

Human iPSCs as Model Systems for BMP-Related Rare Diseases


Abstract
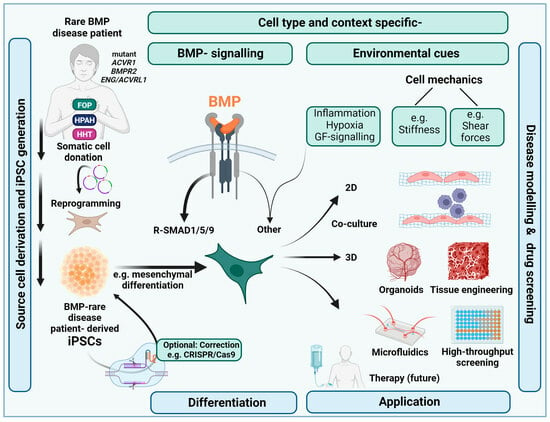
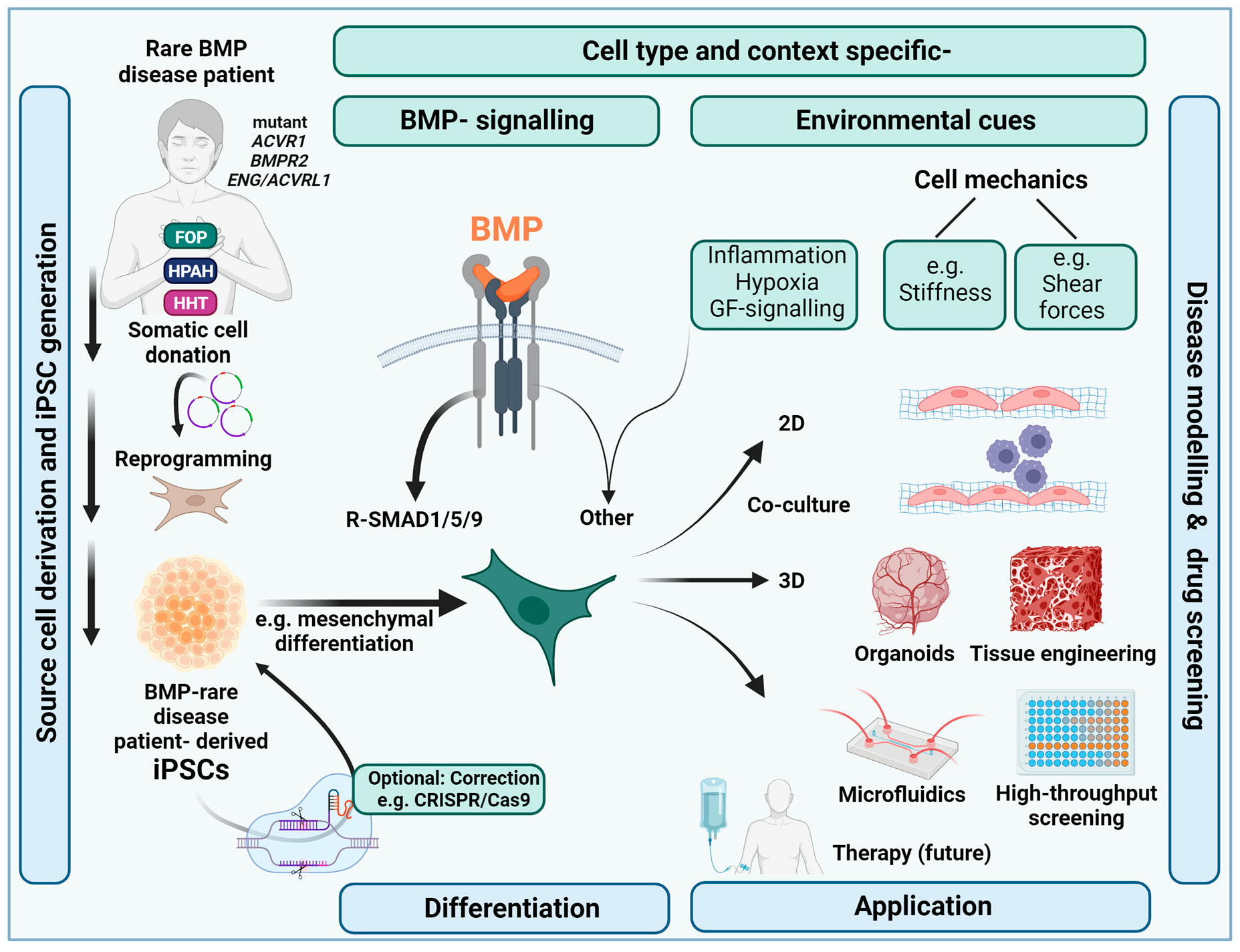
1. Introduction:
1.1. Use of iPSCs for Efficient BMP Rare Disease Research and Orphan Drug Discovery
1.2. Basics in BMP Signalling

1.3. Cell-Type Specific BMP Activity
1.4. BMPs in iPSC Stemness and Differentiation
2. iPSC Technology and BMP-Related Rare Diseases
2.1. Fibrodysplasia Ossificans Progressiva and hiPSCs
2.2. Hereditary Pulmonary Arterial Hypertension and hiPSCs

2.3. Hereditary Haemorrhagic Telangiectasia and hiPSCs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rare Disease | Mutation | Somatic Source Cell | Age and Sex of Donor (s) | Re-Programming Method | Differentiation Protocol | Reference |
|---|---|---|---|---|---|---|
| FOP | ACVR1 c.617G>A (p.R206H) c.1067G>A (p.G356D) | Dermal fibroblasts | 18-M, 59-F, 22-M, 66-F | Sendai | Spontaneous differentiation into mesodermal and endodermal lineages | [114] |
| FOP | ACVR1 c.617G>A (p.R206H) | Dermal fibroblasts | 16-F, 16-F, ∅-M ∅-M 12-M, 12-M, 50-M | Retroviral, Episomal | Osteoblast-like mineralization, chondrogenic EC: [121] 2D and 3D chondrogenic: [130] Presomitic mesoderm and somites [125] Muscle stem/progenitor cells: [126] | [115] |
| FOP | ACVR1 c.617G>A (p.R206H) | Kidney cells isolated from urine | ∅-M ∅-M | Episomal | EC | [116] |
| FOP | ACVR1 c.617G>A (p.R206H) | Urinary cells | ∅-M ∅-F | Sendai | EC: [133] | [231] |
| FOP | ACVR1 c.617G>A (p.R206H) | Periodontal ligament fibroblasts | 23 years | Sendai | Spontaneous differentiation into EBs | [136] |
| HPAH | BMPR2 c.27G>A (p.W9X) c.1039T>C (p.C347R) | Late-outgrowth endothelial progenitor cells from vein blood | ∅-∅ ∅-∅ | Retroviral | Differentiation into 3 germ layers and teratoma | [183] |
| HPAH | BMPR2 c.354T>G (p.C118W) c.2504del (p.T835fs) c.350G>A p.C117>Y | Dermal fibroblasts | Lentiviral/ Sendai | EC | [184] | |
| HPAH | BMPR2 c.1471C>T (p.Arg491Trp) | Dermal fibroblasts | 37-M, 33-F | EC | [187] | |
| HPAH | BMPR2 c.1471C>T (p.Arg491Trp) | Dermal fibroblasts | 37-M, 33-F | Sendai | EC | [185] |
| HPAH | BMPR2 W9X+/− C2 ΔExon1 | Parental wt iPSC line genome edited via CRISPR/Cas9 | ∅-∅ | SMC EC | [191] | |
| HPAH | BMPR2 c.1598A>G (p.His533Arg) c.248-1–418+1del171 (p.Cys84_Ser140del) c.1471C>T (p.Arg491Trp) | CD34+ blood cells | 38-M, 17-F, 5-F | Sendai | In vitro spontaneous differentiation into 3 germ layers | [232] |
| HHT2 | ACVRL1 (ALK1) | Dermal fibroblasts | ∅-∅ | Retroviral | Cardiomyocytes by END-2 co-culture | [221] |
| HHT2 | ACVRL1 c.1120del18 | Dermal fibroblasts | 42-M | Episomal | In vitro spontaneous differentiation and directed trilineage differentiation | [222] |
| HHT2 | ACVRL1 c.772 + 3_772 + 4dup | PBMCs | 61-F | Sendai | In vitro directed trilineage differentiation | [233] |
| HHT2 | ACVRL1 heterozygous frameshift deletion of 17 base pairs in exon 8 | Fibroblast-derived parental iPSC line ATCC; CRL-2097 genome edited via CRISPR/Cas9 | Neonate (<1 month)-M | Sendai | Embryoid bodies and ECs | [225] |
| HHT-1 | ENG c.88T>C (p.C30R) | PBMNCs | 62-F | Episomal | EC | [223] |
| HHT-1 | ENG c.1678C>T p.(G560∗) | PBMCs | ∅-M | Episomal | EC co-culture with primary human brain vascular pericytes | [226] |
3. Culturing iPSCs under (Patho)Physiologically Relevant Microenvironmental Conditions
3.1. Microenvironmental Aspects Relevant for Mimicking BMP Rare Diseases with iPSCs
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | Two dimensional |
| 3D | Three dimensional |
| ACVR1 | Activin A receptor type 1 |
| ALK2 | Activin receptor-like kinase-2 |
| ALPL | Alkaline phosphatase |
| BMP | Bone morphogenetic protein |
| BMPR1 | Bone morphogenetic protein receptor type 1 |
| BMPR2 | Bone morphogenetic protein receptor type 2 |
| Cas9 | CRISPR-associated protein 9 |
| CAV1 | Caveolin 1 |
| CD31 | Platelet endothelial cell adhesion molecule 1 |
| CD45 | Tyrosin-proteinphosphatase C |
| C-MYC | c-myelocytomatosis oncogene product |
| COL1A1 | Collagen type I1 alpha 1 chain |
| COL2A1 | Collagen type II alpha 1 chain |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| EC | Endothelial cell |
| ECM | Extracellular matrix |
| EIF2AK4 | Eukaryotic translation initiation factor 2 alpha kinase 4 |
| EndMT | Endothelial-to-mesenchymal transition |
| ENG | Endoglin |
| EPC | Endothelial progenitor cell |
| ERK | Extracellular signal-regulated kinase |
| FAP | Fibro/adipogenic progenitor |
| FDA | U.S. Food and Drug Administration |
| FGF | Fibroblast growth factor |
| FKBP12 | FK506 binding protein 1a |
| FOP | Fibrodysplasia ossificans progressiva |
| FSP-1 | Ferroptosis suppressor protein 1 |
| GSK3-β | Glycogen synthase kinase 3 beta |
| hESC | Human embryonic stem cell |
| HHT | Hereditary haemorrhagic telangiectasia |
| HHT1 | Hereditary haemorrhagic telangiectasia type 1 |
| HHT2 | Hereditary haemorrhagic telangiectasia type 2 |
| hiPSCs | Human induced pluripotent stem cells |
| HO | Heterotopic ossification |
| HPAH | Hereditary pulmonary arterial hypertension |
| MuSCs | Human primary satellite cells |
| ID | Inhibitor-of-differentiation |
| iECs | iPSC-derived endothelial cells |
| IFN-g | Interferon-gamma |
| IL-6 | Interleukin 6 |
| iMAC | iPSC-derived macrophages |
| iMPCs | iPSC-derived muscle stem/progenitor cells |
| iMSCs | iPSC-derived mesenchymal stromal cells |
| IPAH | Idiopathic pulmonary arterial hypertension |
| iPSC | Induced pluripotent stem cell |
| I-SMADS | Inhibitory SMADs |
| JNK | c-Jun N-terminal kinase |
| KCNK3 | Potassium two pore domain channel subfamily K member 3 |
| KLF-4 | Kruppel-like factor 4 |
| L-EPC | Late-outgrowth endothelial progenitor cell |
| MAPK | Mitogen activated protein kinase |
| M-CSF | Macrophage colony-stimulating factor |
| MDM2 | Mouse double minute 2 homolog |
| mESC | Mouse embryonic stem cell |
| mTOR | Mammalian target of rapamycin |
| NMD | Nonsense mediated decay |
| NOTCH | Neurogenic locus notch homolog protein 1 |
| OCT-4 | Octamer-binding transcription factor 4 |
| PAEC | Pulmonary arterial endothelial cells |
| PAH | Pulmonary arterial hypertension |
| PASMCs | Pulmonary arterial smooth muscle cells |
| PBMNC | Peripheral blood mononuclear cells |
| PDGF | Platelet-derived growth factor |
| PDGFRa | Platelet-derived growth factor receptor A |
| PI3K | Phosphoinositide-3-kinase |
| R-SMADS | Receptor regulated SMADs |
| SCA1 | Spinocerebellar ataxia type 1 |
| Scx | Scleraxis bHLH transcription factor |
| SMAD | Mothers against decapentaplegic homolog |
| SOX-2 | SRY-box transcription factor 2 |
| TFM | Traction force microscopy |
| TGF-β | Transforming growth factor beta |
| TGFBR | TGF-b receptor |
| Tie2 | Angiopoietin-1 receptor |
| VEGF | Vascular endothelial growth factor |
| VSMC | Vascular smooth muscle cell |
References
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R. The burden of rare diseases. Am. J. Med. Genet. A 2019, 179, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Taruscio, D.; Capozzoli, F.; Frank, C. Rare diseases and orphan drugs. Ann. Dell’istituto Super. Sanita 2011, 47, 83–93. [Google Scholar]
- Edwards, D.S.; Clasper, J.C. Heterotopic ossification: A systematic review. J. R. Army Med. Corps 2015, 161, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, D.M.; Ramirez, M.R.; Reginato, A.M.; Medici, D. Molecular and cellular mechanisms of heterotopic ossification. Histol. Histopathol. 2014, 29, 1281–1285. [Google Scholar]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
- Happe, C.; Kurakula, K.; Sun, X.Q.; da Silva Goncalves Bos, D.; Rol, N.; Guignabert, C.; Tu, L.; Schalij, I.; Wiesmeijer, K.C.; Tura-Ceide, O.; et al. The BMP Receptor 2 in Pulmonary Arterial Hypertension: When and Where the Animal Model Matches the Patient. Cells 2020, 9, 1422. [Google Scholar] [CrossRef]
- Wang, H.; Brown, P.C.; Chow, E.C.Y.; Ewart, L.; Ferguson, S.S.; Fitzpatrick, S.; Freedman, B.S.; Guo, G.L.; Hedrich, W.; Heyward, S.; et al. 3D cell culture models: Drug pharmacokinetics, safety assessment, and regulatory consideration. Clin. Transl. Sci. 2021, 14, 1659–1680. [Google Scholar] [CrossRef]
- Urist, M.R. Bone: Formation by autoinduction. Science 1965, 150, 893–899. [Google Scholar] [CrossRef]
- Urist, M.R.; Strates, B.S. Bone morphogenetic protein. J. Dent. Res. 1971, 50, 1392–1406. [Google Scholar] [CrossRef] [PubMed]
- Wozney, J.M.; Rosen, V.; Celeste, A.J.; Mitsock, L.M.; Whitters, M.J.; Kriz, R.W.; Hewick, R.M.; Wang, E.A. Novel regulators of bone formation: Molecular clones and activities. Science 1988, 242, 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Duffhues, G.; Williams, E.; Goumans, M.J.; Heldin, C.H.; Ten Dijke, P. Bone morphogenetic protein receptors: Structure, function and targeting by selective small molecule kinase inhibitors. Bone 2020, 138, 115472. [Google Scholar] [CrossRef] [PubMed]
- Yadin, D.; Knaus, P.; Mueller, T.D. Structural insights into BMP receptors: Specificity, activation and inhibition. Cytokine Growth Factor Rev. 2016, 27, 13–34. [Google Scholar] [CrossRef]
- Castonguay, R.; Werner, E.D.; Matthews, R.G.; Presman, E.; Mulivor, A.W.; Solban, N.; Sako, D.; Pearsall, R.S.; Underwood, K.W.; Seehra, J.; et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J. Biol. Chem. 2011, 286, 30034–30046. [Google Scholar] [CrossRef]
- Garcia de Vinuesa, A.; Sanchez-Duffhues, G.; Blaney-Davidson, E.; van Caam, A.; Lodder, K.; Ramos, Y.; Kloppenburg, M.; Meulenbelt, I.; van der Kraan, P.; Goumans, M.J.; et al. Cripto favors chondrocyte hypertrophy via TGF-beta SMAD1/5 signaling during development of osteoarthritis. J. Pathol. 2021, 255, 330–342. [Google Scholar] [CrossRef]
- Perez-Gomez, E.; Villa-Morales, M.; Santos, J.; Fernandez-Piqueras, J.; Gamallo, C.; Dotor, J.; Bernabeu, C.; Quintanilla, M. A role for endoglin as a suppressor of malignancy during mouse skin carcinogenesis. Cancer Res. 2007, 67, 10268–10277. [Google Scholar] [CrossRef]
- Watanabe, K.; Bianco, C.; Strizzi, L.; Hamada, S.; Mancino, M.; Bailly, V.; Mo, W.; Wen, D.; Miatkowski, K.; Gonzales, M.; et al. Growth factor induction of Cripto-1 shedding by glycosylphosphatidylinositol-phospholipase D and enhancement of endothelial cell migration. J. Biol. Chem. 2007, 282, 31643–31655. [Google Scholar] [CrossRef]
- Siebold, C.; Yamashita, T.; Monnier, P.P.; Mueller, B.K.; Pasterkamp, R.J. RGMs: Structural Insights, Molecular Regulation, and Downstream Signaling. Trends Cell Biol. 2017, 27, 365–378. [Google Scholar] [CrossRef]
- Onichtchouk, D.; Chen, Y.G.; Dosch, R.; Gawantka, V.; Delius, H.; Massague, J.; Niehrs, C. Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature 1999, 401, 480–485. [Google Scholar] [CrossRef]
- Hagihara, M.; Endo, M.; Hata, K.; Higuchi, C.; Takaoka, K.; Yoshikawa, H.; Yamashita, T. Neogenin, a receptor for bone morphogenetic proteins. J. Biol. Chem. 2011, 286, 5157–5165. [Google Scholar] [CrossRef] [PubMed]
- Bragdon, B.; Thinakaran, S.; Moseychuk, O.; King, D.; Young, K.; Litchfield, D.W.; Petersen, N.O.; Nohe, A. Casein kinase 2 beta-subunit is a regulator of bone morphogenetic protein 2 signaling. Biophys. J. 2010, 99, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Dannewitz Prosseda, S.; Tian, X.; Kuramoto, K.; Boehm, M.; Sudheendra, D.; Miyagawa, K.; Zhang, F.; Solow-Cordero, D.; Saldivar, J.C.; Austin, E.D.; et al. FHIT, a Novel Modifier Gene in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2019, 199, 83–98. [Google Scholar] [CrossRef]
- Andruska, A.M.; Ali, M.K.; Tian, X.; Spiekerkoetter, E. Selective Src-Family B Kinase Inhibition Promotes Pulmonary Artery Endothelial Cell Dysfunction. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ali, M.K.; Tian, X.; Zhao, L.; Schimmel, K.; Rhodes, C.J.; Wilkins, M.R.; Nicolls, M.R.; Spiekerkoetter, E.F. PTPN1 Deficiency Modulates BMPR2 Signaling and Induces Endothelial Dysfunction in Pulmonary Arterial Hypertension. Cells 2023, 12, 316. [Google Scholar] [CrossRef]
- Wilkinson, L.; Kolle, G.; Wen, D.; Piper, M.; Scott, J.; Little, M. CRIM1 regulates the rate of processing and delivery of bone morphogenetic proteins to the cell surface. J. Biol. Chem. 2003, 278, 34181–34188. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.M.; Hubbell, J.A. The 12th–14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 2010, 24, 4711–4721. [Google Scholar]
- Hauff, K.; Zambarda, C.; Dietrich, M.; Halbig, M.; Grab, A.L.; Medda, R.; Cavalcanti-Adam, E.A. Matrix-Immobilized BMP-2 on Microcontact Printed Fibronectin as an in vitro Tool to Study BMP-Mediated Signaling and Cell Migration. Front. Bioeng. Biotechnol. 2015, 3, 62. [Google Scholar] [CrossRef]
- Xu, E.R.; Blythe, E.E.; Fischer, G.; Hyvonen, M. Structural analyses of von Willebrand factor C domains of collagen 2A and CCN3 reveal an alternative mode of binding to bone morphogenetic protein-2. J. Biol. Chem. 2017, 292, 12516–12527. [Google Scholar] [CrossRef]
- Sieron, A.L.; Louneva, N.; Fertala, A. Site-specific interaction of bone morphogenetic protein 2 with procollagen II. Cytokine 2002, 18, 214–221. [Google Scholar] [CrossRef]
- Sengle, G.; Charbonneau, N.L.; Ono, R.N.; Sasaki, T.; Alvarez, J.; Keene, D.R.; Bachinger, H.P.; Sakai, L.Y. Targeting of bone morphogenetic protein growth factor complexes to fibrillin. J. Biol. Chem. 2008, 283, 13874–13888. [Google Scholar] [CrossRef]
- Wohl, A.P.; Troilo, H.; Collins, R.F.; Baldock, C.; Sengle, G. Extracellular Regulation of Bone Morphogenetic Protein Activity by the Microfibril Component Fibrillin-1. J. Biol. Chem. 2016, 291, 12732–12746. [Google Scholar] [CrossRef] [PubMed]
- Spanou, C.E.S.; Wohl, A.P.; Doherr, S.; Correns, A.; Sonntag, N.; Lutke, S.; Morgelin, M.; Imhof, T.; Gebauer, J.M.; Baumann, U.; et al. Targeting of bone morphogenetic protein complexes to heparin/heparan sulfate glycosaminoglycans in bioactive conformation. FASEB J. 2023, 37, e22717. [Google Scholar] [CrossRef]
- Kanzaki, S.; Takahashi, T.; Kanno, T.; Ariyoshi, W.; Shinmyouzu, K.; Tujisawa, T.; Nishihara, T. Heparin inhibits BMP-2 osteogenic bioactivity by binding to both BMP-2 and BMP receptor. J. Cell. Physiol. 2008, 216, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, S.; Ariyoshi, W.; Takahashi, T.; Okinaga, T.; Kaneuji, T.; Mitsugi, S.; Nakashima, K.; Tsujisawa, T.; Nishihara, T. Dual effects of heparin on BMP-2-induced osteogenic activity in MC3T3-E1 cells. Pharmacol. Rep. 2011, 63, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Rider, C.C. Heparin/heparan sulphate binding in the TGF-beta cytokine superfamily. Biochem. Soc. Trans. 2006, 34 Pt 3, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Lee, J.Y.; Park, J.H.; Park, J.B.; Suh, J.S.; Choi, Y.S.; Lee, S.J.; Chung, C.P.; Park, Y.J. The identification of a heparin binding domain peptide from bone morphogenetic protein-4 and its role on osteogenesis. Biomaterials 2010, 31, 7226–7238. [Google Scholar] [CrossRef] [PubMed]
- Correns, A.; Zimmermann, L.A.; Baldock, C.; Sengle, G. BMP antagonists in tissue development and disease. Matrix Biol. Plus 2021, 11, 100071. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Runyan, C.E.; Schnaper, H.W.; Poncelet, A.C. The role of internalization in transforming growth factor beta1-induced Smad2 association with Smad anchor for receptor activation (SARA) and Smad2-dependent signaling in human mesangial cells. J. Biol. Chem. 2005, 280, 8300–8308. [Google Scholar] [CrossRef]
- Shi, W.; Chang, C.; Nie, S.; Xie, S.; Wan, M.; Cao, X. Endofin acts as a Smad anchor for receptor activation in BMP signaling. J. Cell Sci. 2007, 120 Pt 7, 1216–1224. [Google Scholar] [CrossRef] [PubMed]
- Hanyu, A.; Ishidou, Y.; Ebisawa, T.; Shimanuki, T.; Imamura, T.; Miyazono, K. The N domain of Smad7 is essential for specific inhibition of transforming growth factor-beta signaling. J. Cell Biol. 2001, 155, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Park, J.S.; Kim, J.H.; Jung, S.M.; Lee, J.Y.; Kim, S.J.; Park, S.H. Smad6-specific recruitment of Smurf E3 ligases mediates TGF-beta1-induced degradation of MyD88 in TLR4 signalling. Nat. Commun. 2011, 2, 460. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Kamiya, Y.; Imamura, T.; Miyazono, K.; Miyazawa, K. Selective inhibitory effects of Smad6 on bone morphogenetic protein type I receptors. J. Biol. Chem. 2007, 282, 20603–20611. [Google Scholar] [CrossRef] [PubMed]
- Murakami, G.; Watabe, T.; Takaoka, K.; Miyazono, K.; Imamura, T. Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol. Biol. Cell 2003, 14, 2809–2817. [Google Scholar] [CrossRef]
- Sieber, C.; Kopf, J.; Hiepen, C.; Knaus, P. Recent advances in BMP receptor signaling. Cytokine Growth Factor Rev. 2009, 20, 343–355. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Wakayama, Y.; Fukuhara, S.; Ando, K.; Matsuda, M.; Mochizuki, N. Cdc42 mediates Bmp-induced sprouting angiogenesis through Fmnl3-driven assembly of endothelial filopodia in zebrafish. Dev. Cell 2015, 32, 109–122. [Google Scholar] [CrossRef]
- Hiepen, C.; Benn, A.; Denkis, A.; Lukonin, I.; Weise, C.; Boergermann, J.H.; Knaus, P. BMP2-induced chemotaxis requires PI3K p55gamma/p110alpha-dependent phosphatidylinositol (3,4,5)-triphosphate production and LL5beta recruitment at the cytocortex. BMC Biol. 2014, 12, 43. [Google Scholar] [CrossRef]
- Sapkota, G.; Alarcon, C.; Spagnoli, F.M.; Brivanlou, A.H.; Massague, J. Balancing BMP signaling through integrated inputs into the Smad1 linker. Mol. Cell 2007, 25, 441–454. [Google Scholar] [CrossRef]
- Fuentealba, L.C.; Eivers, E.; Ikeda, A.; Hurtado, C.; Kuroda, H.; Pera, E.M.; De Robertis, E.M. Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 2007, 131, 980–993. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, M.; Mendez, P.L.; da Silva Madaleno, C.; Ugorets, V.; Rikeit, P.; Boerno, S.; Jatzlau, J.; Knaus, P. Differential Impact of Fluid Shear Stress and YAP/TAZ on BMP/TGF-beta Induced Osteogenic Target Genes. Adv. Biol. 2021, 5, e2000051. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.F.; Harvey, S.A.; Thies, R.S.; Olson, M.S. Bone morphogenetic protein-9. An autocrine/paracrine cytokine in the liver. J. Biol. Chem. 2000, 275, 17937–17945. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, H.; Rosen, V.; Thies, R.S. Heart specific expression of mouse BMP-10 a novel member of the TGF-beta superfamily. Mech. Dev. 1999, 80, 181–184. [Google Scholar] [CrossRef]
- Chen, H.; Shi, S.; Acosta, L.; Li, W.; Lu, J.; Bao, S.; Chen, Z.; Yang, Z.; Schneider, M.D.; Chien, K.R.; et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 2004, 131, 2219–2231. [Google Scholar] [CrossRef]
- Jones, C.M.; Lyons, K.M.; Hogan, B.L. Involvement of Bone Morphogenetic Protein-4 (BMP-4) and Vgr-1 in morphogenesis and neurogenesis in the mouse. Development 1991, 111, 531–542. [Google Scholar] [CrossRef]
- Ozkaynak, E.; Schnegelsberg, P.N.; Oppermann, H. Murine osteogenic protein (OP-1): High levels of mRNA in kidney. Biochem. Biophys. Res. Commun. 1991, 179, 116–123. [Google Scholar] [CrossRef]
- Wang, L.; Rice, M.; Swist, S.; Kubin, T.; Wu, F.; Wang, S.; Kraut, S.; Weissmann, N.; Bottger, T.; Wheeler, M.; et al. BMP9 and BMP10 Act Directly on Vascular Smooth Muscle Cells for Generation and Maintenance of the Contractile State. Circulation 2021, 143, 1394–1410. [Google Scholar] [CrossRef]
- Astrom, A.K.; Jin, D.; Imamura, T.; Roijer, E.; Rosenzweig, B.; Miyazono, K.; ten Dijke, P.; Stenman, G. Chromosomal localization of three human genes encoding bone morphogenetic protein receptors. Mamm. Genome 1999, 10, 299–302. [Google Scholar]
- Ide, H.; Katoh, M.; Sasaki, H.; Yoshida, T.; Aoki, K.; Nawa, Y.; Osada, Y.; Sugimura, T.; Terada, M. Cloning of human bone morphogenetic protein type IB receptor (BMPR-IB) and its expression in prostate cancer in comparison with other BMPRs. Oncogene 1997, 14, 1377–1382. [Google Scholar] [CrossRef]
- Bidart, M.; Ricard, N.; Levet, S.; Samson, M.; Mallet, C.; David, L.; Subileau, M.; Tillet, E.; Feige, J.J.; Bailly, S. BMP9 is produced by hepatocytes and circulates mainly in an active mature form complexed to its prodomain. Cell. Mol. Life Sci. 2012, 69, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Brock, M.; Trenkmann, M.; Gay, R.E.; Michel, B.A.; Gay, S.; Fischler, M.; Ulrich, S.; Speich, R.; Huber, L.C. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ. Res. 2009, 104, 1184–1191. [Google Scholar] [CrossRef]
- Hurst, L.A.; Dunmore, B.J.; Long, L.; Crosby, A.; Al-Lamki, R.; Deighton, J.; Southwood, M.; Yang, X.; Nikolic, M.Z.; Herrera, B.; et al. TNFalpha drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat. Commun. 2017, 8, 14079. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Song, H.; Kumar, S.; Nam, D.; Kwon, H.S.; Chang, K.H.; Son, D.J.; Kang, D.W.; Brodie, S.A.; Weiss, D.; et al. Anti-inflammatory and antiatherogenic role of BMP receptor II in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Mendez, P.L.; Obendorf, L.; Jatzlau, J.; Burdzinski, W.; Reichenbach, M.; Nageswaran, V.; Haghikia, A.; Stangl, V.; Hiepen, C.; Knaus, P. Atheroprone fluid shear stress-regulated ALK1-Endoglin-SMAD signaling originates from early endosomes. BMC Biol. 2022, 20, 210. [Google Scholar] [CrossRef]
- Baeyens, N.; Larrivee, B.; Ola, R.; Hayward-Piatkowskyi, B.; Dubrac, A.; Huang, B.; Ross, T.D.; Coon, B.G.; Min, E.; Tsarfati, M.; et al. Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. J. Cell Biol. 2016, 214, 807–816. [Google Scholar] [CrossRef]
- Rui, Y.F.; Lui, P.P.; Ni, M.; Chan, L.S.; Lee, Y.W.; Chan, K.M. Mechanical loading increased BMP-2 expression which promoted osteogenic differentiation of tendon-derived stem cells. J. Orthop. Res. 2011, 29, 390–396. [Google Scholar] [CrossRef]
- Gilde, F.; Fourel, L.; Guillot, R.; Pignot-Paintrand, I.; Okada, T.; Fitzpatrick, V.; Boudou, T.; Albiges-Rizo, C.; Picart, C. Stiffness-dependent cellular internalization of matrix-bound BMP-2 and its relation to Smad and non-Smad signaling. Acta Biomater. 2016, 46, 55–67. [Google Scholar] [CrossRef]
- Wei, Q.; Holle, A.; Li, J.; Posa, F.; Biagioni, F.; Croci, O.; Benk, A.S.; Young, J.; Noureddine, F.; Deng, J.; et al. BMP-2 Signaling and Mechanotransduction Synergize to Drive Osteogenic Differentiation via YAP/TAZ. Adv. Sci. 2020, 7, 1902931. [Google Scholar] [CrossRef]
- De Robertis, E.M.; Kuroda, H. Dorsal-ventral patterning and neural induction in Xenopus embryos. Annu. Rev. Cell Dev. Biol. 2004, 20, 285–308. [Google Scholar] [CrossRef]
- Zinski, J.; Tajer, B.; Mullins, M.C. TGF-beta Family Signaling in Early Vertebrate Development. Cold Spring Harb. Perspect. Biol. 2018, 10, a033274. [Google Scholar] [CrossRef] [PubMed]
- Nishimatsu, S.; Thomsen, G.H. Ventral mesoderm induction and patterning by bone morphogenetic protein heterodimers in Xenopus embryos. Mech. Dev. 1998, 74, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, J.; Tan, Z.; Wang, C.; Liu, T.; Chen, L.; Yong, J.; Jiang, W.; Sun, X.; Du, L.; et al. Short-term BMP-4 treatment initiates mesoderm induction in human embryonic stem cells. Blood 2008, 111, 1933–1941. [Google Scholar] [CrossRef] [PubMed]
- Sporle, R.; Schughart, K. Neural tube morphogenesis. Curr. Opin. Genet. Dev. 1997, 7, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Shroyer, N.F.; Wong, M.H. BMP signaling in the intestine: Cross-talk is key. Gastroenterology 2007, 133, 1035–1038. [Google Scholar] [CrossRef]
- Herrera, B.; Sanchez, A.; Fabregat, I. BMPS and liver: More questions than answers. Curr. Pharm. Des. 2012, 18, 4114–4125. [Google Scholar] [CrossRef]
- Godin, R.E.; Robertson, E.J.; Dudley, A.T. Role of BMP family members during kidney development. Int. J. Dev. Biol. 1999, 43, 405–411. [Google Scholar]
- Zhang, J.; Li, L. BMP signaling and stem cell regulation. Dev. Biol. 2005, 284, 1–11. [Google Scholar] [CrossRef]
- Qi, X.; Li, T.G.; Hao, J.; Hu, J.; Wang, J.; Simmons, H.; Miura, S.; Mishina, Y.; Zhao, G.Q. BMP4 supports self-renewal of embryonic stem cells by inhibiting mitogen-activated protein kinase pathways. Proc. Natl. Acad. Sci. USA 2004, 101, 6027–6032. [Google Scholar] [CrossRef]
- Ying, Q.L.; Nichols, J.; Chambers, I.; Smith, A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 2003, 115, 281–292. [Google Scholar] [CrossRef]
- Tomizawa, M.; Shinozaki, F.; Sugiyama, T.; Yamamoto, S.; Sueishi, M.; Yoshida, T. Activin A maintains pluripotency markers and proliferative potential of human induced pluripotent stem cells. Exp. Ther. Med. 2011, 2, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Hsiao, E.C.; Sami, S.; Lancero, M.; Schlieve, C.R.; Nguyen, T.; Yano, K.; Nagahashi, A.; Ikeya, M.; Matsumoto, Y.; et al. BMP-SMAD-ID promotes reprogramming to pluripotency by inhibiting p16/INK4A-dependent senescence. Proc. Natl. Acad. Sci. USA 2016, 113, 13057–13062. [Google Scholar] [CrossRef] [PubMed]
- Ali, G.; Abdelalim, E.M. Directed differentiation of human pluripotent stem cells into epidermal keratinocyte-like cells. STAR Protoc. 2022, 3, 101613. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani-Dalini, S.; Azarpira, N.; Sangtarash, M.H.; Soleimanpour-Lichaei, H.R.; Yaghobi, R.; Lorzadeh, S.; Sabet, A.; Sarshar, M.; Al-Abdullah, I.H. Optimization of activin-A: A breakthrough in differentiation of human induced pluripotent stem cell into definitive endoderm. 3 Biotech 2020, 10, 215. [Google Scholar] [CrossRef] [PubMed]
- Bogacheva, M.S.; Harjumaki, R.; Flander, E.; Taalas, A.; Bystriakova, M.A.; Yliperttula, M.; Xiang, X.; Leung, A.W.; Lou, Y.R. Differentiation of Human Pluripotent Stem Cells into Definitive Endoderm Cells in Various Flexible Three-Dimensional Cell Culture Systems: Possibilities and Limitations. Front. Cell Dev. Biol. 2021, 9, 726499. [Google Scholar] [CrossRef]
- Patsch, C.; Challet-Meylan, L.; Thoma, E.C.; Urich, E.; Heckel, T.; O’Sullivan, J.F.; Grainger, S.J.; Kapp, F.G.; Sun, L.; Christensen, K.; et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 2015, 17, 994–1003. [Google Scholar] [CrossRef]
- Kim, M.S.; Horst, A.; Blinka, S.; Stamm, K.; Mahnke, D.; Schuman, J.; Gundry, R.; Tomita-Mitchell, A.; Lough, J. Activin-A and Bmp4 levels modulate cell type specification during CHIR-induced cardiomyogenesis. PLoS ONE 2015, 10, e0118670. [Google Scholar] [CrossRef]
- Park, S.W.; Jun Koh, Y.; Jeon, J.; Cho, Y.H.; Jang, M.J.; Kang, Y.; Kim, M.J.; Choi, C.; Sook Cho, Y.; Chung, H.M.; et al. Efficient differentiation of human pluripotent stem cells into functional CD34+ progenitor cells by combined modulation of the MEK/ERK and BMP4 signaling pathways. Blood 2010, 116, 5762–5772. [Google Scholar] [CrossRef]
- Gai, H.; Leung, E.L.; Costantino, P.D.; Aguila, J.R.; Nguyen, D.M.; Fink, L.M.; Ward, D.C.; Ma, Y. Generation and characterization of functional cardiomyocytes using induced pluripotent stem cells derived from human fibroblasts. Cell Biol. Int. 2009, 33, 1184–1193. [Google Scholar] [CrossRef]
- Guzzo, R.M.; Drissi, H. Differentiation of Human Induced Pluripotent Stem Cells to Chondrocytes. Methods Mol. Biol. 2015, 1340, 79–95. [Google Scholar]
- Song, B.; Smink, A.M.; Jones, C.V.; Callaghan, J.M.; Firth, S.D.; Bernard, C.A.; Laslett, A.L.; Kerr, P.G.; Ricardo, S.D. The directed differentiation of human iPS cells into kidney podocytes. PLoS ONE 2012, 7, e46453. [Google Scholar] [CrossRef] [PubMed]
- Tanigawa, S.; Naganuma, H.; Kaku, Y.; Era, T.; Sakuma, T.; Yamamoto, T.; Taguchi, A.; Nishinakamura, R. Activin Is Superior to BMP7 for Efficient Maintenance of Human iPSC-Derived Nephron Progenitors. Stem Cell Rep. 2019, 13, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Lichtner, B.; Knaus, P.; Lehrach, H.; Adjaye, J. BMP10 as a potent inducer of trophoblast differentiation in human embryonic and induced pluripotent stem cells. Biomaterials 2013, 34, 9789–9802. [Google Scholar] [CrossRef] [PubMed]
- Williams, I.M.; Wu, J.C. Generation of Endothelial Cells from Human Pluripotent Stem Cells. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1317–1329. [Google Scholar] [CrossRef]
- Jansen, M.H.; van Vuurden, D.G.; Vandertop, W.P.; Kaspers, G.J. Diffuse intrinsic pontine gliomas: A systematic update on clinical trials and biology. Cancer Treat. Rev. 2012, 38, 27–35. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef]
- D’Alessandro, L.C.; Al Turki, S.; Manickaraj, A.K.; Manase, D.; Mulder, B.J.; Bergin, L.; Rosenberg, H.C.; Mondal, T.; Gordon, E.; Lougheed, J.; et al. Exome sequencing identifies rare variants in multiple genes in atrioventricular septal defect. Genet. Med. 2016, 18, 189–198. [Google Scholar] [CrossRef]
- Gaussin, V.; Morley, G.E.; Cox, L.; Zwijsen, A.; Vance, K.M.; Emile, L.; Tian, Y.; Liu, J.; Hong, C.; Myers, D.; et al. Alk3/Bmpr1a receptor is required for development of the atrioventricular canal into valves and annulus fibrosus. Circ. Res. 2005, 97, 219–226. [Google Scholar] [CrossRef]
- Howe, J.R.; Bair, J.L.; Sayed, M.G.; Anderson, M.E.; Mitros, F.A.; Petersen, G.M.; Velculescu, V.E.; Traverso, G.; Vogelstein, B. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat. Genet. 2001, 28, 184–187. [Google Scholar] [CrossRef]
- Howe, J.R.; Roth, S.; Ringold, J.C.; Summers, R.W.; Jarvinen, H.J.; Sistonen, P.; Tomlinson, I.P.; Houlston, R.S.; Bevan, S.; Mitros, F.A.; et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998, 280, 1086–1088. [Google Scholar] [CrossRef] [PubMed]
- Demirhan, O.; Turkmen, S.; Schwabe, G.C.; Soyupak, S.; Akgul, E.; Tastemir, D.; Karahan, D.; Mundlos, S.; Lehmann, K. A homozygous BMPR1B mutation causes a new subtype of acromesomelic chondrodysplasia with genital anomalies. J. Med. Genet. 2005, 42, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Umair, M.; Muhammad, D.; Bilal, M.; Lee, K.; Leal, S.M.; Ahmad, W. A novel homozygous variant in BMPR1B underlies acromesomelic dysplasia Hunter-Thompson type. Ann. Hum. Genet. 2018, 82, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Shore, E.M.; Kaplan, F.S. Fibrodysplasia ossificans progressiva: Clinical and genetic aspects. Orphanet J. Rare Dis. 2011, 6, 80. [Google Scholar] [CrossRef]
- Shore, E.M.; Kaplan, F.S. Inherited human diseases of heterotopic bone formation. Nat. Rev. Rheumatol. 2010, 6, 518–527. [Google Scholar] [CrossRef]
- Kou, S.; De Cunto, C.; Baujat, G.; Wentworth, K.L.; Grogan, D.R.; Brown, M.A.; Di Rocco, M.; Keen, R.; Al Mukaddam, M.; le Quan Sang, K.H.; et al. Patients with ACVR1(R206H) mutations have an increased prevalence of cardiac conduction abnormalities on electrocardiogram in a natural history study of Fibrodysplasia Ossificans Progressiva. Orphanet J. Rare Dis. 2020, 15, 193. [Google Scholar] [CrossRef]
- Marseglia, L.; D’Angelo, G.; Manti, S.; Manganaro, A.; Calabro, M.P.; Salpietro, C.; Gitto, E. Fibrodysplasia ossificans progressiva in a newborn with cardiac involvement. Pediatr. Int. 2015, 57, 719–721. [Google Scholar] [CrossRef]
- Ware, A.D.; Brewer, N.; Meyers, C.; Morris, C.; McCarthy, E.; Shore, E.M.; James, A.W. Differential Vascularity in Genetic and Nonhereditary Heterotopic Ossification. Int. J. Surg. Pathol. 2019, 27, 859–867. [Google Scholar] [CrossRef]
- el-Labban, N.G.; Hopper, C.; Barber, P. Ultrastructural finding of vascular degeneration in fibrodysplasia ossificans progressiva (FOP). J. Oral Pathol. Med. 1995, 24, 125–129. [Google Scholar] [CrossRef]
- Hegyi, L.; Gannon, F.H.; Glaser, D.L.; Shore, E.M.; Kaplan, F.S.; Shanahan, C.M. Stromal cells of fibrodysplasia ossificans progressiva lesions express smooth muscle lineage markers and the osteogenic transcription factor Runx2/Cbfa-1: Clues to a vascular origin of heterotopic ossification? J. Pathol. 2003, 201, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Duffhues, G.; Williams, E.; Benderitter, P.; Orlova, V.; van Wijhe, M.; Garcia de Vinuesa, A.; Kerr, G.; Caradec, J.; Lodder, K.; de Boer, H.C.; et al. Development of Macrocycle Kinase Inhibitors for ALK2 Using Fibrodysplasia Ossificans Progressiva-Derived Endothelial Cells. JBMR Plus 2019, 3, e10230. [Google Scholar] [CrossRef] [PubMed]
- Wentworth, K.L.; Bigay, K.; Chan, T.V.; Ho, J.P.; Morales, B.M.; Connor, J.; Brooks, E.; Shahriar Salamat, M.; Sanchez, H.C.; Wool, G.; et al. Clinical-pathological correlations in three patients with fibrodysplasia ossificans progressiva. Bone 2018, 109, 104–110. [Google Scholar] [CrossRef]
- Hamasaki, M.; Hashizume, Y.; Yamada, Y.; Katayama, T.; Hohjoh, H.; Fusaki, N.; Nakashima, Y.; Furuya, H.; Haga, N.; Takami, Y.; et al. Pathogenic mutation of ALK2 inhibits induced pluripotent stem cell reprogramming and maintenance: Mechanisms of reprogramming and strategy for drug identification. Stem Cells 2012, 30, 2437–2449. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Hayashi, Y.; Schlieve, C.R.; Ikeya, M.; Kim, H.; Nguyen, T.D.; Sami, S.; Baba, S.; Barruet, E.; Nasu, A.; et al. Induced pluripotent stem cells from patients with human fibrodysplasia ossificans progressiva show increased mineralization and cartilage formation. Orphanet J. Rare Dis. 2013, 8, 190. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Orlova, V.V.; Cai, X.; Eekhoff, E.M.W.; Zhang, K.; Pei, D.; Pan, G.; Mummery, C.L.; Ten Dijke, P. Induced Pluripotent Stem Cells to Model Human Fibrodysplasia Ossificans Progressiva. Stem Cell Rep. 2015, 5, 963–970. [Google Scholar] [CrossRef]
- Orlova, V.V.; van den Hil, F.E.; Petrus-Reurer, S.; Drabsch, Y.; Ten Dijke, P.; Mummery, C.L. Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nat. Protoc. 2014, 9, 1514–1531. [Google Scholar] [CrossRef]
- Orlova, V.V.; Drabsch, Y.; Freund, C.; Petrus-Reurer, S.; van den Hil, F.E.; Muenthaisong, S.; Dijke, P.T.; Mummery, C.L. Functionality of endothelial cells and pericytes from human pluripotent stem cells demonstrated in cultured vascular plexus and zebrafish xenografts. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 177–186. [Google Scholar] [CrossRef]
- Sanchez-Duffhues, G.; Garcia de Vinuesa, A.; Ten Dijke, P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn. 2018, 247, 492–508. [Google Scholar] [CrossRef]
- Medici, D.; Shore, E.M.; Lounev, V.Y.; Kaplan, F.S.; Kalluri, R.; Olsen, B.R. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 2010, 16, 1400–1406. [Google Scholar] [CrossRef]
- Barruet, E.; Morales, B.M.; Lwin, W.; White, M.P.; Theodoris, C.V.; Kim, H.; Urrutia, A.; Wong, S.A.; Srivastava, D.; Hsiao, E.C. The ACVR1 R206H mutation found in fibrodysplasia ossificans progressiva increases human induced pluripotent stem cell-derived endothelial cell formation and collagen production through BMP-mediated SMAD1/5/8 signaling. Stem Cell Res. Ther. 2016, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Lees-Shepard, J.B.; Yamamoto, M.; Biswas, A.A.; Stoessel, S.J.; Nicholas, S.E.; Cogswell, C.A.; Devarakonda, P.M.; Schneider, M.J., Jr.; Cummins, S.M.; Legendre, N.P.; et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 2018, 9, 471. [Google Scholar] [CrossRef] [PubMed]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef]
- Dey, D.; Bagarova, J.; Hatsell, S.J.; Armstrong, K.A.; Huang, L.; Ermann, J.; Vonner, A.J.; Shen, Y.; Mohedas, A.H.; Lee, A.; et al. Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci. Transl. Med. 2016, 8, 366ra163. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Shibata, M.; Nishio, M.; Nagata, S.; Alev, C.; Sakurai, H.; Toguchida, J.; Ikeya, M. Modeling human somite development and fibrodysplasia ossificans progressiva with induced pluripotent stem cells. Development 2018, 145, dev165431. [Google Scholar] [CrossRef] [PubMed]
- Barruet, E.; Garcia, S.M.; Wu, J.; Morales, B.M.; Tamaki, S.; Moody, T.; Pomerantz, J.H.; Hsiao, E.C. Modeling the ACVR1(R206H) mutation in human skeletal muscle stem cells. Elife 2021, 10, e66107. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Lepinski, A.; Chavez, R.D.; Barruet, E.; Pereira, A.; Moody, T.A.; Ton, A.N.; Sharma, A.; Hellman, J.; Tomoda, K.; et al. ACVR1(R206H) extends inflammatory responses in human induced pluripotent stem cell-derived macrophages. Bone 2021, 153, 116129. [Google Scholar] [CrossRef]
- Maekawa, H.; Jin, Y.; Nishio, M.; Kawai, S.; Nagata, S.; Kamakura, T.; Yoshitomi, H.; Niwa, A.; Saito, M.K.; Matsuda, S.; et al. Recapitulation of pro-inflammatory signature of monocytes with ACVR1A mutation using FOP patient-derived iPSCs. Orphanet J. Rare Dis. 2022, 17, 364. [Google Scholar] [CrossRef]
- Yanagimachi, M.D.; Niwa, A.; Tanaka, T.; Honda-Ozaki, F.; Nishimoto, S.; Murata, Y.; Yasumi, T.; Ito, J.; Tomida, S.; Oshima, K.; et al. Robust and highly-efficient differentiation of functional monocytic cells from human pluripotent stem cells under serum- and feeder cell-free conditions. PLoS ONE 2013, 8, e59243. [Google Scholar]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef]
- Hino, K.; Horigome, K.; Nishio, M.; Komura, S.; Nagata, S.; Zhao, C.; Jin, Y.; Kawakami, K.; Yamada, Y.; Ohta, A.; et al. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J. Clin. Investig. 2017, 127, 3339–3352. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Zhao, C.; Horigome, K.; Nishio, M.; Okanishi, Y.; Nagata, S.; Komura, S.; Yamada, Y.; Toguchida, J.; Ohta, A.; et al. An mTOR Signaling Modulator Suppressed Heterotopic Ossification of Fibrodysplasia Ossificans Progressiva. Stem Cell Rep. 2018, 11, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, S.; Kampfrath, B.; Fischer, K.; Hildebrand, L.; Haupt, J.; Stachelscheid, H.; Knaus, P. ActivinA Induced SMAD1/5 Signaling in an iPSC Derived EC Model of Fibrodysplasia Ossificans Progressiva (FOP) Can Be Rescued by the Drug Candidate Saracatinib. Stem Cell Rev. Rep. 2021, 17, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Micha, D.; Voermans, E.; Eekhoff, M.E.W.; van Essen, H.W.; Zandieh-Doulabi, B.; Netelenbos, C.; Rustemeyer, T.; Sistermans, E.A.; Pals, G.; Bravenboer, N. Inhibition of TGFbeta signaling decreases osteogenic differentiation of fibrodysplasia ossificans progressiva fibroblasts in a novel in vitro model of the disease. Bone 2016, 84, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Del Zotto, G.; Antonini, F.; Azzari, I.; Ortolani, C.; Tripodi, G.; Giacopelli, F.; Cappato, S.; Moretta, L.; Ravazzolo, R.; Bocciardi, R. Peripheral Blood Mononuclear Cell Immunophenotyping in Fibrodysplasia Ossificans Progressiva Patients: Evidence for Monocyte DNAM1 Up-regulation. Cytom. B Clin. Cytom. 2018, 94, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Duffhues, G.; Mikkers, H.; de Jong, D.; Szuhai, K.; de Vries, T.J.; Freund, C.; Bravenboer, N.; van Es, R.J.J.; Netelenbos, J.C.; Goumans, M.J.; et al. Generation of Fibrodysplasia ossificans progressiva and control integration free iPSC lines from periodontal ligament fibroblasts. Stem Cell Res. 2019, 41, 101639. [Google Scholar] [CrossRef]
- Schoenmaker, T.; Mokry, M.; Micha, D.; Netelenbos, C.; Bravenboer, N.; Gilijamse, M.; Eekhoff, E.M.W.; de Vries, T.J. Activin-A Induces Early Differential Gene Expression Exclusively in Periodontal Ligament Fibroblasts from Fibrodysplasia Ossificans Progressiva Patients. Biomedicines 2021, 9, 629. [Google Scholar] [CrossRef]
- de Ruiter, R.D.; Wisse, L.E.; Schoenmaker, T.; Yaqub, M.; Sanchez-Duffhues, G.; Eekhoff, E.M.W.; Micha, D. TGF-Beta Induces Activin A Production in Dermal Fibroblasts Derived from Patients with Fibrodysplasia Ossificans Progressiva. Int. J. Mol. Sci. 2023, 24, 2299. [Google Scholar] [CrossRef]
- Evans, J.D.; Girerd, B.; Montani, D.; Wang, X.J.; Galie, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grunig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801899. [Google Scholar] [CrossRef]
- Machado, R.D.; Southgate, L.; Eichstaedt, C.A.; Aldred, M.A.; Austin, E.D.; Best, D.H.; Chung, W.K.; Benjamin, N.; Elliott, C.G.; Eyries, M.; et al. Pulmonary Arterial Hypertension: A Current Perspective on Established and Emerging Molecular Genetic Defects. Hum. Mutat. 2015, 36, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Soubrier, F.; Chung, W.K.; Machado, R.; Grunig, E.; Aldred, M.; Geraci, M.; Loyd, J.E.; Elliott, C.G.; Trembath, R.C.; Newman, J.H.; et al. Genetics and genomics of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2013, 62, D13–D21. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.H.; Trembath, R.C.; Morse, J.A.; Grunig, E.; Loyd, J.E.; Adnot, S.; Coccolo, F.; Ventura, C.; Phillips, J.A., 3rd; Knowles, J.A.; et al. Genetic basis of pulmonary arterial hypertension: Current understanding and future directions. J. Am. Coll. Cardiol. 2004, 43, 33S–39S. [Google Scholar] [CrossRef] [PubMed]
- Chida, A.; Shintani, M.; Nakayama, T.; Furutani, Y.; Hayama, E.; Inai, K.; Saji, T.; Nonoyama, S.; Nakanishi, T. Missense mutations of the BMPR1B (ALK6) gene in childhood idiopathic pulmonary arterial hypertension. Circ. J. 2012, 76, 1501–1508. [Google Scholar] [CrossRef]
- Frump, A.L.; Lowery, J.W.; Hamid, R.; Austin, E.D.; de Caestecker, M. Abnormal trafficking of endogenously expressed BMPR2 mutant allelic products in patients with heritable pulmonary arterial hypertension. PLoS ONE 2013, 8, e80319. [Google Scholar]
- Yang, Y.M.; Lane, K.B.; Sehgal, P.B. Subcellular mechanisms in pulmonary arterial hypertension: Combinatorial modalities that inhibit anterograde trafficking and cause bone morphogenetic protein receptor type 2 mislocalization. Pulm. Circ. 2013, 3, 533–550. [Google Scholar] [CrossRef]
- Machado, R.D.; Aldred, M.A.; James, V.; Harrison, R.E.; Patel, B.; Schwalbe, E.C.; Gruenig, E.; Janssen, B.; Koehler, R.; Seeger, W.; et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum. Mutat. 2006, 27, 121–132. [Google Scholar] [CrossRef]
- Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Lane, K.B.; Morgan, N.V.; Wheeler, L.; Phillips, J.A., 3rd; Newman, J.; Williams, D.; Galie, N.; et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am. J. Hum. Genet. 2001, 68, 92–102. [Google Scholar] [CrossRef]
- Newman, J.H.; Wheeler, L.; Lane, K.B.; Loyd, E.; Gaddipati, R.; Phillips, J.A., 3rd; Loyd, J.E. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N. Engl. J. Med. 2001, 345, 319–324. [Google Scholar] [CrossRef]
- Cogan, J.D.; Pauciulo, M.W.; Batchman, A.P.; Prince, M.A.; Robbins, I.M.; Hedges, L.K.; Stanton, K.C.; Wheeler, L.A.; Phillips, J.A., 3rd; Loyd, J.E.; et al. High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2006, 174, 590–598. [Google Scholar] [CrossRef]
- Cogan, J.; Austin, E.; Hedges, L.; Womack, B.; West, J.; Loyd, J.; Hamid, R. Role of BMPR2 alternative splicing in heritable pulmonary arterial hypertension penetrance. Circulation 2012, 126, 1907–1916. [Google Scholar] [CrossRef]
- Seki, T.; Hong, K.H.; Oh, S.P. Nonoverlapping expression patterns of ALK1 and ALK5 reveal distinct roles of each receptor in vascular development. Lab. Investig. 2006, 86, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Yun, J.; Oh, S.P. Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circ. Res. 2003, 93, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Alt, A.; Miguel-Romero, L.; Donderis, J.; Aristorena, M.; Blanco, F.J.; Round, A.; Rubio, V.; Bernabeu, C.; Marina, A. Structural and functional insights into endoglin ligand recognition and binding. PLoS ONE 2012, 7, e29948. [Google Scholar]
- Tazat, K.; Pomeraniec-Abudy, L.; Hector-Greene, M.; Szilagyi, S.S.; Sharma, S.; Cai, E.M.; Corona, A.L.; Ehrlich, M.; Blobe, G.C.; Henis, Y.I. ALK1 regulates the internalization of endoglin and the type III TGF-beta receptor. Mol. Biol. Cell 2021, 32, 605–621. [Google Scholar] [CrossRef] [PubMed]
- Sugden, W.W.; Meissner, R.; Aegerter-Wilmsen, T.; Tsaryk, R.; Leonard, E.V.; Bussmann, J.; Hamm, M.J.; Herzog, W.; Jin, Y.; Jakobsson, L.; et al. Endoglin controls blood vessel diameter through endothelial cell shape changes in response to haemodynamic cues. Nat. Cell Biol. 2017, 19, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Seghers, L.; de Vries, M.R.; Pardali, E.; Hoefer, I.E.; Hierck, B.P.; ten Dijke, P.; Goumans, M.J.; Quax, P.H. Shear induced collateral artery growth modulated by endoglin but not by ALK1. J. Cell. Mol. Med. 2012, 16, 2440–2450. [Google Scholar] [CrossRef]
- David, L.; Mallet, C.; Mazerbourg, S.; Feige, J.J.; Bailly, S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007, 109, 1953–1961. [Google Scholar] [CrossRef]
- David, L.; Mallet, C.; Keramidas, M.; Lamande, N.; Gasc, J.M.; Dupuis-Girod, S.; Plauchu, H.; Feige, J.J.; Bailly, S. Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ. Res. 2008, 102, 914–922. [Google Scholar] [CrossRef]
- Desroches-Castan, A.; Tillet, E.; Bouvard, C.; Bailly, S. BMP9 and BMP10: Two close vascular quiescence partners that stand out. Dev. Dyn. 2022, 251, 178–197. [Google Scholar] [CrossRef]
- Akla, N.; Viallard, C.; Popovic, N.; Lora Gil, C.; Sapieha, P.; Larrivee, B. BMP9 (Bone Morphogenetic Protein-9)/Alk1 (Activin-Like Kinase Receptor Type I) Signaling Prevents Hyperglycemia-Induced Vascular Permeability. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1821–1836. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Ormiston, M.L.; Yang, X.; Southwood, M.; Graf, S.; Machado, R.D.; Mueller, M.; Kinzel, B.; Yung, L.M.; Wilkinson, J.M.; et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 2015, 21, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Long, L.; Yang, X.; Tong, Z.; Southwood, M.; King, R.; Caruso, P.; Upton, P.D.; Yang, P.; Bocobo, G.A.; et al. Circulating BMP9 Protects the Pulmonary Endothelium during Inflammation-induced Lung Injury in Mice. Am. J. Respir. Crit. Care Med. 2021, 203, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Ponomarev, L.C.; Ksiazkiewicz, J.; Staring, M.W.; Luttun, A.; Zwijsen, A. The BMP Pathway in Blood Vessel and Lymphatic Vessel Biology. Int. J. Mol. Sci. 2021, 22, 6364. [Google Scholar] [CrossRef]
- Gorelova, A.; Berman, M.; Al Ghouleh, I. Endothelial-to-Mesenchymal Transition in Pulmonary Arterial Hypertension. Antioxid. Redox Signal. 2021, 34, 891–914. [Google Scholar] [CrossRef]
- Yun, E.; Kook, Y.; Yoo, K.H.; Kim, K.I.; Lee, M.S.; Kim, J.; Lee, A. Endothelial to Mesenchymal Transition in Pulmonary Vascular Diseases. Biomedicines 2020, 8, 639. [Google Scholar] [CrossRef]
- Ihida-Stansbury, K.; McKean, D.M.; Lane, K.B.; Loyd, J.E.; Wheeler, L.A.; Morrell, N.W.; Jones, P.L. Tenascin-C is induced by mutated BMP type II receptors in familial forms of pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L694–L702. [Google Scholar] [CrossRef]
- Hiepen, C.; Jatzlau, J.; Hildebrandt, S.; Kampfrath, B.; Goktas, M.; Murgai, A.; Cuellar Camacho, J.L.; Haag, R.; Ruppert, C.; Sengle, G.; et al. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFbeta responses and altered cell mechanics. PLoS Biol. 2019, 17, e3000557. [Google Scholar] [CrossRef]
- Thenappan, T.; Chan, S.Y.; Weir, E.K. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1322–H1331. [Google Scholar] [CrossRef]
- Hiepen, C.; Jatzlau, J.; Knaus, P. Biomechanical stress provides a second hit in the establishment of BMP/TGFbeta-related vascular disorders. Cell Stress 2020, 4, 44–47. [Google Scholar] [CrossRef]
- Wang, L.; Moonen, J.R.; Cao, A.; Isobe, S.; Li, C.G.; Tojais, N.F.; Taylor, S.; Marciano, D.P.; Chen, P.I.; Gu, M.; et al. Dysregulated Smooth Muscle Cell BMPR2-ARRB2 Axis Causes Pulmonary Hypertension. Circ. Res. 2023, 132, 545–564. [Google Scholar] [CrossRef] [PubMed]
- Tada, Y.; Majka, S.; Carr, M.; Harral, J.; Crona, D.; Kuriyama, T.; West, J. Molecular effects of loss of BMPR2 signaling in smooth muscle in a transgenic mouse model of PAH. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1556–L1563. [Google Scholar] [CrossRef] [PubMed]
- Andruska, A.; Spiekerkoetter, E. Consequences of BMPR2 Deficiency in the Pulmonary Vasculature and Beyond: Contributions to Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 2499. [Google Scholar] [CrossRef] [PubMed]
- West, J.D.; Austin, E.D.; Gaskill, C.; Marriott, S.; Baskir, R.; Bilousova, G.; Jean, J.C.; Hemnes, A.R.; Menon, S.; Bloodworth, N.C.; et al. Identification of a common Wnt-associated genetic signature across multiple cell types in pulmonary arterial hypertension. Am. J. Physiol. Cell Physiol. 2014, 307, C415–C430. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.H.; Ma, J.L.; Ding, D.; Ma, Y.J.; Wei, Y.P.; Jing, Z.C. Experimental animal models of pulmonary hypertension: Development and challenges. Anim. Model Exp. Med. 2022, 5, 207–216. [Google Scholar] [CrossRef]
- Jasinska-Stroschein, M. A review of genetically-driven rodent models of pulmonary hypertension. Vasc. Pharmacol. 2022, 144, 106970. [Google Scholar] [CrossRef]
- Spiekerkoetter, E.; Tian, X.; Cai, J.; Hopper, R.K.; Sudheendra, D.; Li, C.G.; El-Bizri, N.; Sawada, H.; Haghighat, R.; Chan, R.; et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J. Clin. Investig. 2013, 123, 3600–3613. [Google Scholar] [CrossRef]
- Yung, L.M.; Yang, P.; Joshi, S.; Augur, Z.M.; Kim, S.S.J.; Bocobo, G.A.; Dinter, T.; Troncone, L.; Chen, P.S.; McNeil, M.E.; et al. ACTRIIA-Fc rebalances activin/GDF versus BMP signaling in pulmonary hypertension. Sci. Transl. Med. 2020, 12, eaaz5660. [Google Scholar] [CrossRef]
- Yndestad, A.; Larsen, K.O.; Oie, E.; Ueland, T.; Smith, C.; Halvorsen, B.; Sjaastad, I.; Skjonsberg, O.H.; Pedersen, T.M.; Anfinsen, O.G.; et al. Elevated levels of activin A in clinical and experimental pulmonary hypertension. J. Appl. Physiol. 2009, 106, 1356–1364. [Google Scholar] [CrossRef]
- Rosa, S.; Praca, C.; Pitrez, P.R.; Gouveia, P.J.; Aranguren, X.L.; Ricotti, L.; Ferreira, L.S. Functional characterization of iPSC-derived arterial- and venous-like endothelial cells. Sci. Rep. 2019, 9, 3826. [Google Scholar] [CrossRef]
- Nguyen, J.; Lin, Y.Y.; Gerecht, S. The next generation of endothelial differentiation: Tissue-specific ECs. Cell Stem Cell 2021, 28, 1188–1204. [Google Scholar] [CrossRef] [PubMed]
- Hamid, R.; Yan, L. Induced Pluripotent Stem Cells in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2017, 195, 852–853. [Google Scholar] [CrossRef] [PubMed]
- Geti, I.; Ormiston, M.L.; Rouhani, F.; Toshner, M.; Movassagh, M.; Nichols, J.; Mansfield, W.; Southwood, M.; Bradley, A.; Rana, A.A.; et al. A practical and efficient cellular substrate for the generation of induced pluripotent stem cells from adults: Blood-derived endothelial progenitor cells. Stem Cells Transl. Med. 2012, 1, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Shao, N.Y.; Sa, S.; Li, D.; Termglinchan, V.; Ameen, M.; Karakikes, I.; Sosa, G.; Grubert, F.; Lee, J.; et al. Patient-Specific iPSC-Derived Endothelial Cells Uncover Pathways that Protect against Pulmonary Hypertension in BMPR2 Mutation Carriers. Cell Stem Cell 2017, 20, 490–504.e5. [Google Scholar] [CrossRef] [PubMed]
- Sa, S.; Gu, M.; Chappell, J.; Shao, N.Y.; Ameen, M.; Elliott, K.A.; Li, D.; Grubert, F.; Li, C.G.; Taylor, S.; et al. Induced Pluripotent Stem Cell Model of Pulmonary Arterial Hypertension Reveals Novel Gene Expression and Patient Specificity. Am. J. Respir. Crit. Care Med. 2017, 195, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Tang, Y.; Tai, Y.Y.; Handen, A.; Zhao, J.; Speyer, G.; Al Aaraj, Y.; Watson, A.; Romanelli, M.E.; Sembrat, J.; et al. SCUBE1 Controls BMPR2-Relevant Pulmonary Endothelial Function: Implications for Diagnostic Marker Development in Pulmonary Arterial Hypertension. JACC Basic Transl. Sci. 2020, 5, 1073–1092. [Google Scholar] [CrossRef]
- Gu, M.; Donato, M.; Guo, M.; Wary, N.; Miao, Y.; Mao, S.; Saito, T.; Otsuki, S.; Wang, L.; Harper, R.L.; et al. iPSC-endothelial cell phenotypic drug screening and in silico analyses identify tyrphostin-AG1296 for pulmonary arterial hypertension. Sci. Transl. Med. 2021, 13, eaba6480. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Liu, Q.; Wang, A. Tyrphostin AG1296, a platelet-derived growth factor receptor inhibitor, induces apoptosis, and reduces viability and migration of PLX4032-resistant melanoma cells. OncoTargets Ther. 2015, 8, 1043–1051. [Google Scholar] [CrossRef]
- Lasota, M.; Bentke-Imiolek, A.; Skrzypek, K.; Bobrowska, J.; Jagusiak, A.; Bryniarska-Kubiak, N.; Zagajewski, J.; Kot, M.; Szydlak, R.; Lekka, M.; et al. Small-molecule inhibitor-tyrphostin AG1296 regulates proliferation, survival and migration of rhabdomyosarcoma cells. J. Physiol. Pharmacol. 2021, 72, 881–893. [Google Scholar]
- Ueda, S.; Ikeda, H.; Mizuki, M.; Ishiko, J.; Matsumura, I.; Tanaka, H.; Shibayama, H.; Sugahara, H.; Takai, E.; Zhang, X.; et al. Constitutive activation of c-kit by the juxtamembrane but not the catalytic domain mutations is inhibited selectively by tyrosine kinase inhibitors STI571 and AG1296. Int. J. Hematol. 2002, 76, 427–435. [Google Scholar] [CrossRef]
- Kiskin, F.N.; Chang, C.H.; Huang, C.J.Z.; Kwieder, B.; Cheung, C.; Dunmore, B.J.; Serrano, F.; Sinha, S.; Morrell, N.W.; Rana, A.A. Contributions of BMPR2 Mutations and Extrinsic Factors to Cellular Phenotypes of Pulmonary Arterial Hypertension Revealed by Induced Pluripotent Stem Cell Modeling. Am. J. Respir. Crit. Care Med. 2018, 198, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W. Finding the needle in the haystack: BMP9 and 10 emerge from the genome in pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1900078. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.-P.; Zhang, S.-J.; Ding, Y.-J.; Fang, J.; Zhou, L.; Xie, S.-M.; Ge, X.; Fu, L.-J.; Li, Q.-Y.; Qu, J.-M.; et al. Progesterone is an Inducement of Heritable Pulmonary Arterial Hypertension with BMPR2 Mutation. bioRxiv 2023. [Google Scholar] [CrossRef]
- Chen, X.; Talati, M.; Fessel, J.P.; Hemnes, A.R.; Gladson, S.; French, J.; Shay, S.; Trammell, A.; Phillips, J.A.; Hamid, R.; et al. Estrogen Metabolite 16alpha-Hydroxyestrone Exacerbates Bone Morphogenetic Protein Receptor Type II-Associated Pulmonary Arterial Hypertension Through MicroRNA-29-Mediated Modulation of Cellular Metabolism. Circulation 2016, 133, 82–97. [Google Scholar] [CrossRef]
- Singla, S.; Machado, R.F. The imitation game in pulmonary arterial hypertension. Sex, bone morphogenetic protein receptor, and the estrogen paradox. Am. J. Respir. Crit. Care Med. 2015, 191, 612–613. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, W.H.; Mao, Y.M.; Yuan, P.; Zhang, R.; Ju, F.L.; Jing, Z.C. BMPR2 mutations influence phenotype more obviously in male patients with pulmonary arterial hypertension. Circ. Cardiovasc. Genet. 2012, 5, 511–518. [Google Scholar] [CrossRef]
- Devendran, A.; Kar, S.; Bailey, R.; Trivieri, M.G. The Role of Bone Morphogenetic Protein Receptor Type 2 (BMPR2) and the Prospects of Utilizing Induced Pluripotent Stem Cells (iPSCs) in Pulmonary Arterial Hypertension Disease Modeling. Cells 2022, 11, 3823. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Ke, M.W.; Cheng, C.C.; Chiou, S.H.; Wann, S.R.; Shu, C.W.; Chiou, K.R.; Tseng, C.J.; Pan, H.W.; Mar, G.Y.; et al. Therapeutic Benefits of Induced Pluripotent Stem Cells in Monocrotaline-Induced Pulmonary Arterial Hypertension. PLoS ONE 2016, 11, e0142476. [Google Scholar] [CrossRef]
- Oh, S.; Jung, J.H.; Ahn, K.J.; Jang, A.Y.; Byun, K.; Yang, P.C.; Chung, W.J. Stem Cell and Exosome Therapy in Pulmonary Hypertension. Korean Circ. J. 2022, 52, 110–122. [Google Scholar] [CrossRef]
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrell, J.; et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 1994, 8, 345–351. [Google Scholar] [CrossRef]
- Gallione, C.J.; Pasyk, K.A.; Boon, L.M.; Lennon, F.; Johnson, D.W.; Helmbold, E.A.; Markel, D.S.; Vikkula, M.; Mulliken, J.B.; Warman, M.L.; et al. A gene for familial venous malformations maps to chromosome 9p in a second large kindred. J. Med. Genet. 1995, 32, 197–199. [Google Scholar] [CrossRef]
- Johnson, D.W.; Berg, J.N.; Baldwin, M.A.; Gallione, C.J.; Marondel, I.; Yoon, S.J.; Stenzel, T.T.; Speer, M.; Pericak-Vance, M.A.; Diamond, A.; et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat. Genet. 1996, 13, 189–195. [Google Scholar] [CrossRef]
- Abdalla, S.A.; Cymerman, U.; Johnson, R.M.; Deber, C.M.; Letarte, M. Disease-associated mutations in conserved residues of ALK-1 kinase domain. Eur. J. Hum. Genet. 2003, 11, 279–287. [Google Scholar] [CrossRef]
- McAllister, K.A.; Baldwin, M.A.; Thukkani, A.K.; Gallione, C.J.; Berg, J.N.; Porteous, M.E.; Guttmacher, A.E.; Marchuk, D.A. Six novel mutations in the endoglin gene in hereditary hemorrhagic telangiectasia type 1 suggest a dominant-negative effect of receptor function. Hum. Mol. Genet. 1995, 4, 1983–1985. [Google Scholar] [CrossRef]
- Wooderchak-Donahue, W.L.; McDonald, J.; O’Fallon, B.; Upton, P.D.; Li, W.; Roman, B.L.; Young, S.; Plant, P.; Fulop, G.T.; Langa, C.; et al. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am. J. Hum. Genet. 2013, 93, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Dupuis-Girod, S.; Bailly, S.; Plauchu, H. Hereditary hemorrhagic telangiectasia: From molecular biology to patient care. J. Thromb. Haemost. 2010, 8, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L. Hereditary haemorrhagic telangiectasia: Pathophysiology, diagnosis and treatment. Blood Rev. 2010, 24, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Thalgott, J.H.; Dos-Santos-Luis, D.; Hosman, A.E.; Martin, S.; Lamande, N.; Bracquart, D.; Srun, S.; Galaris, G.; de Boer, H.C.; Tual-Chalot, S.; et al. Decreased Expression of Vascular Endothelial Growth Factor Receptor 1 Contributes to the Pathogenesis of Hereditary Hemorrhagic Telangiectasia Type 2. Circulation 2018, 138, 2698–2712. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, R.L.; Jonker, L.; Goumans, M.J.; Larsson, J.; Bouwman, P.; Karlsson, S.; Dijke, P.T.; Arthur, H.M.; Mummery, C.L. Defective paracrine signalling by TGFbeta in yolk sac vasculature of endoglin mutant mice: A paradigm for hereditary haemorrhagic telangiectasia. Development 2004, 131, 6237–6247. [Google Scholar] [CrossRef] [PubMed]
- Lebrin, F.; Srun, S.; Raymond, K.; Martin, S.; van den Brink, S.; Freitas, C.; Breant, C.; Mathivet, T.; Larrivee, B.; Thomas, J.L.; et al. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat. Med. 2010, 16, 420–428. [Google Scholar] [CrossRef]
- Gerhardt, H. VEGF and endothelial guidance in angiogenic sprouting. Organogenesis 2008, 4, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Larrivee, B.; Prahst, C.; Gordon, E.; del Toro, R.; Mathivet, T.; Duarte, A.; Simons, M.; Eichmann, A. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev. Cell 2012, 22, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Kim, Y.H.; Choe, S.W.; Tak, Y.G.; Garrido-Martin, E.M.; Chang, M.; Lee, Y.J.; Oh, S.P. Enhanced responses to angiogenic cues underlie the pathogenesis of hereditary hemorrhagic telangiectasia 2. PLoS ONE 2013, 8, e63138. [Google Scholar] [CrossRef] [PubMed]
- Vion, A.C.; Alt, S.; Klaus-Bergmann, A.; Szymborska, A.; Zheng, T.; Perovic, T.; Hammoutene, A.; Oliveira, M.B.; Bartels-Klein, E.; Hollfinger, I.; et al. Primary cilia sensitize endothelial cells to BMP and prevent excessive vascular regression. J. Cell Biol. 2018, 217, 1651–1665. [Google Scholar] [CrossRef]
- Corti, P.; Young, S.; Chen, C.Y.; Patrick, M.J.; Rochon, E.R.; Pekkan, K.; Roman, B.L. Interaction between alk1 and blood flow in the development of arteriovenous malformations. Development 2011, 138, 1573–1582. [Google Scholar] [CrossRef]
- Hiepen, C.; Mendez, P.L.; Knaus, P. It Takes Two to Tango: Endothelial TGFbeta/BMP Signaling Crosstalk with Mechanobiology. Cells 2020, 9, 1965. [Google Scholar] [CrossRef]
- Eisa-Beygi, S.; Burrows, P.E.; Link, B.A. Endothelial cilia dysfunction in pathogenesis of hereditary hemorrhagic telangiectasia. Front. Cell Dev. Biol. 2022, 10, 1037453. [Google Scholar] [CrossRef]
- Riss, D.; Burian, M.; Wolf, A.; Kranebitter, V.; Kaider, A.; Arnoldner, C. Intranasal submucosal bevacizumab for epistaxis in hereditary hemorrhagic telangiectasia: A double-blind, randomized, placebo-controlled trial. Head Neck 2015, 37, 783–787. [Google Scholar] [CrossRef]
- Robert, F.; Desroches-Castan, A.; Bailly, S.; Dupuis-Girod, S.; Feige, J.J. Future treatments for hereditary hemorrhagic telangiectasia. Orphanet. J. Rare Dis. 2020, 15, 4. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kim, M.J.; Choe, S.W.; Sprecher, D.; Lee, Y.J.; Oh, S.P. Selective effects of oral antiangiogenic tyrosine kinase inhibitors on an animal model of hereditary hemorrhagic telangiectasia. J. Thromb. Haemost. 2017, 15, 1095–1102. [Google Scholar] [CrossRef]
- Freund, C.; Davis, R.P.; Gkatzis, K.; Ward-van Oostwaard, D.; Mummery, C.L. The first reported generation of human induced pluripotent stem cells (iPS cells) and iPS cell-derived cardiomyocytes in the Netherlands. Neth. Heart J. 2010, 18, 51–54. [Google Scholar] [PubMed]
- Bouma, M.J.; Orlova, V.; van den Hil, F.E.; Mager, H.J.; Baas, F.; de Knijff, P.; Mummery, C.L.; Mikkers, H.; Freund, C. Generation and genetic repair of 2 iPSC clones from a patient bearing a heterozygous c.1120del18 mutation in the ACVRL1 gene leading to Hereditary Hemorrhagic Telangiectasia (HHT) type 2. Stem Cell Res. 2020, 46, 101786. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Zhao, X.; Liu, X.; Liu, Y.; Ma, F.; Liu, B.; Yang, J. Autologous correction in patient induced pluripotent stem cell-endothelial cells to identify a novel pathogenic mutation of hereditary hemorrhagic telangiectasia. Pulm. Circ. 2020, 10, 2045894019885357. [Google Scholar] [CrossRef]
- Fernandez, L.A.; Sanz-Rodriguez, F.; Zarrabeitia, R.; Perez-Molino, A.; Hebbel, R.P.; Nguyen, J.; Bernabeu, C.; Botella, L.M. Blood outgrowth endothelial cells from Hereditary Haemorrhagic Telangiectasia patients reveal abnormalities compatible with vascular lesions. Cardiovasc. Res. 2005, 68, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Xiang-Tischhauser, L.; Bette, M.; Rusche, J.R.; Roth, K.; Kasahara, N.; Stuck, B.A.; Bakowsky, U.; Wartenberg, M.; Sauer, H.; Geisthoff, U.W.; et al. Generation of a Syngeneic Heterozygous ACVRL1((wt/mut)) Knockout iPS Cell Line for the in Vitro Study of HHT2-Associated Angiogenesis. Cells 2023, 12, 1600. [Google Scholar] [CrossRef]
- Orlova, V.V.; Nahon, D.M.; Cochrane, A.; Cao, X.; Freund, C.; van den Hil, F.; Westermann, C.J.J.; Snijder, R.J.; Ploos van Amstel, J.K.; Ten Dijke, P.; et al. Vascular defects associated with hereditary hemorrhagic telangiectasia revealed in patient-derived isogenic iPSCs in 3D vessels on chip. Stem Cell Rep. 2022, 17, 1536–1545. [Google Scholar] [CrossRef]
- Girerd, B.; Montani, D.; Coulet, F.; Sztrymf, B.; Yaici, A.; Jais, X.; Tregouet, D.; Reis, A.; Drouin-Garraud, V.; Fraisse, A.; et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am. J. Respir. Crit. Care Med. 2010, 181, 851–861. [Google Scholar] [CrossRef]
- Walsh, L.J.; Collins, C.; Ibrahim, H.; Kerins, D.M.; Brady, A.P.; TM, O.C. Pulmonary arterial hypertension in hereditary hemorrhagic telangiectasia associated with ACVRL1 mutation: A case report. J. Med. Case Rep. 2022, 16, 99. [Google Scholar] [CrossRef]
- Kularatne, M.; Eyries, M.; Savale, L.; Humbert, M.; Montani, D. Isolated Pulmonary Arteriovenous Malformations Associated With BMPR2 Pathogenic Variants. Chest 2023, 164, e23–e26. [Google Scholar] [CrossRef]
- Harrison, R.E.; Flanagan, J.A.; Sankelo, M.; Abdalla, S.A.; Rowell, J.; Machado, R.D.; Elliott, C.G.; Robbins, I.M.; Olschewski, H.; McLaughlin, V.; et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J. Med. Genet. 2003, 40, 865–871. [Google Scholar] [CrossRef]
- Hildebrand, L.; Rossbach, B.; Kuhnen, P.; Gossen, M.; Kurtz, A.; Reinke, P.; Seemann, P.; Stachelscheid, H. Generation of integration free induced pluripotent stem cells from fibrodysplasia ossificans progressiva (FOP) patients from urine samples. Stem Cell Res. 2016, 16, 54–58. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Usman, A.; Haase, A.; Merkert, S.; Gohring, G.; Hansmann, G.; Gall, H.; Schermuly, R.; Martin, U.; Olmer, R. Generation of pulmonary arterial hypertension patient-specific induced pluripotent stem cell lines from three unrelated patients with a heterozygous missense mutation in exon 12, a heterozygous in-frame deletion in exon 3 and a missense mutation in exon 11 of the BMPR2 gene. Stem Cell Res. 2021, 55, 102488. [Google Scholar] [PubMed]
- Ura, H.; Togi, S.; Iwata, Y.; Ozaki, M.; Niida, Y. Establishment of a human induced pluripotent stem cell line, KMUGMCi001-A, from a patient bearing a heterozygous c.772 + 3_772 + 4dup mutation in the ACVRL1 gene leading Telangiectasia, hereditary hemorrhagic, type 2 (HHT2). Stem Cell Res. 2022, 61, 102743. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.A.; Boghdady, C.M.; Campbell, B.E.; Moraes, C. Integrating mechanical sensor readouts into organ-on-a-chip platforms. Front. Bioeng. Biotechnol. 2022, 10, 1060895. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Aleman, J.; Shin, S.R.; Kilic, T.; Kim, D.; Mousavi Shaegh, S.A.; Massa, S.; Riahi, R.; Chae, S.; Hu, N.; et al. Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc. Natl. Acad. Sci. USA 2017, 114, E2293–E2302. [Google Scholar] [CrossRef]
- Fanizza, F.; Campanile, M.; Forloni, G.; Giordano, C.; Albani, D. Induced pluripotent stem cell-based organ-on-a-chip as personalized drug screening tools: A focus on neurodegenerative disorders. J. Tissue Eng. 2022, 13, 20417314221095339. [Google Scholar] [CrossRef]
- Hockney, S.; Parker, J.; Turner, J.E.; Todd, X.; Todryk, S.; Gieling, R.G.; Hilgen, G.; Simoes, D.C.M.; Pal, D. Next generation organoid engineering to replace animals in cancer drug testing. Biochem. Pharmacol. 2023, 213, 115586. [Google Scholar] [CrossRef]
- Fuhr, A.; Kurtz, A.; Hiepen, C.; Müller, S. Organoids as Miniature Twins—Challenges for Comparability and Need for Data Standardization and Access. Organoids 2022, 1, 28–36. [Google Scholar] [CrossRef]
- Yin, X.; Mead, B.E.; Safaee, H.; Langer, R.; Karp, J.M.; Levy, O. Engineering Stem Cell Organoids. Cell Stem Cell 2016, 18, 25–38. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Y.; Qin, J. Multi-organ microphysiological system: A new paradigm for COVID-19 research. Organs Chip 2023, 5, 100029. [Google Scholar] [CrossRef]
- Zommiti, M.; Connil, N.; Tahrioui, A.; Groboillot, A.; Barbey, C.; Konto-Ghiorghi, Y.; Lesouhaitier, O.; Chevalier, S.; Feuilloley, M.G.J. Organs-on-Chips Platforms Are Everywhere: A Zoom on Biomedical Investigation. Bioengineering 2022, 9, 646. [Google Scholar] [CrossRef] [PubMed]
- Koenig, L.; Ramme, A.P.; Faust, D.; Mayer, M.; Flotke, T.; Gerhartl, A.; Brachner, A.; Neuhaus, W.; Appelt-Menzel, A.; Metzger, M.; et al. A Human Stem Cell-Derived Brain-Liver Chip for Assessing Blood-Brain-Barrier Permeation of Pharmaceutical Drugs. Cells 2022, 11, 3295. [Google Scholar] [CrossRef] [PubMed]
- Whelan, I.T.; Burdis, R.; Shahreza, S.; Moeendarbary, E.; Hoey, D.A.; Kelly, D.J. A microphysiological model of bone development and regeneration. Biofabrication 2023, 15, 034103. [Google Scholar] [CrossRef]
- Knowles, H.J.; Chanalaris, A.; Koutsikouni, A.; Cribbs, A.P.; Grover, L.M.; Hulley, P.A. Mature primary human osteocytes in mini organotypic cultures secrete FGF23 and PTH1-34-regulated sclerostin. Front. Endocrinol. 2023, 14, 1167734. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef]
- Yang, P.; Yu, P.B. In Search of the Second Hit in Pulmonary Arterial Hypertension. Circ. Res. 2019, 124, 6–8. [Google Scholar] [CrossRef]
- Eichstaedt, C.A.; Song, J.; Benjamin, N.; Harutyunova, S.; Fischer, C.; Grunig, E.; Hinderhofer, K. EIF2AK4 mutation as “second hit” in hereditary pulmonary arterial hypertension. Respir. Res. 2016, 17, 141. [Google Scholar] [CrossRef] [PubMed]
- Viales, R.R.; Eichstaedt, C.A.; Ehlken, N.; Fischer, C.; Lichtblau, M.; Grunig, E.; Hinderhofer, K. Mutation in BMPR2 Promoter: A ‘Second Hit’ for Manifestation of Pulmonary Arterial Hypertension? PLoS ONE 2015, 10, e0133042. [Google Scholar] [CrossRef]
- Bernabeu, C.; Bayrak-Toydemir, P.; McDonald, J.; Letarte, M. Potential Second-Hits in Hereditary Hemorrhagic Telangiectasia. J. Clin. Med. 2020, 9, 3571. [Google Scholar] [CrossRef]
- Jaliawala, H.A.; Parmar, M.; Summers, K.; Bernardo, R.J. A second hit? Pulmonary arterial hypertension, BMPR2 mutation, and exposure to prescription amphetamines. Pulm. Circ. 2022, 12, e12053. [Google Scholar] [CrossRef]
- Vengethasamy, L.; Hautefort, A.; Tielemans, B.; Belge, C.; Perros, F.; Verleden, S.; Fadel, E.; Van Raemdonck, D.; Delcroix, M.; Quarck, R. BMPRII influences the response of pulmonary microvascular endothelial cells to inflammatory mediators. Pflugers Arch. 2016, 468, 1969–1983. [Google Scholar] [CrossRef]
- Hagen, M.; Fagan, K.; Steudel, W.; Carr, M.; Lane, K.; Rodman, D.M.; West, J. Interaction of interleukin-6 and the BMP pathway in pulmonary smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1473–L1479. [Google Scholar] [CrossRef] [PubMed]
- Arai, K.Y.; Ono, M.; Kudo, C.; Fujioka, A.; Okamura, R.; Nomura, Y.; Nishiyama, T. IL-1beta stimulates activin betaA mRNA expression in human skin fibroblasts through the MAPK pathways, the nuclear factor-kappaB pathway, and prostaglandin E2. Endocrinology 2011, 152, 3779–3790. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, L.; Gaber, T.; Kuhnen, P.; Morhart, R.; Unterborsch, H.; Schomburg, L.; Seemann, P. Trace element and cytokine concentrations in patients with Fibrodysplasia Ossificans Progressiva (FOP): A case control study. J. Trace Elem. Med. Biol. 2017, 39, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Pagani, C.A.; Das, N.; Marini, S.; Huber, A.K.; Xie, L.; Jimenez, J.; Brydges, S.; Lim, W.K.; Nannuru, K.C.; et al. Activin A does not drive post-traumatic heterotopic ossification. Bone 2020, 138, 115473. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.J.; Holmes, A.M.; Deighton, J.; Long, L.; Yang, X.; Barker, L.; Walker, C.; Budd, D.C.; Upton, P.D.; Morrell, N.W. BMP type II receptor deficiency confers resistance to growth inhibition by TGF-beta in pulmonary artery smooth muscle cells: Role of proinflammatory cytokines. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L604–L615. [Google Scholar] [CrossRef] [PubMed]
- Soon, E.; Crosby, A.; Southwood, M.; Yang, P.; Tajsic, T.; Toshner, M.; Appleby, S.; Shanahan, C.M.; Bloch, K.D.; Pepke-Zaba, J.; et al. Bone morphogenetic protein receptor type II deficiency and increased inflammatory cytokine production. A gateway to pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 859–872. [Google Scholar] [CrossRef]
- Furuya, Y.; Satoh, T.; Kuwana, M. Interleukin-6 as a potential therapeutic target for pulmonary arterial hypertension. Int. J. Rheumatol. 2010, 2010, 720305. [Google Scholar] [CrossRef]
- Zhang, R.; Han, Z.; Degos, V.; Shen, F.; Choi, E.J.; Sun, Z.; Kang, S.; Wong, M.; Zhu, W.; Zhan, L.; et al. Persistent infiltration and pro-inflammatory differentiation of monocytes cause unresolved inflammation in brain arteriovenous malformation. Angiogenesis 2016, 19, 451–461. [Google Scholar] [CrossRef]
- Haupt, J.; Stanley, A.; McLeod, C.M.; Cosgrove, B.D.; Culbert, A.L.; Wang, L.; Mourkioti, F.; Mauck, R.L.; Shore, E.M. ACVR1(R206H) FOP mutation alters mechanosensing and tissue stiffness during heterotopic ossification. Mol. Biol. Cell 2019, 30, 17–29. [Google Scholar] [CrossRef]
- Park, J.S.; Chu, J.S.; Tsou, A.D.; Diop, R.; Tang, Z.; Wang, A.; Li, S. The effect of matrix stiffness on the differentiation of mesenchymal stem cells in response to TGF-beta. Biomaterials 2011, 32, 3921–3930. [Google Scholar] [CrossRef] [PubMed]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122 Pt 2, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Ashe, H.L. Modulation of BMP signalling by integrins. Biochem. Soc. Trans. 2016, 44, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Hemnes, A.R.; Perrien, D.S.; Schuster, M.; Robinson, L.J.; Gladson, S.; Loibner, H.; Bai, S.; Blackwell, T.R.; Tada, Y.; et al. Cytoskeletal defects in Bmpr2-associated pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L474–L484. [Google Scholar] [CrossRef]
- Fernandez, L.A.; Sanz-Rodriguez, F.; Blanco, F.J.; Bernabeu, C.; Botella, L.M. Hereditary hemorrhagic telangiectasia, a vascular dysplasia affecting the TGF-beta signaling pathway. Clin. Med. Res. 2006, 4, 66–78. [Google Scholar] [CrossRef]
- Melchionna, R.; Trono, P.; Tocci, A.; Nistico, P. Actin Cytoskeleton and Regulation of TGFbeta Signaling: Exploring Their Links. Biomolecules 2021, 11, 336. [Google Scholar] [CrossRef]
- Ingber, D.E. Mechanobiology and diseases of mechanotransduction. Ann. Med. 2003, 35, 564–577. [Google Scholar] [CrossRef]
- Zuela-Sopilniak, N.; Lammerding, J. Can’t handle the stress? Mechanobiology and disease. Trends Mol. Med. 2022, 28, 710–725. [Google Scholar] [CrossRef]
- Abdelilah-Seyfried, S.; Iruela-Arispe, M.L.; Penninger, J.M.; Tournier-Lasserve, E.; Vikkula, M.; Cleaver, O. Recalibrating vascular malformations and mechanotransduction by pharmacological intervention. J. Clin. Investig. 2022, 132, e160227. [Google Scholar] [CrossRef]
- Tan, W.; Madhavan, K.; Hunter, K.S.; Park, D.; Stenmark, K.R. Vascular stiffening in pulmonary hypertension: Cause or consequence? (2013 Grover Conference series). Pulm. Circ. 2014, 4, 560–580. [Google Scholar] [CrossRef]
- Deng, H.; Min, E.; Baeyens, N.; Coon, B.G.; Hu, R.; Zhuang, Z.W.; Chen, M.; Huang, B.; Afolabi, T.; Zarkada, G.; et al. Activation of Smad2/3 signaling by low fluid shear stress mediates artery inward remodeling. Proc. Natl. Acad. Sci. USA 2021, 118, e2105339118. [Google Scholar] [CrossRef]
- Kopf, J.; Petersen, A.; Duda, G.N.; Knaus, P. BMP2 and mechanical loading cooperatively regulate immediate early signalling events in the BMP pathway. BMC Biol. 2012, 10, 37. [Google Scholar] [CrossRef]
- Papaioannou, T.G.; Stefanadis, C. Vascular wall shear stress: Basic principles and methods. Hellenic. J. Cardiol. 2005, 46, 9–15. [Google Scholar] [PubMed]
- Munoz, C.J.; Lucas, A.; Williams, A.T.; Cabrales, P. A Review on Microvascular Hemodynamics: The Control of Blood Flow Distribution and Tissue Oxygenation. Crit. Care Clin. 2020, 36, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Campinho, P.; Vilfan, A.; Vermot, J. Blood Flow Forces in Shaping the Vascular System: A Focus on Endothelial Cell Behavior. Front. Physiol. 2020, 11, 552. [Google Scholar] [CrossRef] [PubMed]
- Katt, M.E.; Xu, Z.S.; Gerecht, S.; Searson, P.C. Human Brain Microvascular Endothelial Cells Derived from the BC1 iPS Cell Line Exhibit a Blood-Brain Barrier Phenotype. PLoS ONE 2016, 11, e0152105. [Google Scholar] [CrossRef] [PubMed]
- Linville, R.M.; DeStefano, J.G.; Sklar, M.B.; Xu, Z.; Farrell, A.M.; Bogorad, M.I.; Chu, C.; Walczak, P.; Cheng, L.; Mahairaki, V.; et al. Human iPSC-derived blood-brain barrier microvessels: Validation of barrier function and endothelial cell behavior. Biomaterials 2019, 190-191, 24–37. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef]
- Byfield, S.D.; Roberts, A.B. Lateral signaling enhances TGF-beta response complexity. Trends Cell Biol. 2004, 14, 107–111. [Google Scholar] [CrossRef]
- Wadman, M. FDA no longer has to require animal testing for new drugs. Science 2023, 379, 127–128. [Google Scholar] [CrossRef]
- Middelkamp, H.H.T.; Verboven, A.H.A.; De Sa Vivas, A.G.; Schoenmaker, C.; Klein Gunnewiek, T.M.; Passier, R.; Albers, C.A.; t Hoen, P.A.C.; Nadif Kasri, N.; van der Meer, A.D. Cell type-specific changes in transcriptomic profiles of endothelial cells, iPSC-derived neurons and astrocytes cultured on microfluidic chips. Sci. Rep. 2021, 11, 2281. [Google Scholar] [CrossRef] [PubMed]
- Discher, D.E.; Janmey, P.; Wang, Y.L. Tissue cells feel and respond to the stiffness of their substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Zhou, Q.; Liu, C.; Zhang, Y.; Xie, M.; Qiao, W.; Dong, N. Substrate stiffness regulates differentiation of induced pluripotent stem cells into heart valve endothelial cells. Acta Biomater. 2022, 143, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.H.; Kang, D.; Seo, S.J.; Jin, Y. Engineering the Extracellular Matrix for Organoid Culture. Int. J. Stem Cells 2022, 15, 60–69. [Google Scholar] [CrossRef]
- Sedlmeier, G.; Sleeman, J.P. Extracellular regulation of BMP signaling: Welcome to the matrix. Biochem. Soc. Trans. 2017, 45, 173–181. [Google Scholar] [CrossRef]
- Xiang, Y.; Miller, K.; Guan, J.; Kiratitanaporn, W.; Tang, M.; Chen, S. 3D bioprinting of complex tissues in vitro: State-of-the-art and future perspectives. Arch. Toxicol. 2022, 96, 691–710. [Google Scholar] [CrossRef]
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordonez-Moran, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, 560–564. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Duffhues, G.; Hiepen, C. Human iPSCs as Model Systems for BMP-Related Rare Diseases. Cells 2023, 12, 2200. https://doi.org/10.3390/cells12172200
Sánchez-Duffhues G, Hiepen C. Human iPSCs as Model Systems for BMP-Related Rare Diseases. Cells. 2023; 12(17):2200. https://doi.org/10.3390/cells12172200
Chicago/Turabian StyleSánchez-Duffhues, Gonzalo, and Christian Hiepen. 2023. "Human iPSCs as Model Systems for BMP-Related Rare Diseases" Cells 12, no. 17: 2200. https://doi.org/10.3390/cells12172200
APA StyleSánchez-Duffhues, G., & Hiepen, C. (2023). Human iPSCs as Model Systems for BMP-Related Rare Diseases. Cells, 12(17), 2200. https://doi.org/10.3390/cells12172200