1. Introduction
Epithelial homeostasis, the critical balance ensuring proper structure and function of epithelial tissues, is maintained through the coordination of diverse cellular processes and factors [
1]. Essential components, such as cell proliferation, differentiation, and apoptosis, work in tandem to replace damaged or aging cells, generate specialized cell types, and eliminate non-functional cells [
2,
3,
4]. Moreover, cell-cell adhesion through tight junctions, adherens junctions, and desmosomes, as well as interactions with the extracellular matrix, reinforce the structural integrity and regulation of the epithelium [
5,
6,
7]. Disruptions to this finely tuned balance can result in pathological conditions, such as chronic inflammation and cancer, highlighting the importance of understanding and preserving epithelial homeostasis.
Apical extrusion, a crucial mechanism for removing superfluous cells from the epithelial layer, involves the formation of an actomyosin contractile ring by adjacent cells, effectively forcing the aberrant cell out of the epithelial layer [
8,
9]. Pro-apoptotic signals produced by dying cells have been demonstrated to initiate apical extrusion in a caspase-dependent manner [
10]. Recent research also suggests that the expulsion of live cells from an epithelial sheet contributes significantly to maintaining epithelial homeostasis, as exemplified by the rapid turnover of epithelial cells in mammalian gut villi [
11,
12]. Extruded live cells eventually undergo apoptosis or anoikis due to the lack of a conducive environment for continued proliferation [
8]. However, defective cell extrusion plays a critical role in epithelial pathologies, such as cancer [
13]. Cells with oncogenic transformations that are extruded exhibit increased survival signals, enabling them to bypass normal apoptotic processes and continue proliferating. The extrusion of transformed cells has been implicated in tumorigenesis and the metastasis of cancer cells from primary to distant sites. However, the molecular mechanisms behind this phenomenon are not yet fully understood.
E-cadherin, a key component of adherens junctions, plays an essential role in cell extrusion by maintaining cell-cell adhesion and regulating tissue homeostasis [
14]. In the context of normal tissue turnover or cellular stress, E-cadherin’s functions are manipulated to permit the extrusion process. For example, in some cases, E-cadherin expression is downregulated or its localization altered, leading to a decrease in cell-cell adhesion. This downregulation can be induced by various pathways, such as through H-Ras overexpression leading to matrix metalloprotease-dependent cleavage, which weakens the adhesions between oncogenic and wild-type cells [
15]. Consequently, this downregulation of E-cadherin promotes the extrusion of affected cells from the epithelial layer. Interestingly, E-cadherin’s role extends beyond the initial extrusion event. The loss of apical E-cadherin not only promotes cell extrusion but also activates other signaling molecules like Src at the preserved basolateral E-cadherin-containing junctions. This Src activation can lead to increased cell proliferation, contributing to the formation of cell clusters on the epithelial monolayer [
16]. In certain contexts, the loss of E-cadherin in neighboring cells can lead to basal extrusion. This mechanism is particularly important in the context of cancerous transformations, where basal extrusion could potentially lead to the invasive behavior of cancer cells [
17].
Angiomotin-like 2 (AmotL2) is a scaffold protein within the angiomotin protein family, playing an essential role in regulating cell junction formation, actin cytoskeleton organization, and maintenance of cell polarity [
18,
19,
20,
21,
22]. The p100AmotL2 isoform is well-characterized and associated with the VE/E-cadherin/actin mechano-transductive complex [
23,
24]. In contrast, the p60AmotL2 isoform is significantly upregulated in invasive breast and colon cancers, acting as a negative regulator of apical-basal polarity by disrupting the connection between E-cadherin and radial actin filaments [
25,
26]. Activation of this isoform primarily occurs in response to stress signals such as severe hypoxia and is rarely detected in normal or tumor cells in vitro. In this study, we report that p60AmotL2 promotes apical extrusion and propose that tumor cells may exploit this process for delamination and invasion during cancer progression.
2. Materials and Methods
2.1. Antibodies and Reagents
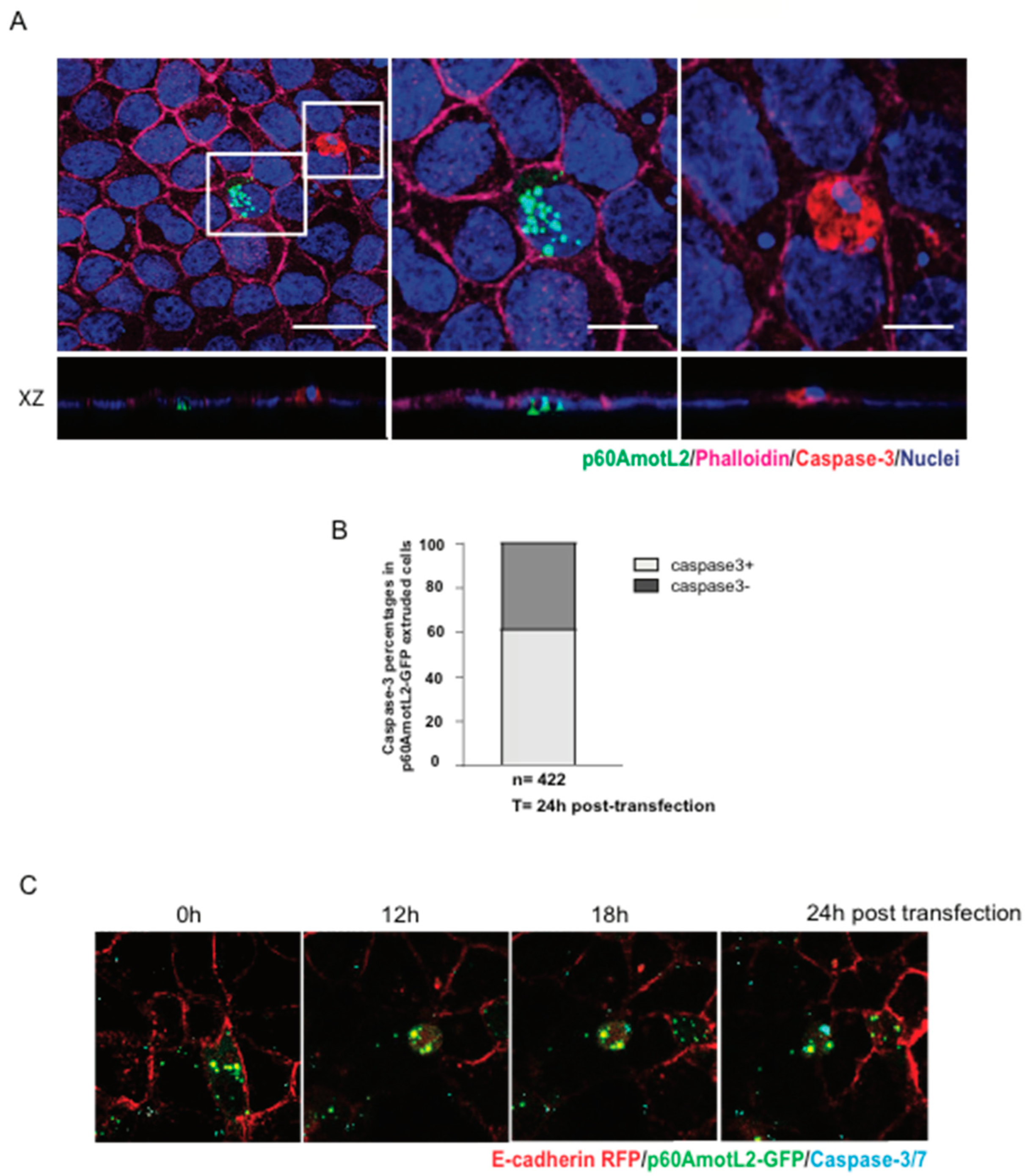
The following primary antibodies were used: LDS-AmotL2 (polyclonal antibodies reactive to human AmotL2 C-terminal peptide and detecting both p60 and p100AmotL2 isoforms), E-cadherin (product number 610193, BD Biosciences, Franklin Lakes, NJ, USA), β-actin (product number ab3280, Abcam, Cambridge, UK), Caspase-3 (product number 9662S, Cell Signaling, Danvers, MA, USA), and cytokeratin (product number ab27988, Abcam, Cambridge, UK). The following secondary antibodies were utilized: Alexa 568 anti-mouse (product number A10037, Cytiva, Marlborough, MA, USA) and Alexa 488 anti-rabbit (product number A11008, Cytiva, Marlborough, MA, USA). Texas Red phalloidin (product number T7471, Life Technologies, Carlsbad, California, USA) and Alexa Fluor™ 647 Phalloidin (product number A22287, Invitrogen, Waltham, MA, USA) were employed to visualize actin filaments. Nuclei were stained with DAPI (product number F6057, Sigma-Aldrich, St. Louis, MO, USA). For western blot detection, the following reagents were used: ECL anti-mouse IgG horseradish peroxidase (product number NA931V, Cytiva, Marlborough, MA, USA) and ECL anti-rabbit IgG horseradish peroxidase (product number NA934V, Cytiva, Marlborough, MA, USA). In live cell imaging, nuclei were visualized using NucBlue™ Live ReadyProbes™ Reagent (product number R37605, Thermo Fisher Scientific, Waltham, MA, USA). Actin was stained with the SiR-actin Kit (product number CY-SC001, Spirochrome, Thurgau, Switzerland).
2.2. Immunohistochemistry of Patient Samples
The patient studies were approved by the Karolinska Hospital, Research Ethics Committee, Dnr 2006/1014-31/4, 2012/90-31/2 and 010/0066-32. The patient sample sections were subjected to deparaffinization using xylene, followed by rehydration through a series of ethanol solutions. To quench endogenous peroxidase activity, the slides were incubated with 3% hydrogen peroxide. For antigen retrieval, the slides were exposed to citrate buffer (pH 6.0) in a microwave oven for 20 min, followed by a cooling period of 20 min at room temperature. To mitigate nonspecific background staining, the slides were treated with horse serum as a blocking agent. Subsequently, the slides were subjected to an overnight incubation at 4 °C with a polyclonal antibody against AmotL2 (LDS-AmotL2, dilution 1:400). Following the incubation, the slides were thoroughly rinsed and then exposed to biotinylated horse anti-mouse secondary antibodies. This was succeeded by another round of rinsing and incubation, during which avidin-biotin-peroxidase complexes were applied to the slides. For visualization of immunostaining, the slides were immersed in a solution containing 0.05% 3,3′-diaminobenzidine tetrahydrochloride. Subsequently, counterstaining was performed using hematoxylin.
2.3. Mouse Experiments
Animal experiments were conducted in compliance with the Stockholm South Ethical Committee in Sweden (N129/15). All experiments utilized wild-type mice. Intestines were harvested at the animal facility at KMB, Karolinska Institutet, and processed separately to isolate the epithelial layer in the lab. All methods adhered to relevant guidelines and regulations, with experimental protocols approved by the Swedish Board of Agriculture.
2.4. Immunohistochemistry
Mouse small intestine samples were fixed in 4% paraformaldehyde (PFA; product number sc-281692, Santa Cruz Biotechnology, Santa Cruz, CA, USA) in 1×PBS and embedded into a cryomold. Tissues were then cut into 10-μm sections and mounted onto glass slides. The slides were rinsed once in 1×PBS, permeabilized with 0.5% TritonX-100 (product number X100, Sigma-Aldrich, St. Louis, MO, USA) for 15 min, and non-specific staining was blocked with 0.5% horse serum diluted in 1×PBS. The slides were incubated with primary antibodies overnight at 4 °C, washed 3 times for 10 min in 1×PBS, and positive signals were detected with a secondary antibody at room temperature for 1 h. The slides were washed 3 times for 10 min in 1×PBS and mounted with DAPI-containing mounting media (product number F6182, Sigma-Aldrich, St. Louis, MO, USA). Immunohistochemistry of AmotL2 in human tissues was performed in collaboration with
http://www.proteinatlas.org (accessed on 15 January 2021). The antibodies were generated in the Holmgren lab, and the results have not yet been made publicly available.
2.5. Western Blot (WB)
For cell lysate preparation, cells were treated with a lysis buffer that contained 50 mM Hepes buffer, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, and 1% TritonX-100, along with a freshly added 1× protease inhibitor (product number 04693159001, Roche, Basel, Switzerland). This process was performed on ice. Following this, the mixture was subjected to centrifugation at 15,000 rpm for a span of 3 min and the supernatant was collected. Lysates were then combined with an SDS sample buffer (4×, product number 1225644, Novex, Wadsworth, OH, USA) that was enhanced with a 10% sample-reducing agent (product number 1176192, Novex). The proteins present were fractionated using a Bis-Tris precast polyacrylamide gel with a gradient of 4–12% (product number NP0322BOX, Novex, Wadsworth, OH, USA). These fractionated proteins were then moved onto a nitrocellulose membrane (product number 10401396, Whatman, Maidstone, UK). To block any non-specific binding, the membrane was treated with a solution of 5% non-fat milk and 0.1% Tween 20 in 1×PBS for an hour at room temperature. Following this, the membrane was incubated overnight at 4 °C with the primary antibody and was then subjected to treatment with the secondary antibody for an additional hour at room temperature. Finally, proteins that were labeled with antibodies were identified using a chemiluminescent substrate (ECL; product number RPN2232, Amersham Biosciences, Amersham, UK) using an iBright imaging system (Thermo Fisher Scientific, Waltham, MA, USA).
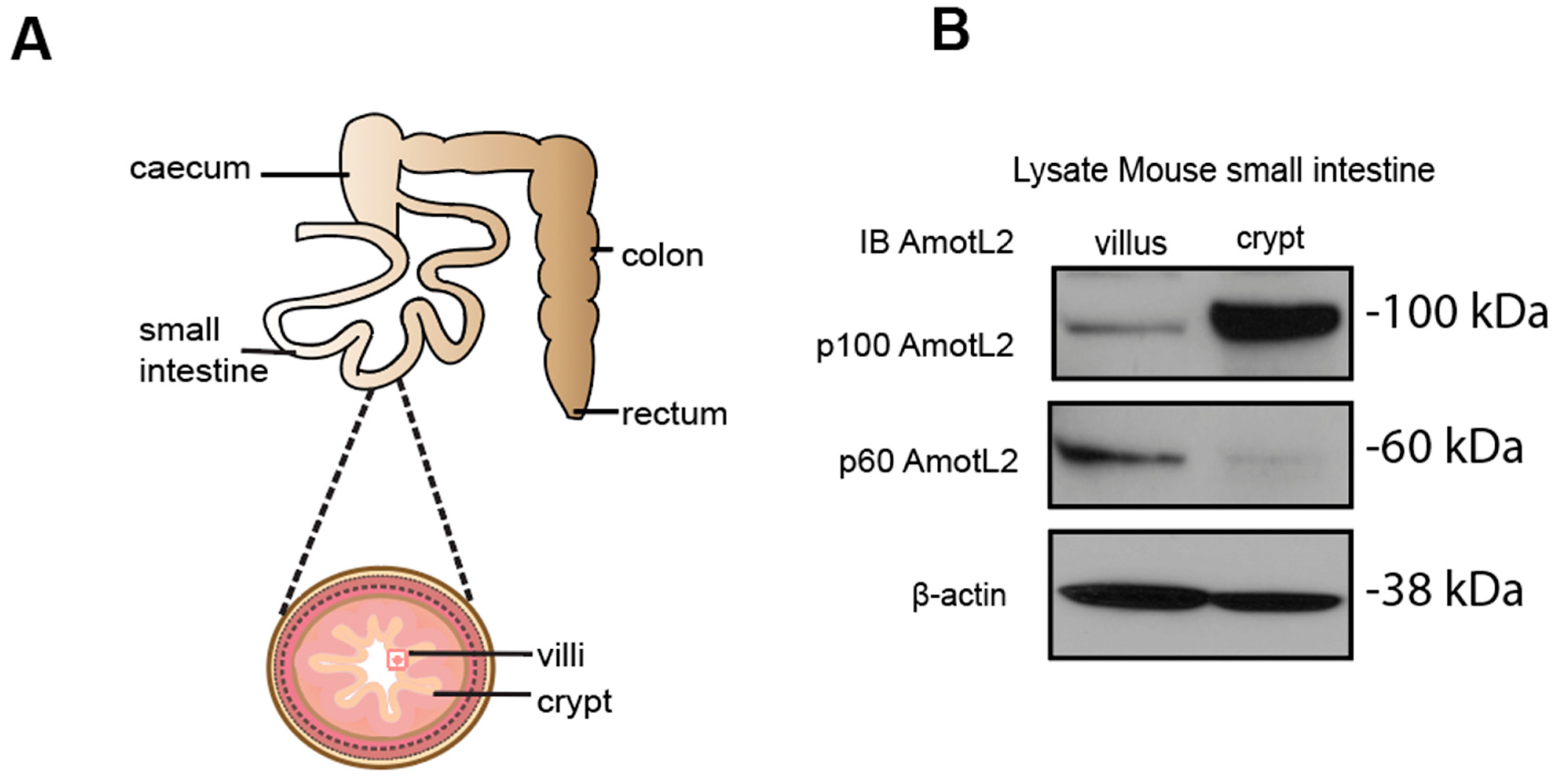
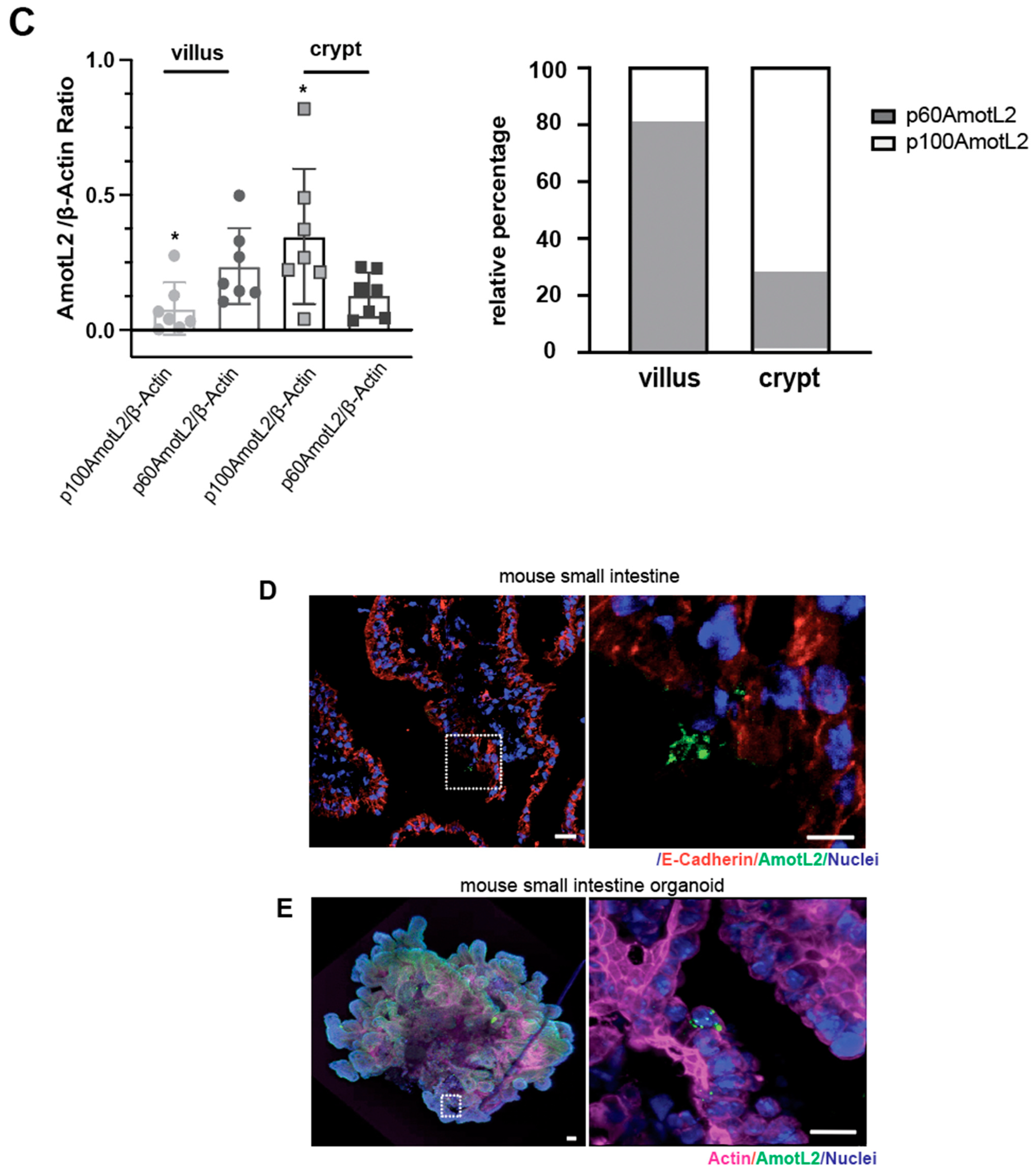
2.6. Western Blot Analysis of Mouse Small Intestine
Small intestine samples were collected from wild-type c57B6/J mice (n = 4), and intestinal villi and crypts were gently scraped using a cell scraper. The villi and crypts were homogenized in lysis buffer and heated at 95 °C for 5 min. Cell extracts were centrifuged at 15,000 rpm for 10 min, and the supernatants were collected for western blot analysis. The 25 μg of protein were loaded on a 10% Tris-Acetate gel at 120 V for 120 min. Detection was performed using a standard ECL kit. AmotL2 protein levels are presented relative to the beta-actin loading control.
2.7. Mouse Small Intestine Organoids
Mouse small intestine organoids were harvested and cultured following previously described methods [
27]. In brief, small intestine tissue from sacrificed mice was sectioned into small fragments and rinsed thoroughly with cold 1×PBS until the supernatant remained clear. Tissue fragments containing crypts were isolated using Gentle Cell Dissociation Reagent (product number 07174, STEMCELL, Vancouver, BC, Canada) and filtered through 70 μm cell strainers. The fractions were embedded in Geltrex matrigel (product number A1413202, Thermo Fisher Scientific, Waltham, MA, USA) and maintained in IntestiCult™ Organoid Growth Medium (product number 06005, STEMCELL, Vancouver, BC, Canada) for the duration of the experiments.
2.8. Cell Culture
Madin-Darby Canine Kidney (MDCK) wild-type cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). MDCK cells stably expressing E-cadherin tagged with a red fluorescent protein (MDCK-E-cad-RFP) were generously provided by Prof. Christopher P. Torret’s lab. All cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (product number 31053028, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, product number 16000044, Thermo Fisher Scientific, Waltham, MA, USA) and penicillin/streptomycin (product number 10378016, Thermo Fisher Scientific, Waltham, MA, USA).
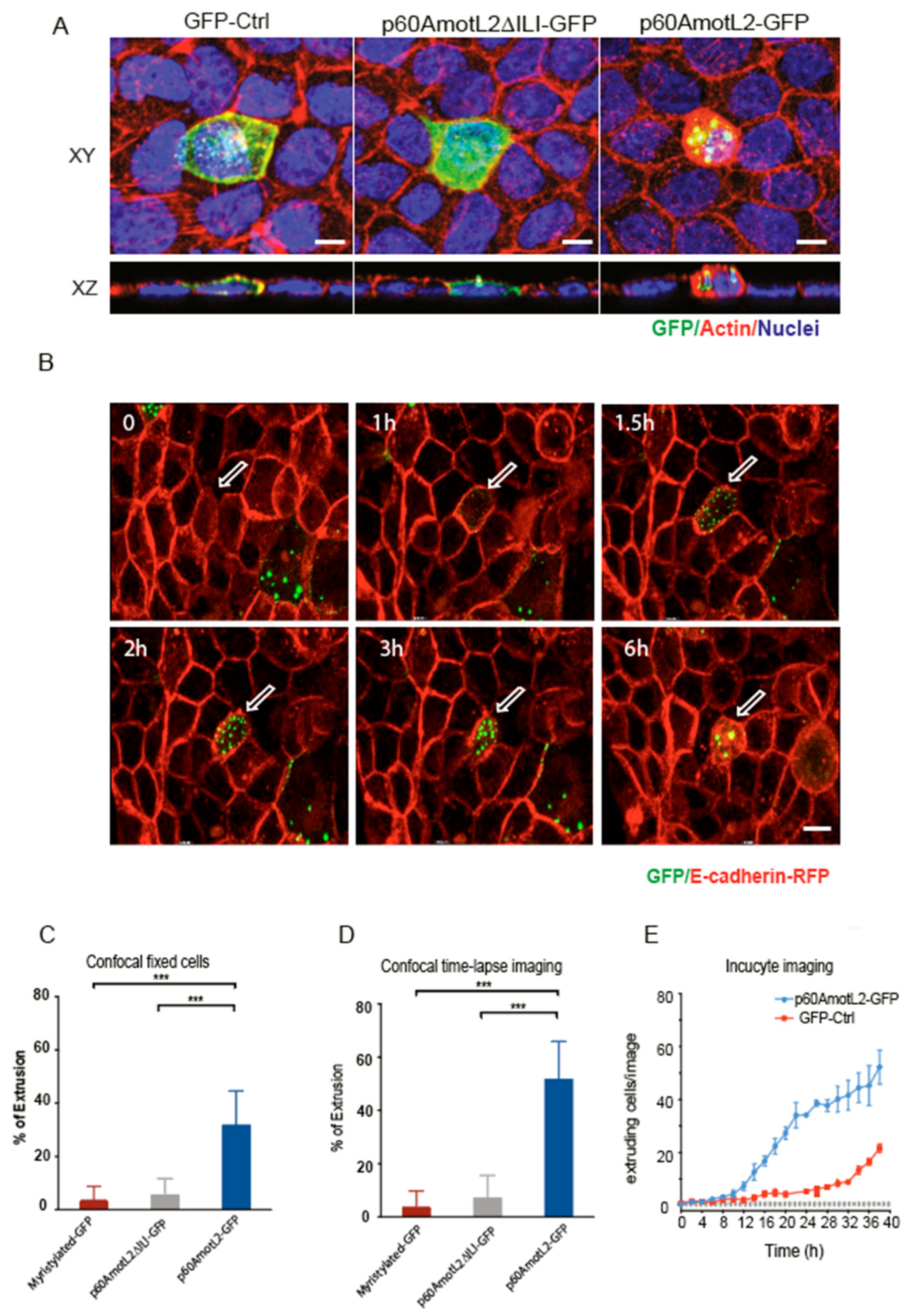
MDCK cell lines stably expressing doxycycline-inducible p60AmotL2 were generated using the Gateway™ system (Thermo Fisher Scientific, Waltham, MA, USA), as previously described [
25]. To induce p60AmotL2 expression, doxycycline (Dox) (10 ng/mL, product number D3447, Sigma-Aldrich, St. Louis, MO, USA) was added to the culture after seeding the cells. For plasmid transfections, MDCK cells were seeded on glass slides (BD Falcon Culture slides, BD Biosciences, Franklin Lakes, NJ, USA) in a growth medium one day prior to transfection. Plasmids p60AmotL2-GFP, p60AmotL2ΔILI-GFP, and myristoylated-GFP were transfected into the cells using the GenJet™ In Vitro DNA transfection reagent for MDCK cells (product number SL100489-MDCK, SignaGen Laboratories, Frederick, MD, USA), following the manufacturer’s protocol.
2.9. Immunofluorescent Staining
MDCK cells were cultured in 8-well chamber slides to confluency and fixed with 4% paraformaldehyde in PBS (product number sc-281692, Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 10 min, permeabilized for 5 min with 0.5% TritonX-100 (product number X100, Sigma-Aldrich, St. Louis, MO, USA) in 1×PBS, washed three times with 1×PBS, and blocked with 5% horse serum in 1×PBS for 1 h before incubating with primary antibodies. After washing 3 times in 1×PBS, the slides were incubated with secondary antibodies diluted in 5% horse serum in 1×PBS. Following incubation with the secondary antibody, the slides were washed 3 times in 1×PBS and then mounted onto coverslips.
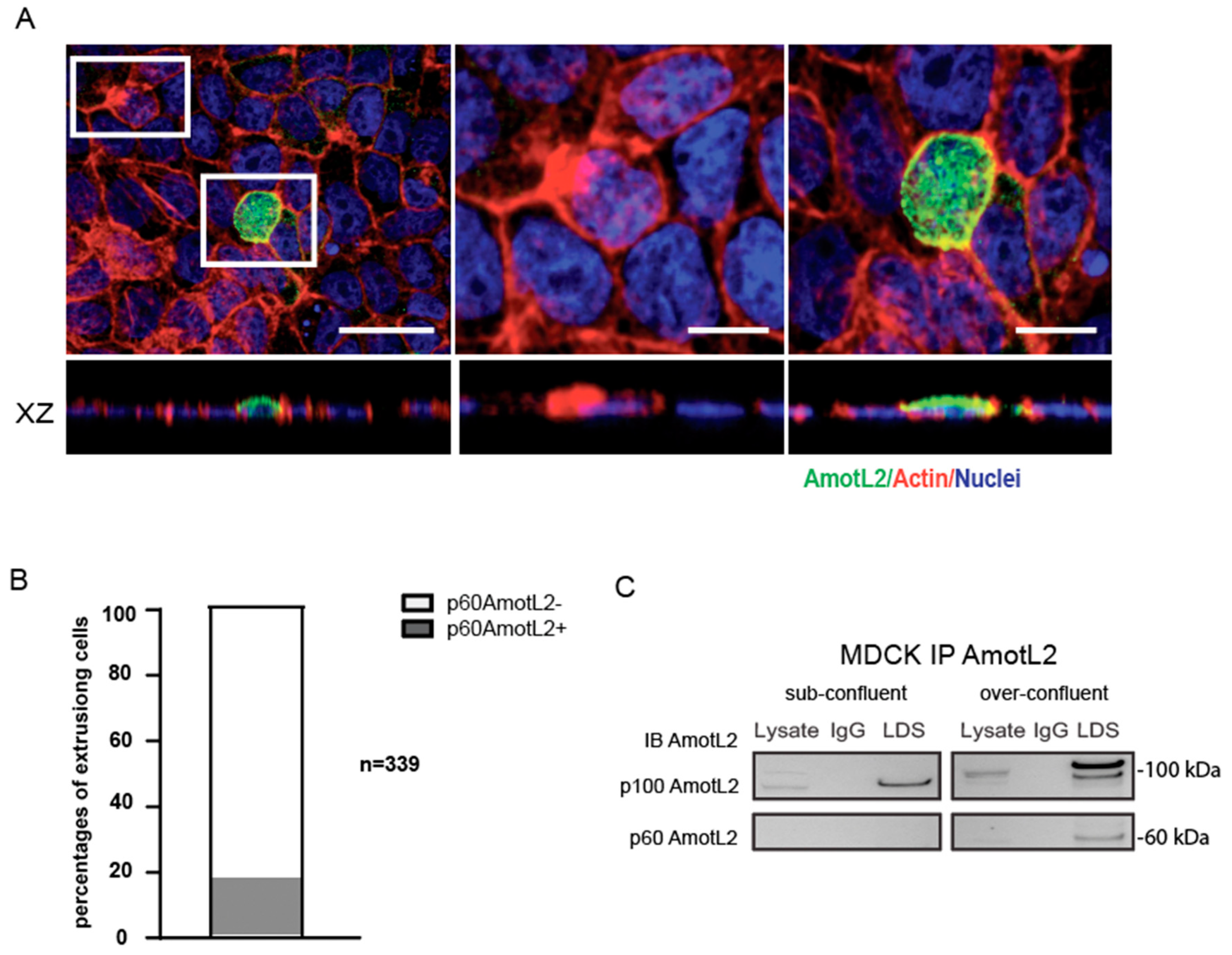
2.10. Overcrowding Assay
MDCK cells were plated at a concentration of 500,000 cells per cm2 and grown to confluency for 4 days in 8-well chamber slides. The slides were then fixed in 4% PFA (product number sc-281692, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and subjected to immunofluorescence staining as described above in the cell immunostaining method. Slides were imaged using a Zeiss AxioObserver with LSM700 confocal module and a 63× objective (Zeiss, Oberkochen, Germany).
2.11. Co-immunoprecipitation(co-IP)
For the co-immunoprecipitation (co-IP) experiment, cells were rinsed with cold PBS and directly scraped off a 10 cm dish into a lysis buffer. The lysis buffer consisted of 50 mM Hepes buffer, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, and 1% TritonX-100. The lysates were then centrifuged at 15,000 rpm for 5 min to remove debris. To minimize non-specific binding to Sepharose beads, cell lysates were pre-cleared by incubating with protein G Sepharose beads (product number ab193259, Abcam, Cambridge, UK) for 1.5 h at 4 °C. After pre-clearing, 2 µg of AmotL2 or control antibodies from the same species were added to the lysates and incubated overnight at 4 °C. Immunocomplexes were precipitated by adding protein G beads to the lysate and incubating for 2 h at 4 °C. The beads were then washed five times by centrifugation at 12,000 rpm for 20 s each time with the lysis buffer to remove non-specifically bound proteins. Finally, the protein-bead complexes were denatured, and the proteins were separated from the beads for analysis by western blotting.
2.12. Confocal Time-Lapse Imaging
MDCK RFP-E-Cadherin cells were seeded at confluency on a 2 mg/mL collagen matrix in a 35 mm Petri dish (Greiner bio-one, Kremsmünster, Germany). Cells were transfected with p60AmotL2-GFP, p60AmotL2ΔILI-GFP plasmid, or GFP-myristylated for 8h or more before obtaining confocal images. NucBlue™ Live ReadyProbes™ Reagent (product number R37605, Thermo Fisher Scientific, Waltham, MA, USA) was used to visualize nuclei. Images were captured using a Zeiss AxioObserver with LSM700 confocal module in 40× or 63× objective (Zeiss, Oberkochen, Germany). During the experiments, Petri dishes were maintained in a confocal integrated incubator (37 °C, 5% CO
2, and a humid atmosphere). Images were captured every 30 min for at least 12 h. Image and video captures were processed and analyzed using the IMARIS software (
https://imaris.oxinst.com (accessed on 14 January 2019)) and ImageJ software (version 1.53t).
2.13. Time-Lapse Imaging Using Incucyte®
MDCK wild-type cells were seeded in a 48-well plate (Thermo Fisher Scientific, Waltham, MA, USA) and allowed to reach 70% confluency. Subsequently, the cells were transfected with p60AmotL2-GFP or GFP-ctrl plasmid using the GenJet™ In Vitro DNA transfection reagent, as previously mentioned. Following transfection, the cells were transferred to the Incucyte® Live-Cell Analysis System (Essen Bioscience, Ann Arbor, MI, USA). Live images were captured at 2-h intervals over a 48-h period using bright field and GFP imaging with 20× objective lenses. The imaging process was performed inside an incubator. To analyze the extruding cell numbers, Incucyte® analysis software (Version 2021B, Essen Bioscience, Ann Arbor, MI, USA) was used.
2.14. Foci Formation Assay
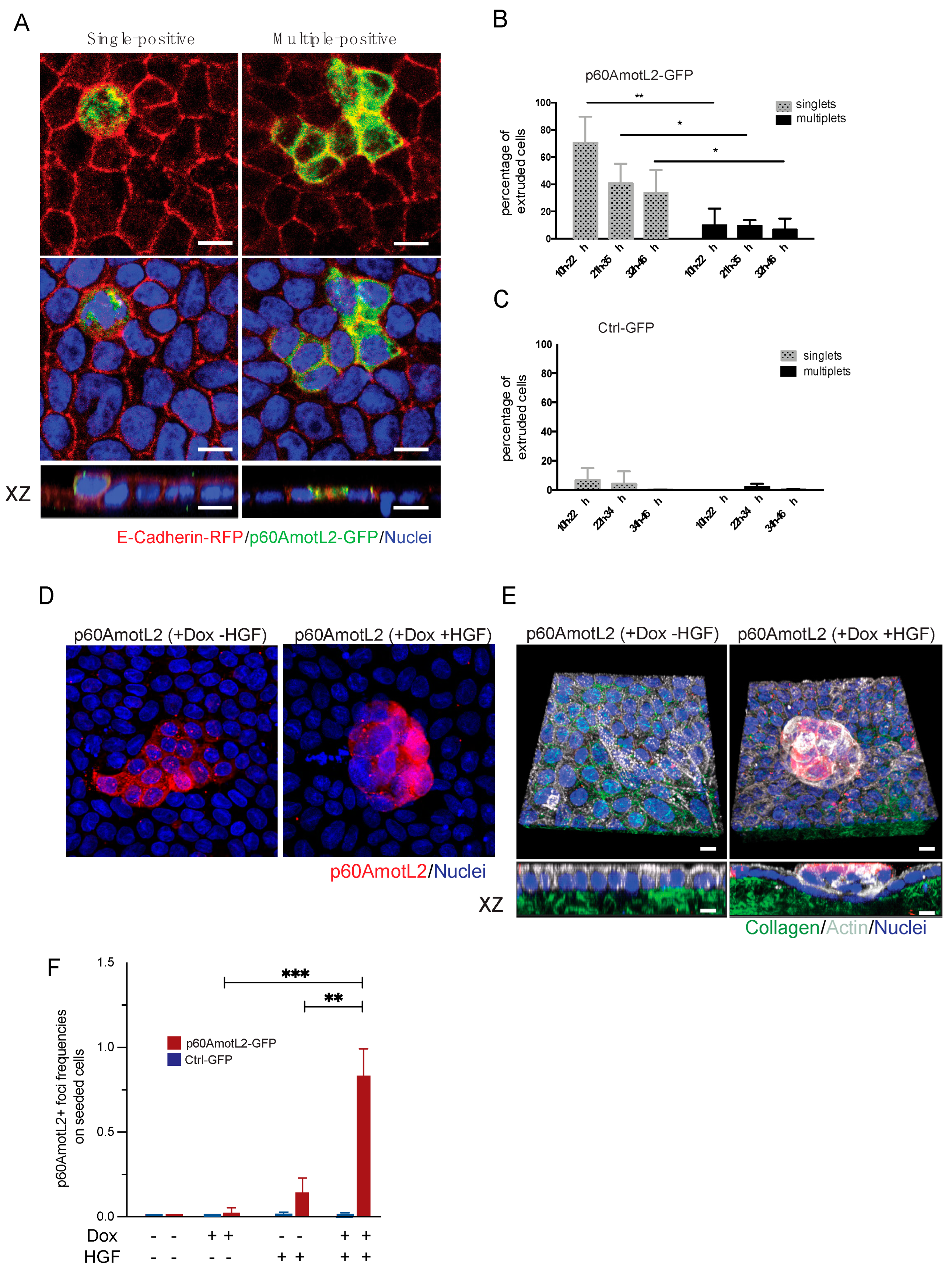
MDCK wild-type cells and MDCK cells with Dox-inducible p60AmotL2 expression were mixed at a ratio of 50:1 and seeded on bovine collagen conjugated with Oregon Green (product number O10241, Thermo Fisher Scientific, Waltham, MA, USA) at a final concentration of 30 µg/mL. The cells were initially treated with Dox for 24 h to induce p60AmotL2 expression. Subsequently, they were treated with Hepatocyte Growth Factor (HGF, product number PHG0254, Thermo Fisher Scientific, Waltham, MA, USA) for the following 3 days. The cells, along with the collagen matrix, were then fixed and stained with LDS-AmotL2 antibody, as previously described. The following secondary antibodies were used: Alexa 568 anti-rabbit (product number A11011, Cytiva, Marlborough, MA, USA). Nuclei were stained with DAPI (product number F6057, Sigma-Aldrich, St. Louis, MO, USA), and Alexa Fluor™ 647 Phalloidin (product number A22287, Invitrogen, Waltham, MA, USA) was utilized for F-actin staining. The images were captured using an LSM700 confocal module with a 40× objective. Manual counting of foci and analysis using ImageJ software were performed to determine the results.
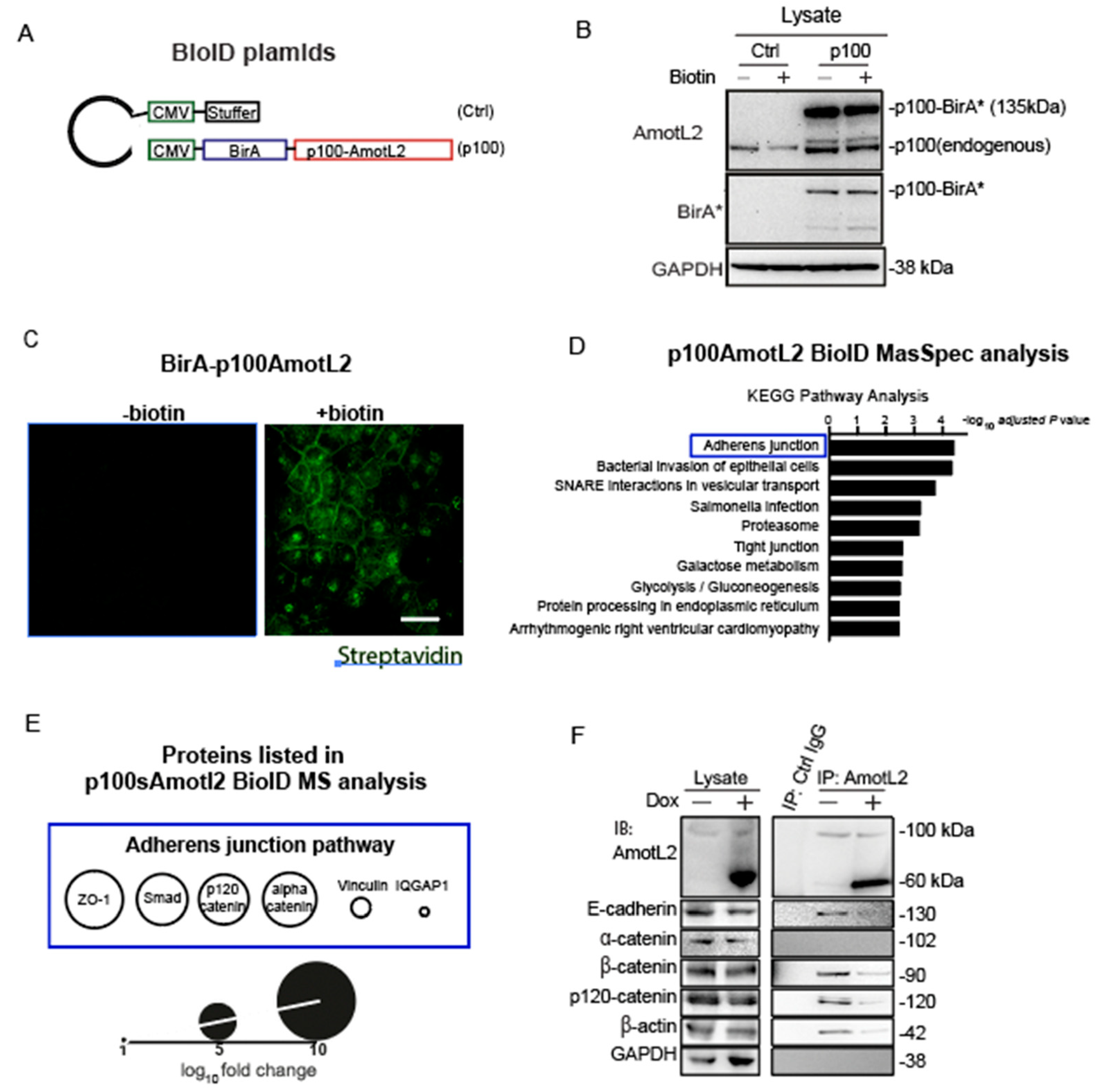
2.15. Proximity-Dependent Biotin Identification (BioID)
BioID plasmids were constructed using a mammalian gene expression lentiviral vector by combining cDNA fragments encoding human p100AmotL2 (Accession No: NM_001278683) with the N-terminus of E. coli biotin ligase (BirA). An empty vector with the same backbone was used as a negative control. All constructs were verified through restriction enzyme digestion. The p100AmotL2 BioID construct, and empty vector were packaged into lentivirus using Lipofectamine 3000 Transfection Reagent according to the manufacturer’s protocol. MDCK cells were transduced with the lentiviral constructs, and stable cell lines were generated using a 0.5 mg/mL geneticin selection. Stably transduced cells were cultured in DMEM medium, supplemented with 10% FBS and 0.5 mg/mL geneticin.
For the BioID experiment, transfected MDCK cells were treated with 50 µM biotin for 16 h, followed by harvesting in lysis buffer containing 50 mM Tris·HCl (pH 7.4), 8 M urea, 1 mM DTT, and protease inhibitors. Lysates were supplemented with 1% Triton X-100 before sonication. Biotinylated proteins were purified using streptavidin beads overnight at 4 °C. After washing five times with 8M urea in 50 mM Tris·HCl (pH 7.4) and once with 50 mM Tris·HCl (pH 7.4), the beads were resuspended in PBS and prepared for further protein analysis (Western Blotting and Mass Spectrometry analysis). Three independent experiments were performed (n = 3 in all groups), including ‘p100AmotL2 ± biotin’, and ‘empty vector ± biotin’.
Protein identification criteria for the BirA p100AmotL2 BioID construct were as follows: (1) at least two of three samples in the ‘p100AmotL2 + biotin’ group had a positive value; (2) the minimum value of the ‘p100AmotL2 + biotin’ group was equal to or higher than the maximum value of the ‘empty vector ± biotin’ and ‘p100AmotL2—biotin’ groups; (3) the average value of the ‘p100AmotL2 + biotin’ group was higher than those in the ‘empty vector ± biotin’ and ‘p100AmotL2—biotin’ groups.
4. Discussion
Here we report for the first time that p60AmotL2 is an inducer of apical extrusion. Our findings are in line with data showing that actomyosin contractility plays an important role in apical extrusion. E-cadherin organizes epithelial cells by coupling cells to contractile actin cytoskeleton [
31]. E-cadherin not only acts as a junctional complex connecting cells but is also of importance for relaying mechanical forces between neighboring cells and preventing the de-lamination of individual cells [
32]. The precise control of tensile activity appears to be critical not only for the function of the epithelium but may also determine which cells are destined for extrusion. For example, the Rho-Kinase (ROCK) is required for spontaneous as well as oncogenic-driven extrusion of cells [
12]. Similar findings have been shown with the small GTPase CDC42, which is involved in the organization of the cytoskeleton [
33]. We have previously shown that E-cadherin is dependent on p100AmotL2 to generate radial actin filaments that relay force between cells [
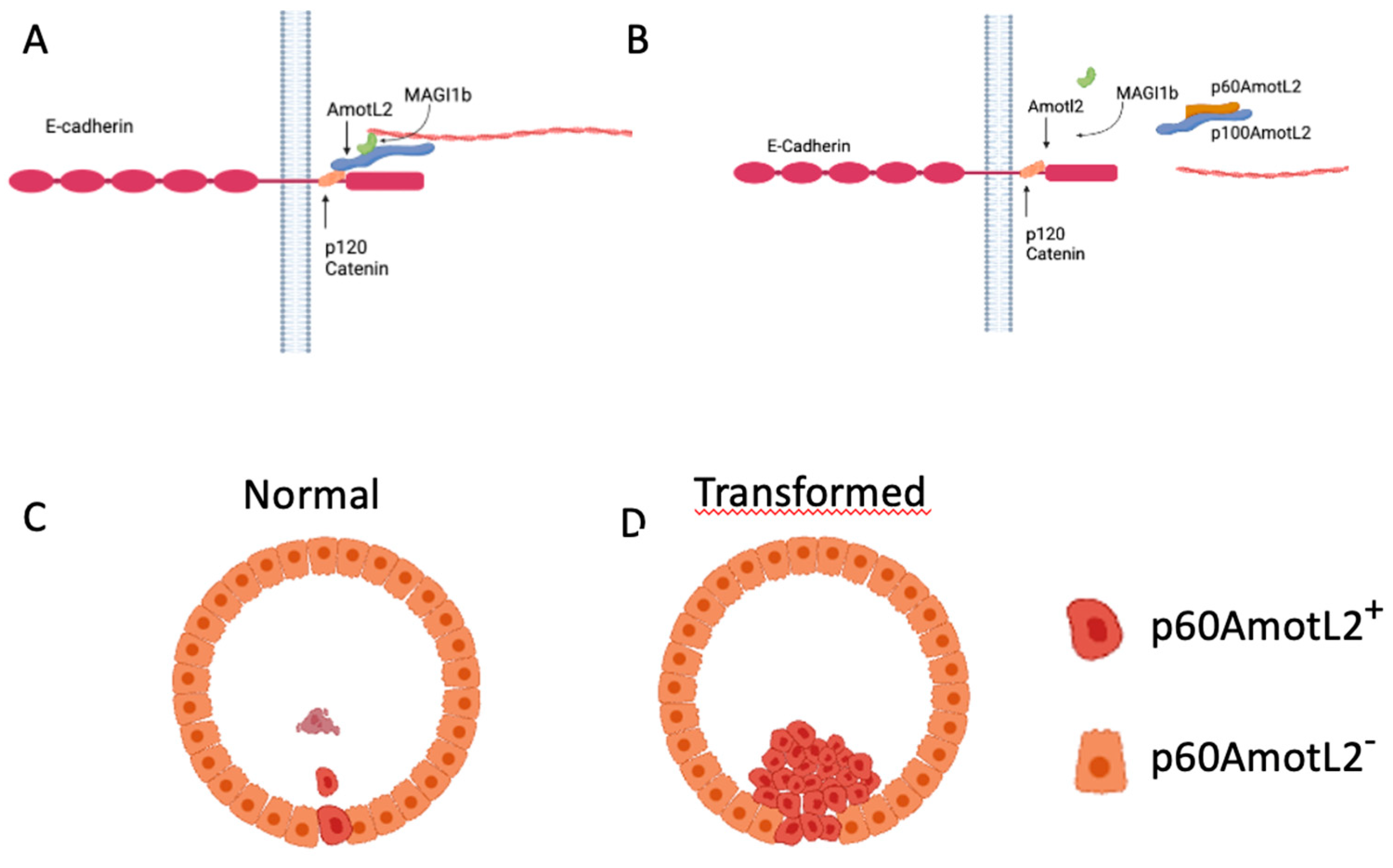
26]. Furthermore, the p60AmotL2 isoform acts as an antagonist to the p100AmotL2/E-cadherin complex, disconnecting radial actin and thereby decreasing tensile force (Schematic in
Figure 8). Here we further report that p100AmotL2 is associated with p120catenin and that this complex is inhibited by the expression of p60AmotL2. This is of interest as p120catenin has previously been shown to play a role in cell extrusion. Research by Kourtidis et al. has illustrated that apical and basal E-cadherin-based junctions have contrasting roles in growth regulation, contingent on the activity of p120catenin and Src, where apical complexes inhibit the growth and EMT-promoting effects of Src and p120catenin via the microRNA miR-30b [
34].
The p60AmotL2 is normally expressed at low or non-detectable levels in culture. Specific stress signals such as hypoxia activate transcription of p60AmotL2 by the binding of c-Fos to its promoter. How p60AmotL2 is activated on a single cell level remains to be elucidated. It does, however, indicate that the initial signal to be extruded comes from the targeted cell. We hypothesize that the decrease in radial actin and a concomitant decrease in tensile strength triggers the formation of a contractile actin ring in the neighboring cells that ultimately squeezes out the redundant cell. However, as we have previously shown that p60AmotL2 is a negative regulator of apical-basal polarity we cannot exclude that this may also affect cell extrusion.
As p60AmotL2 appears to be expressed in extruding cells it is possible that p60AmotL2 expression is pro-apoptotic and thereby triggers extrusion. However, data shown here indicates that cells being extruded by this mechanism are initially live as they have intact membranes, and markers of Caspase-3 activity were not present. Furthermore, p60AmotL2-expressing cells retained an intact capacity to grow in a colony formation assay, indicating that p60AmotL2 expression is not deleterious to the cells. Additionally, it was shown that clusters of p60AmotL2 positive cells were maintained in the epithelial layer without being extruded or undergoing cell death. This is similar to results indicated in glandular cancer such as the prostate where the high intensity of vesicular AmotL2 could be detected without apparent shedding into the lumen. Collectively this indicates that singular cells expressing p60AmotL2 are prone to extrusion, whereas groups of cells remain within the epithelial monolayer (
Figure 8).
This investigation has centered on elucidating the functional role of p60AmotL2 in the apical extrusion mechanism. However, it is noteworthy that in vitro activation of oncogenes results in an opposing basal extrusion phenomenon. Marshall and colleagues demonstrated a transition from apical to basal extrusion through the inactivation of the Adenomatosis Polyposis Coli (APC) tumor suppressor gene [
35]. The implications of this directional shift on the viability of shed tumor cells remain enigmatic. In scenarios such as ductal carcinoma in situ (DCIS) of the breast, where tumor cells colonize ducts, one might speculate on an apical mode of extrusion. As the evidence accumulates, the induction of apical extrusion by p60AmotL2 further supports the notion that tumor cells co-opt ordinary cellular processes to fortify their survival and proliferation [
17]. Subsequent investigations will whether p60AmotL2-expressing cells exhibit distinct susceptibility to cancer therapies. In conclusion, our findings provide insights into the intricate interplay between normal and malignant cell dynamics, shedding light on potential avenues for therapeutic intervention.


 and
and 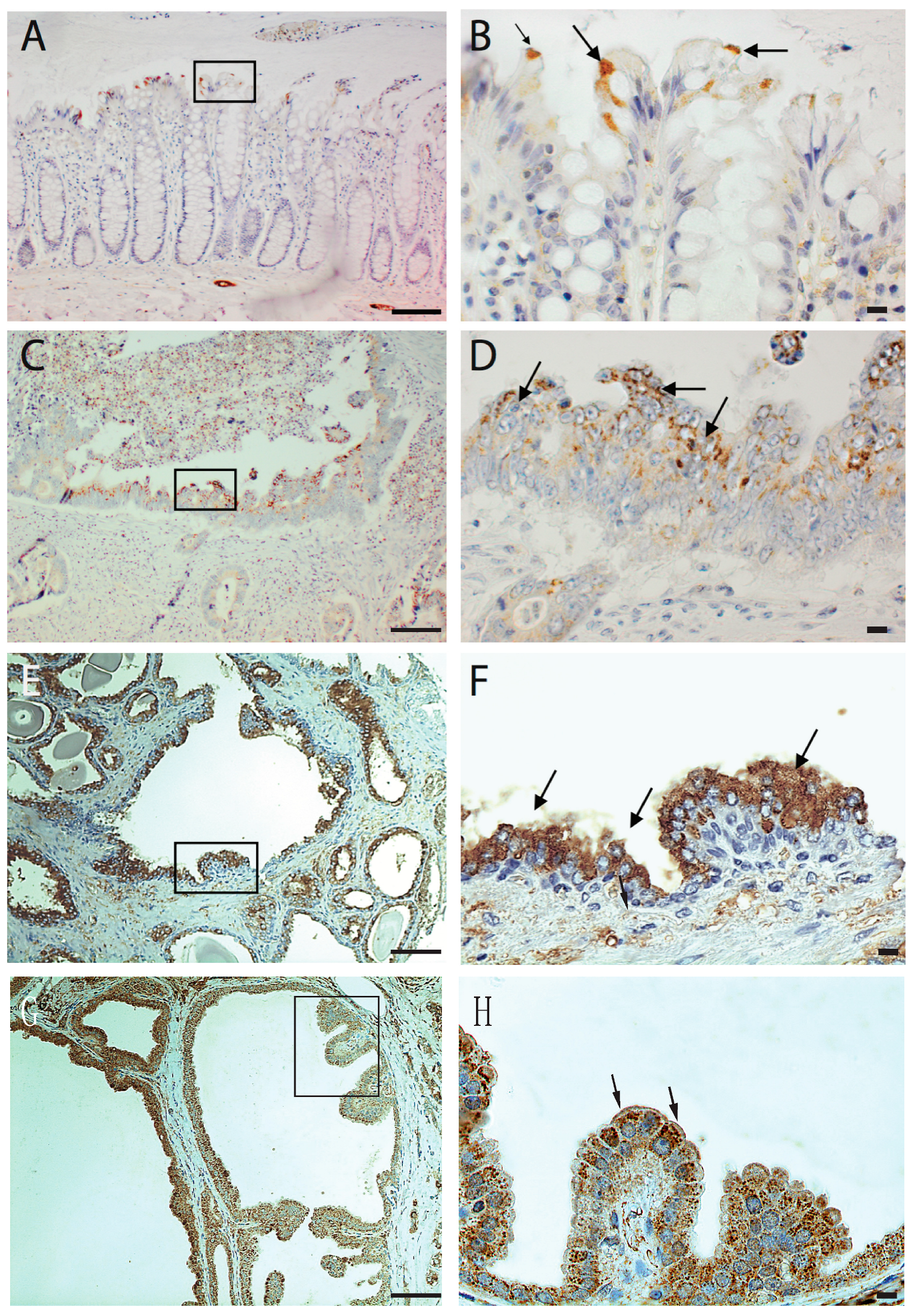{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








