholar] [CrossRef]
- Smith, C.W. Endothelial adhesion molecules and their role in inflammation. Can. J. Physiol. Pharmacol. 1993, 71, 76–87. [Google Scholar] [CrossRef]
- Guagnozzi, D.; Caprilli, R. Natalizumab in the treatment of Crohn’s disease. Biol. Targets Ther. 2008, 2, 275–284. [Google Scholar]
- McLean, L.P.; Shea-Donohue, T.; Cross, R.K.; Khanna, R.; Feagan, B.G.; Renna, S.; Orlando, A.; Cottone, M.; Rogler, G.; Kawalec, P.; et al. Vedolizumab for the treatment of ulcerative colitis and Crohn’s disease. Immunotherapy 2012, 4, 883–898. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Crizanlizumab: First Approval. Drugs 2020, 80, 79–84. [Google Scholar] [CrossRef]
- Wordeman, L.; Vicente, J.J. Microtubule Targeting Agents in Disease: Classic Drugs, Novel Roles. Cancers 2021, 13, 5650. [Google Scholar] [CrossRef]
- Schlesinger, N.; Schumacher, R.; Catton, M.; Maxwell, L. Colchicine for acute gout. Cochrane Database Syst. Rev. 2006, 8, CD006190. [Google Scholar] [CrossRef]
- Cerquaglia, C.; Diaco, M.; Nucera, G.; Regina, M.; Montalto, M.; Manna, R. Pharmacological and Clinical Basis of Treatment of Familial Mediterranean Fever (FMF) with Colchicine or Analogues: An Update. Curr. Drug Targets-Inflamm. Allergy 2005, 4, 117–124. [Google Scholar] [CrossRef]
- Leung, Y.Y.; Yao Hui, L.L.; Kraus, V.B. Colchicine—Update on mechanisms of action and therapeutic uses. Semin. Arthritis Rheum. 2015, 45, 341–350. [Google Scholar] [CrossRef]
- Robertson, S.; Martínez, G.J.; Payet, C.A.; Barraclough, J.Y.; Celermajer, D.S.; Bursill, C.; Patel, S. Colchicine therapy in acute coronary syndrome patients acts on caspase-1 to suppress NLRP3 inflammasome monocyte activation. Clin. Sci. 2016, 130, 1237–1246. [Google Scholar] [CrossRef]
- Ben-Chetrit, E.; Bergmann, S.; Sood, R. Mechanism of the anti-inflammatory effect of colchicine in rheumatic diseases: A possible new outlook through microarray analysis. Rheumatology 2005, 45, 274–282. [Google Scholar] [CrossRef]
- Meyer-Lindemann, U.; Mauersberger, C.; Schmidt, A.-C.; Moggio, A.; Hinterdobler, J.; Li, X.; Khangholi, D.; Hettwer, J.; Gräßer, C.; Dutsch, A.; et al. Colchicine Impacts Leukocyte Trafficking in Atherosclerosis and Reduces Vascular Inflammation. Front. Immunol. 2022, 13, 898690. [Google Scholar] [CrossRef]
- Masson, W.; Lobo, M.; Molinero, G.; Masson, G.; Lavalle-Cobo, A. Role of Colchicine in Stroke Prevention: An Updated Meta-Analysis. J. Stroke Cerebrovasc. Dis. 2020, 29, 104756. [Google Scholar] [CrossRef]
- Sandmann, A.; Sasse, F.; Müller, R. Identification and Analysis of the Core Biosynthetic Machinery of Tubulysin, a Potent Cytotoxin with Potential Anticancer Activity. Chem. Biol. 2004, 11, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Elnakady, Y.A.; Wiedmann, R.M.; Ullrich, A.; Rohde, M.; Kazmaier, U.; Vollmar, A.M.; Müller, R. Pretubulysin: From Hypothetical Biosynthetic Intermediate to Potential Lead in Tumor Therapy. PLOS ONE 2012, 7, e37416. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Liebl, J.; Fürst, R.; Ullrich, A.; Burkhart, J.; Kazmaier, U.; Herrmann, J.; Müller, R.; Günther, M.; Schreiner, L.; et al. Anti-angiogenic effects of the tubulysin precursor pretubulysin and of simplified pretubulysin derivatives. Br. J. Pharmacol. 2012, 167, 1048–1061. [Google Scholar] [CrossRef]
- Braig, S.; Wiedmann, R.M.; Liebl, J.; Singer, M.; Kubisch, R.; Schreiner, L.; A Abhari, B.; Wagner, E.; Kazmaier, U.; Fulda, S.; et al. Pretubulysin: A new option for the treatment of metastatic cancer. Cell Death Dis. 2014, 5, e1001. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, R.; Stehning, T.; Bischoff, I.; Ullrich, A.; Kazmaier, U.; Fürst, R. The pretubulysin-induced exposure of collagen is caused by endothelial cell retraction that results in an increased adhesion and decreased transmigration of tumor cells. Oncotarget 2017, 8, 77622–77633. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, A.; Chai, Y.; Pistorius, D.; Elnakady, Y.A.; Herrmann, J.E.; Weissman, K.J.; Kazmaier, U.; Müller, R. Pretubulysin, a Potent and Chemically Accessible Tubulysin Precursor from Angiococcus disciformis. Angew. Chem. Int. Ed. 2009, 48, 4422–4425. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.A.; Nachman, R.L.; Becker, C.G.; Minick, C.R. Culture of Human Endothelial Cells Derived from Umbilical Veins. Identification by Morphologic and Immunologic Criteria. J. Clin. Investig. 1973, 52, 2745–2756. [Google Scholar] [CrossRef]
- van der Fits, L.; Mourits, S.; Voerman, J.S.A.; Kant, M.; Boon, L.; Laman, J.D.; Cornelissen, F.; Mus, A.-M.; Florencia, E.; Prens, E.P.; et al. Imiquimod-Induced Psoriasis-Like Skin Inflammation in Mice Is Mediated via the IL-23/IL-17 Axis. J. Immunol. 2009, 182, 5836–5845. [Google Scholar] [CrossRef]
- Baez, S. An open cremaster muscle preparation for the study of blood vessels by in vivo microscopy. Microvasc. Res. 1973, 5, 384–394. [Google Scholar] [CrossRef]
- Mempel, T.R.; Moser, C.; Hutter, J.; Kuebler, W.M.; Krombach, F. Visualization of Leukocyte Transendothelial and Interstitial Migration Using Reflected Light Oblique Transillumination in Intravital Video Microscopy. J. Vasc. Res. 2003, 40, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.C.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Sakurai, H. IKKbeta phosphorylates p65 at S468 in transactivaton domain 2. FASEB J. 2005, 19, 1758–1760. [Google Scholar] [CrossRef]
- Pradère, J.-P.; Hernandez, C.; Koppe, C.; Friedman, R.A.; Luedde, T.; Schwabe, R.F. Negative regulation of NF-κB p65 activity by serine 536 phosphorylation. Sci. Signal. 2016, 9, ra85. [Google Scholar] [CrossRef]
- Wang, H.; Huang, W.; Liang, M.; Shi, Y.; Zhang, C.; Li, Q.; Liu, M.; Shou, Y.; Yin, H.; Zhu, X.; et al. (+)-JQ1 attenuated LPS-induced microglial inflammation via MAPK/NFκB signaling. Cell Biosci. 2018, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Filipčík, P.; Latham, S.L.; Cadell, A.L.; Day, C.L.; Croucher, D.R.; Mace, P.D. A cryptic tubulin-binding domain links MEKK1 to curved tubulin protomers. Proc. Natl. Acad. Sci. USA 2020, 117, 21308–21318. [Google Scholar] [CrossRef]
- Rosette, C.; Karin, M. Cytoskeletal control of gene expression: Depolymerization of microtubules activates NF-kappa B. J. Cell Biol. 1995, 128, 1111–1119. [Google Scholar] [CrossRef]
- Zou, Z.; Huang, B.; Wu, X.; Zhang, H.; Qi, J.; Bradner, J.; Nair, S.; Chen, L.-F. Brd4 maintains constitutively active NF-κB in cancer cells by binding to acetylated RelA. Oncogene 2013, 33, 2395–2404. [Google Scholar] [CrossRef]
- Nishiyama, A.; Dey, A.; Miyazaki, J.-I.; Ozato, K.; Karpova, T.; McNally, J.; Zheng, M.E.Y.; Vanoosthuyse, V.; Prykhozhij, S.; Hardwick, K.G. Brd4 Is Required for Recovery from Antimicrotubule Drug-induced Mitotic Arrest: Preservation of Acetylated Chromatin. Mol. Biol. Cell 2006, 17, 814–823. [Google Scholar] [CrossRef]
- Song, Y.; Hu, G.; Jia, J.; Yao, M.; Wang, X.; Lu, W.; Hutchins, A.P.; Chen, J.; Ozato, K.; Yao, H. DNA Damage Induces Dynamic Associations of BRD4/P-TEFb With Chromatin and Modulates Gene Transcription in a BRD4-Dependent and -Independent Manner. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Ouchida, R.; Kusuhara, M.; Shimizu, N.; Hisada, T.; Makino, Y.; Morimoto, C.; Handa, H.; Ohsuzu, F.; Tanaka, H. Suppression of NF-κB-dependent gene expression by a hexamethylene bisacetamide-inducible protein HEXIM1 in human vascular smooth muscle cells. Genes Cells 2003, 8, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-Y.; Lee, A.-Y.; Hou, S.Y.; Kemper, J.K.; Erdjument-Bromage, H.; Tempst, P.; Chiang, C.-M. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006, 20, 2383–2396. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Primke, T.F.; Ingelfinger, R.; Elewa, M.A.F.; Macinkovic, I.; Weigert, A.; Fabritius, M.P.; Reichel, C.A.; Ullrich, A.; Kazmaier, U.; Burgers, L.D.; et al. The Microtubule-Targeting Agent Pretubulysin Impairs the Inflammatory Response in Endothelial Cells by a JNK-Dependent Deregulation of the Histone Acetyltransferase Brd4. Cells 2023, 12, 2112. https://doi.org/10.3390/cells12162112
Primke TF, Ingelfinger R, Elewa MAF, Macinkovic I, Weigert A, Fabritius MP, Reichel CA, Ullrich A, Kazmaier U, Burgers LD, et al. The Microtubule-Targeting Agent Pretubulysin Impairs the Inflammatory Response in Endothelial Cells by a JNK-Dependent Deregulation of the Histone Acetyltransferase Brd4. Cells. 2023; 12(16):2112. https://doi.org/10.3390/cells12162112
Chicago/Turabian StylePrimke, Tobias F., Rebecca Ingelfinger, Mohammed A. F. Elewa, Igor Macinkovic, Andreas Weigert, Matthias P. Fabritius, Christoph A. Reichel, Angelika Ullrich, Uli Kazmaier, Luisa D. Burgers, and et al. 2023. "The Microtubule-Targeting Agent Pretubulysin Impairs the Inflammatory Response in Endothelial Cells by a JNK-Dependent Deregulation of the Histone Acetyltransferase Brd4" Cells 12, no. 16: 2112. https://doi.org/10.3390/cells12162112