
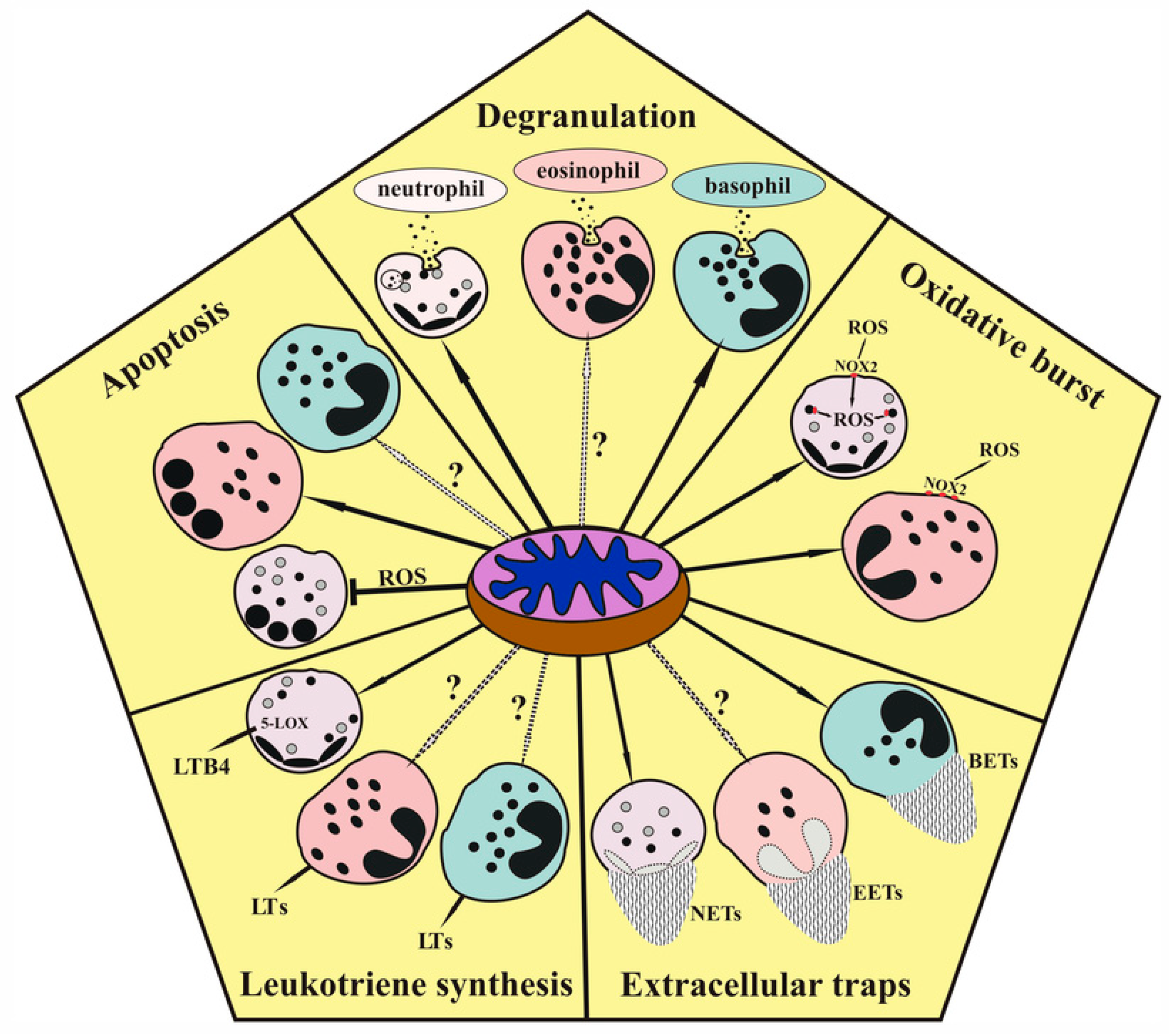
Role of Mitochondria in the Regulation of Effector Functions of Granulocytes

 ,
,  ,
,  , and
, and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Degranulation
3. Oxidative Burst
4. The Role of Mitochondria in Extracellular Trap Formation
4.1. Neutrophil Extracellular Traps (NETs) and NETosis
4.2. The Mechanisms of Eosinophil and Basophil Extracellular Trap Formation
5. Leukotriene Synthesis in Granulocytes
6. Apoptosis in Granulocytes
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Karnovsky, M.L. The Metabolism of Leukocytes. Semin. Hematol. 1968, 5, 156–165. [Google Scholar] [PubMed]
- Furuno, T.; Ohyama, N.; Nakanishi, M. The Relation between Degranulation and Rapid Metabolic Responses in RBL-2H3 Cells. Biol. Pharm. Bull. 1999, 22, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Espinosa, O.; Rojas-Espinosa, O.; Moreno-Altamirano, M.M.B.; López-Villegas, E.O.; Sánchez-García, F.J. Metabolic Requirements for Neutrophil Extracellular Traps Formation. Immunology 2015, 145, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Porter, L.; Toepfner, N.; Bashant, K.R.; Guck, J.; Ashcroft, M.; Farahi, N.; Chilvers, E.R. Metabolic Profiling of Human Eosinophils. Front. Immunol. 2018, 9, 1404. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Ledderose, C.; Seier, T.; Graf, A.F.; Brix, B.; Chong, E.; Junger, W.G. Mitochondria Regulate Neutrophil Activation by Generating ATP for Autocrine Purinergic Signaling. J. Biol. Chem. 2014, 289, 26794–26803. [Google Scholar] [CrossRef]
- Sumbayev, V.V.; Nicholas, S.A.; Streatfield, C.L.; Gibbs, B.F. Involvement of Hypoxia-Inducible Factor-1 HiF(1alpha) in IgE-Mediated Primary Human Basophil Responses. Eur. J. Immunol. 2009, 39, 3511–3519. [Google Scholar] [CrossRef]
- Sharkia, I.; Erlich, T.H.; Landolina, N.; Assayag, M.; Motzik, A.; Rachmin, I.; Kay, G.; Porat, Z.; Tshori, S.; Berkman, N.; et al. Pyruvate Dehydrogenase Has a Major Role in Mast Cell Function, and Its Activity Is Regulated by Mitochondrial Microphthalmia Transcription Factor. J. Allergy Clin. Immunol. 2017, 140, 204–214.e8. [Google Scholar] [CrossRef]
- Pavlyuchenkova, A.N.; Zinovkin, R.A.; Makievskaya, C.I.; Galkin, I.I.; Chelombitko, M.A. Mitochondria-Targeted Triphenylphosphonium-Based Compounds Inhibit FcεRI-Dependent Degranulation of Mast Cells by Preventing Mitochondrial Dysfunction through Erk1/2. Life Sci. 2022, 288, 120174. [Google Scholar] [CrossRef]
- Korchak, H.M.; Rich, A.M.; Wilkenfeld, C.; Rutherford, L.E.; Weissmann, G. A Carbocyanine Dye, DiOC6(3), Acts as a Mitochondrial Probe in Human Neutrophils. Biochem. Biophys. Res. Commun. 1982, 108, 1495–1501. [Google Scholar] [CrossRef]
- van Raam, B.J.; Sluiter, W.; de Wit, E.; Roos, D.; Verhoeven, A.J.; Kuijpers, T.W. Mitochondrial Membrane Potential in Human Neutrophils Is Maintained by Complex III Activity in the Absence of Supercomplex Organisation. PLoS ONE 2008, 3, e2013. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, M.; Meng, X.; Chu, X.; Cai, C.; Zou, F. Phosphorylation of Dynamin-Related Protein 1 at Ser616 Regulates Mitochondrial Fission and Is Involved in Mitochondrial Calcium Uniporter-Mediated Neutrophil Polarization and Chemotaxis. Mol. Immunol. 2017, 87, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Bonjour, K.; Palazzi, C.; Silva, T.P.; Malta, K.K.; Neves, V.H.; Oliveira-Barros, E.G.; Neves, I.; Kersten, V.A.; Fortuna, B.T.; Samarasinghe, A.E.; et al. Mitochondrial Population in Mouse Eosinophils: Ultrastructural Dynamics in Cell Differentiation and Inflammatory Diseases. Front. Cell Dev. Biol. 2022, 10, 836755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Alysandratos, K.-D.; Angelidou, A.; Asadi, S.; Sismanopoulos, N.; Delivanis, D.-A.; Weng, Z.; Miniati, A.; Vasiadi, M.; Katsarou-Katsari, A.; et al. Human Mast Cell Degranulation and Preformed TNF Secretion Require Mitochondrial Translocation to Exocytosis Sites: Relevance to Atopic Dermatitis. J. Allergy Clin. Immunol. 2011, 127, 1522–1531.e8. [Google Scholar] [CrossRef]
- Grivennikova, V.G.; Vinogradov, A.D. Mitochondrial Production of Reactive Oxygen Species. Biochemistry 2013, 78, 1490–1511. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.C. The Phagocyte Respiratory Burst: Historical Perspectives and Recent Advances. Immunol. Lett. 2017, 192, 88–96. [Google Scholar] [CrossRef]
- de Boer, M.; Roos, D. Metabolic Comparison between Basophils and Other Leukocytes from Human Blood. J. Immunol. 1986, 136, 3447–3454. [Google Scholar] [CrossRef]
- Morshed, M.; Hlushchuk, R.; Simon, D.; Walls, A.F.; Obata-Ninomiya, K.; Karasuyama, H.; Djonov, V.; Eggel, A.; Kaufmann, T.; Simon, H.-U.; et al. NADPH Oxidase-Independent Formation of Extracellular DNA Traps by Basophils. J. Immunol. 2014, 192, 5314–5323. [Google Scholar] [CrossRef]
- Petreccia, D.C.; Nauseef, W.M.; Clark, R.A. Respiratory Burst of Normal Human Eosinophils. J. Leukoc. Biol. 1987, 41, 283–288. [Google Scholar] [CrossRef]
- Lacy, P.; Abdel-Latif, D.; Steward, M.; Musat-Marcu, S.; Man, S.F.P.; Moqbel, R. Divergence of Mechanisms Regulating Respiratory Burst in Blood and Sputum Eosinophils and Neutrophils from Atopic Subjects. J. Immunol. 2003, 170, 2670–2679. [Google Scholar] [CrossRef]
- LeSuer, W.E.; Kienzl, M.; Ochkur, S.I.; Schicho, R.; Doyle, A.D.; Wright, B.L.; Rank, M.A.; Krupnick, A.S.; Kita, H.; Jacobsen, E.A. Eosinophils Promote Effector Functions of Lung Group 2 Innate Lymphoid Cells in Allergic Airway Inflammation in Mice. J. Allergy Clin. Immunol. 2023, 152, 469–485.e10. [Google Scholar] [CrossRef]
- Tecchio, C.; Micheletti, A.; Cassatella, M.A. Neutrophil-Derived Cytokines: Facts beyond Expression. Front. Immunol. 2014, 5, 508. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-Derived Suppressor Cells Coming of Age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Zhao, Y.; Rahmy, S.; Liu, Z.; Zhang, C.; Lu, X. Rational Targeting of Immunosuppressive Neutrophils in Cancer. Pharmacol. Ther. 2020, 212, 107556. [Google Scholar] [CrossRef]
- Cheng, G.; Hardy, M.; Topchyan, P.; Zander, R.; Volberding, P.; Cui, W.; Kalyanaraman, B. Mitochondria-Targeted Hydroxyurea Inhibits OXPHOS and Induces Antiproliferative and Immunomodulatory Effects. iScience 2021, 24, 102673. [Google Scholar] [CrossRef]
- Miyake, K.; Shibata, S.; Yoshikawa, S.; Karasuyama, H. Basophils and Their Effector Molecules in Allergic Disorders. Allergy 2021, 76, 1693–1706. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, V.V.; Pletjushkina, O.Y.; Galkin, I.I.; Zinovkin, R.A.; Chernyak, B.V.; Krysko, D.V.; Bachert, C.; Krysko, O.; Skulachev, V.P.; Popova, E.N. Low Concentration of Uncouplers of Oxidative Phosphorylation Decreases the TNF-Induced Endothelial Permeability and Lethality in Mice. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Tani, Y.; Nakanishi, H.; Taguchi, R.; Arita, M.; Arai, H. Eosinophils Promote Resolution of Acute Peritonitis by Producing Proresolving Mediators in Mice. FASEB J. 2011, 25, 561–568. [Google Scholar] [CrossRef]
- Ogawa, M.; Ishihara, T.; Isobe, Y.; Kato, T.; Kuba, K.; Imai, Y.; Uchino, Y.; Tsubota, K.; Arita, M. Eosinophils Promote Corneal Wound Healing via the 12/15-Lipoxygenase Pathway. FASEB J. 2020, 34, 12492–12501. [Google Scholar] [CrossRef] [PubMed]
- Sud’ina, G.F.; Golenkina, E.A.; Prikhodko, A.S.; Kondratenko, N.D.; Gaponova, T.V.; Chernyak, B.V. Mitochondria-Targeted Antioxidant SkQ1 Inhibits Leukotriene Synthesis in Human Neutrophils. Front. Pharmacol. 2022, 13, 1023517. [Google Scholar] [CrossRef]
- Koenderman, L.; Tesselaar, K.; Vrisekoop, N. Human Neutrophil Kinetics: A Call to Revisit Old Evidence. Trends Immunol. 2022, 43, 868–876. [Google Scholar] [CrossRef]
- Park, Y.M.; Bochner, B.S. Eosinophil Survival and Apoptosis in Health and Disease. Allergy Asthma Immunol. Res. 2010, 2, 87–101. [Google Scholar] [CrossRef]
- Ohnmacht, C.; Voehringer, D. Basophil Effector Function and Homeostasis during Helminth Infection. Blood 2009, 113, 2816–2825. [Google Scholar] [CrossRef]
- Wedi, B.; Straede, J.; Wieland, B.; Kapp, A. Eosinophil Apoptosis Is Mediated by Stimulators of Cellular Oxidative Metabolisms and Inhibited by Antioxidants: Involvement of a Thiol-Sensitive Redox Regulation in Eosinophil Cell Death. Blood J. Am. Soc. Hematol. 1999, 94, 2365–2373. [Google Scholar] [CrossRef]
- Ilmarinen, P.; Moilanen, E.; Kankaanranta, H. Mitochondria in the Center of Human Eosinophil Apoptosis and Survival. Int. J. Mol. Sci. 2014, 15, 3952–3969. [Google Scholar] [CrossRef] [PubMed]
- Ramadass, M.; Catz, S.D. Molecular Mechanisms Regulating Secretory Organelles and Endosomes in Neutrophils and Their Implications for Inflammation. Immunol. Rev. 2016, 273, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N.; Cowland, J.B. Granules of the Human Neutrophilic Polymorphonuclear Leukocyte. Blood 1997, 89, 3503–3521. [Google Scholar] [CrossRef] [PubMed]
- Rørvig, S.; Honore, C.; Larsson, L.-I.; Ohlsson, S.; Pedersen, C.C.; Jacobsen, L.C.; Cowland, J.B.; Garred, P.; Borregaard, N. Ficolin-1 Is Present in a Highly Mobilizable Subset of Human Neutrophil Granules and Associates with the Cell Surface after Stimulation with fMLP. J. Leukoc. Biol. 2009, 86, 1439–1449. [Google Scholar] [CrossRef]
- Simon, H.-U.; Yousefi, S.; Germic, N.; Arnold, I.C.; Haczku, A.; Karaulov, A.V.; Simon, D.; Rosenberg, H.F. The Cellular Functions of Eosinophils: Collegium Internationale Allergologicum (CIA) Update 2020. Int. Arch. Allergy Immunol. 2020, 181, 11–23. [Google Scholar] [CrossRef]
- Gigon, L.; Yousefi, S.; Karaulov, A.; Simon, H.-U. Mechanisms of Toxicity Mediated by Neutrophil and Eosinophil Granule Proteins. Allergol. Int. 2021, 70, 30–38. [Google Scholar] [CrossRef]
- Varricchi, G.; Raap, U.; Rivellese, F.; Marone, G.; Gibbs, B.F. Human Mast Cells and Basophils-How Are They Similar How Are They Different? Immunol. Rev. 2018, 282, 8–34. [Google Scholar] [CrossRef]
- Lollike, K.; Lindau, M.; Calafat, J.; Borregaard, N. Compound Exocytosis of Granules in Human Neutrophils. J. Leukoc. Biol. 2002, 71, 973–980. [Google Scholar] [CrossRef]
- Spencer, L.A.; Bonjour, K.; Melo, R.C.N.; Weller, P.F. Eosinophil Secretion of Granule-Derived Cytokines. Front. Immunol. 2014, 5, 496. [Google Scholar] [CrossRef]
- Belambri, S.A.; Rolas, L.; Raad, H.; Hurtado-Nedelec, M.; Dang, P.M.-C.; El-Benna, J. NADPH Oxidase Activation in Neutrophils: Role of the Phosphorylation of Its Subunits. Eur. J. Clin. Investig. 2018, 48 (Suppl. S2), e12951. [Google Scholar] [CrossRef]
- Sengeløv, H.; Kjeldsen, L.; Borregaard, N. Control of Exocytosis in Early Neutrophil Activation. J. Immunol. 1993, 150, 1535–1543. [Google Scholar] [CrossRef]
- Fumagalli, L.; Zhang, H.; Baruzzi, A.; Lowell, C.A.; Berton, G. The Src Family Kinases Hck and Fgr Regulate Neutrophil Responses to N-Formyl-Methionyl-Leucyl-Phenylalanine. J. Immunol. 2007, 178, 3874–3885. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, H.; Xie, W.; Zhang, Z.; Smrcka, A.V.; Wu, D. Roles of PLC-beta2 and -beta3 and PI3Kgamma in Chemoattractant-Mediated Signal Transduction. Science 2000, 287, 1046–1049. [Google Scholar] [CrossRef]
- Adachi, T.; Choudhury, B.K.; Stafford, S.; Sur, S.; Alam, R. The Differential Role of Extracellular Signal-Regulated Kinases and p38 Mitogen-Activated Protein Kinase in Eosinophil Functions. J. Immunol. 2000, 165, 2198–2204. [Google Scholar] [CrossRef]
- Kämpe, M.; Lampinen, M.; Stolt, I.; Janson, C.; Stålenheim, G.; Carlson, M. PI3-Kinase Regulates Eosinophil and Neutrophil Degranulation in Patients with Allergic Rhinitis and Allergic Asthma Irrespective of Allergen Challenge Model. Inflammation 2012, 35, 230–239. [Google Scholar] [CrossRef]
- Fossati, G.; Moulding, D.A.; Spiller, D.G.; Moots, R.J.; White, M.R.H.; Edwards, S.W. The Mitochondrial Network of Human Neutrophils: Role in Chemotaxis, Phagocytosis, Respiratory Burst Activation, and Commitment to Apoptosis. J. Immunol. 2003, 170, 1964–1972. [Google Scholar] [CrossRef]
- Antonenko, Y.N.; Avetisyan, A.V.; Bakeeva, L.E.; Chernyak, B.V.; Chertkov, V.A.; Domnina, L.V.; Ivanova, O.Y.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; et al. Mitochondria-Targeted Plastoquinone Derivatives as Tools to Interrupt Execution of the Aging Program. 1. Cationic Plastoquinone Derivatives: Synthesis and In Vitro Studies. Biochemistry 2008, 73, 1273–1287. [Google Scholar] [CrossRef]
- Vorobjeva, N.; Prikhodko, A.; Galkin, I.; Pletjushkina, O.; Zinovkin, R.; Sud’ina, G.; Chernyak, B.; Pinegin, B. Mitochondrial Reactive Oxygen Species Are Involved in Chemoattractant-Induced Oxidative Burst and Degranulation of Human Neutrophils in Vitro. Eur. J. Cell Biol. 2017, 96, 254–265. [Google Scholar] [CrossRef]
- Henderson, W.R.; Kaliner, M. Immunologic and Nonimmunologic Generation of Superoxide from Mast Cells and Basophils. J. Clin. Investig. 1978, 61, 187–196. [Google Scholar] [CrossRef]
- Kitagawa, S.; Takaku, F.; Sakamoto, S. Serine Protease Inhibitors Inhibit Superoxide Production by Human Basophils Stimulated by Anti-IgE. Biochem. Biophys. Res. Commun. 1980, 95, 801–806. [Google Scholar] [CrossRef]
- Falcone, F.H.; Wan, D.; Barwary, N.; Sagi-Eisenberg, R. RBL Cells as Models for in Vitro Studies of Mast Cells and Basophils. Immunol. Rev. 2018, 282, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Yoshimaru, T.; Suzuki, Y.; Matsui, T.; Yamashita, K.; Ochiai, T.; Yamaki, M.; Shimizu, K. Blockade of Superoxide Generation Prevents High-Affinity Immunoglobulin E Receptor-Mediated Release of Allergic Mediators by Rat Mast Cell Line and Human Basophils. Clin. Exp. Allergy 2002, 32, 612–618. [Google Scholar] [CrossRef]
- Inoue, T.; Suzuki, Y.; Yoshimaru, T.; Ra, C. Reactive Oxygen Species Produced up- or Downstream of Calcium Influx Regulate Proinflammatory Mediator Release from Mast Cells: Role of NADPH Oxidase and Mitochondria. Biochim. Biophys. Acta 2008, 1783, 789–802. [Google Scholar] [CrossRef]
- Pletjushkina, O.Y.; Lyamzaev, K.G.; Popova, E.N.; Nepryakhina, O.K.; Ivanova, O.Y.; Domnina, L.V.; Chernyak, B.V.; Skulachev, V.P. Effect of Oxidative Stress on Dynamics of Mitochondrial Reticulum. Biochim. Biophys. Acta 2006, 1757, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P.; Antonenko, Y.N.; Cherepanov, D.A.; Chernyak, B.V.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; Korshunova, G.A.; Lyamzaev, K.G.; Pletjushkina, O.Y.; et al. Prevention of Cardiolipin Oxidation and Fatty Acid Cycling as Two Antioxidant Mechanisms of Cationic Derivatives of Plastoquinone (SkQs). Biochim. Biophys. Acta 2010, 1797, 878–889. [Google Scholar] [CrossRef]
- Pavlyuchenkova, A.N.; Chelombitko, M.A.; Fedorov, A.V.; Kuznetsova, M.K.; Zinovkin, R.A.; Razin, E. The Distinct Effects of the Mitochondria-Targeted STAT3 Inhibitors Mitocur-1 and Mitocur-3 on Mast Cell and Mitochondrial Functions. Int. J. Mol. Sci. 2023, 24, 1471. [Google Scholar] [CrossRef]
- Steinberg, S.F. Mechanisms for Redox-Regulation of Protein Kinase C. Front. Pharmacol. 2015, 6, 128. [Google Scholar] [CrossRef]
- Yoo, S.K.; Starnes, T.W.; Deng, Q.; Huttenlocher, A. Lyn Is a Redox Sensor That Mediates Leukocyte Wound Attraction in Vivo. Nature 2011, 480, 109–112. [Google Scholar] [CrossRef]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular Mechanisms of Angiotensin II-Mediated Mitochondrial Dysfunction: Linking Mitochondrial Oxidative Damage and Vascular Endothelial Dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef]
- Nazarewicz, R.R.; Dikalova, A.E.; Bikineyeva, A.; Dikalov, S.I. Nox2 as a Potential Target of Mitochondrial Superoxide and Its Role in Endothelial Oxidative Stress. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1131–H1140. [Google Scholar] [CrossRef]
- El Jamali, A.; Valente, A.J.; Clark, R.A. Regulation of Phagocyte NADPH Oxidase by Hydrogen Peroxide through a Ca(2+)/c-Abl Signaling Pathway. Free Radic. Biol. Med. 2010, 48, 798–810. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Li, W.; Doughan, A.K.; Blanco, R.R.; Zafari, A.M. Mitochondrial Reactive Oxygen Species and Calcium Uptake Regulate Activation of Phagocytic NADPH Oxidase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R1134–R1142. [Google Scholar] [CrossRef]
- Kröller-Schön, S.; Steven, S.; Kossmann, S.; Scholz, A.; Daub, S.; Oelze, M.; Xia, N.; Hausding, M.; Mikhed, Y.; Zinssius, E.; et al. Molecular Mechanisms of the Crosstalk between Mitochondria and NADPH Oxidase through Reactive Oxygen Species-Studies in White Blood Cells and in Animal Models. Antioxid. Redox Signal. 2014, 20, 247–266. [Google Scholar] [CrossRef]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef]
- Itani, H.A.; Dikalova, A.E.; McMaster, W.G.; Nazarewicz, R.R.; Bikineyeva, A.T.; Harrison, D.G.; Dikalov, S.I. Mitochondrial Cyclophilin D in Vascular Oxidative Stress and Hypertension. Hypertension 2016, 67, 1218–1227. [Google Scholar] [CrossRef]
- Vorobjeva, N.; Galkin, I.; Pletjushkina, O.; Golyshev, S.; Zinovkin, R.; Prikhodko, A.; Pinegin, V.; Kondratenko, I.; Pinegin, B.; Chernyak, B. Mitochondrial Permeability Transition Pore Is Involved in Oxidative Burst and NETosis of Human Neutrophils. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165664. [Google Scholar] [CrossRef]
- Dumas, J.F.; Argaud, L.; Cottet-Rousselle, C.; Vial, G.; Gonzalez, C.; Detaille, D.; Leverve, X.; Fontaine, E. Effect of Transient and Permanent Permeability Transition Pore Opening on NAD(P)H Localization in Intact Cells. J. Biol. Chem. 2009, 284, 15117–15125. [Google Scholar] [CrossRef]
- Trevelin, S.C.; Lopes, L.R. Protein Disulfide Isomerase and Nox: New Partners in Redox Signaling. Curr. Pharm. Des. 2015, 21, 5951–5963. [Google Scholar] [CrossRef] [PubMed]
- Marciano, B.E.; Spalding, C.; Fitzgerald, A.; Mann, D.; Brown, T.; Osgood, S.; Yockey, L.; Darnell, D.N.; Barnhart, L.; Daub, J.; et al. Common Severe Infections in Chronic Granulomatous Disease. Clin. Infect. Dis. 2015, 60, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Lok, L.S.C.; Clatworthy, M.R. Neutrophils in Secondary Lymphoid Organs. Immunology 2021, 164, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Bode, K.; Hauri-Hohl, M.; Jaquet, V.; Weyd, H. Unlocking the Power of NOX2: A Comprehensive Review on Its Role in Immune Regulation. Redox Biol. 2023, 64, 102795. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Steinberg, B.E.; Grinstein, S. Unconventional Roles of the NADPH Oxidase: Signaling, Ion Homeostasis, and Cell Death. Sci. STKE 2007, 2007, e11. [Google Scholar] [CrossRef]
- Vorobjeva, N.V.; Pinegin, B.V. Neutrophil Extracellular Traps: Mechanisms of Formation and Role in Health and Disease. Biochemistry 2014, 79, 1286–1296. [Google Scholar] [CrossRef]
- Pinegin, B.; Vorobjeva, N.; Pinegin, V. Neutrophil Extracellular Traps and Their Role in the Development of Chronic Inflammation and Autoimmunity. Autoimmun. Rev. 2015, 14, 633–640. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil Extracellular Traps in Immunity and Disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Vorobjeva, N.V.; Chernyak, B.V. NETosis: Molecular Mechanisms, Role in Physiology and Pathology. Biochemistry 2020, 85, 1178–1190. [Google Scholar] [CrossRef]
- Vorobjeva, N.V. Neutrophil Extracellular Traps: New Aspects. Moscow Univ. Biol. Sci. Bull. 2020, 75, 173–188. [Google Scholar] [CrossRef]
- Svistushkin, V.M.; Nikiforova, G.N.; Vorobjeva, N.V.; Dekhanov, A.S.; Dagil, Y.A.; Bredova, O.Y.; Eremeeva, K.V. Neutrophil extracellular traps in the pathogenesis of chronic rhinosinusitis. Vestn. Otorinolaringol. 2021, 86, 105–112. [Google Scholar] [CrossRef]
- Yousefi, S.; Gold, J.A.; Andina, N.; Lee, J.J.; Kelly, A.M.; Kozlowski, E.; Schmid, I.; Straumann, A.; Reichenbach, J.; Gleich, G.J.; et al. Catapult-like Release of Mitochondrial DNA by Eosinophils Contributes to Antibacterial Defense. Nat. Med. 2008, 14, 949–953. [Google Scholar] [CrossRef]
- von Köckritz-Blickwede, M.; Goldmann, O.; Thulin, P.; Heinemann, K.; Norrby-Teglund, A.; Rohde, M.; Medina, E. Phagocytosis-Independent Antimicrobial Activity of Mast Cells by Means of Extracellular Trap Formation. Blood 2008, 111, 3070–3080. [Google Scholar] [CrossRef] [PubMed]
- Ingelsson, B.; Söderberg, D.; Strid, T.; Söderberg, A.; Bergh, A.-C.; Loitto, V.; Lotfi, K.; Segelmark, M.; Spyrou, G.; Rosén, A. Lymphocytes Eject Interferogenic Mitochondrial DNA Webs in Response to CpG and Non-CpG Oligodeoxynucleotides of Class C. Proc. Natl. Acad. Sci. USA 2018, 115, E478–E487. [Google Scholar] [CrossRef] [PubMed]
- Granger, V.; Faille, D.; Marani, V.; Noël, B.; Gallais, Y.; Szely, N.; Flament, H.; Pallardy, M.; Chollet-Martin, S.; de Chaisemartin, L. Human Blood Monocytes Are Able to Form Extracellular Traps. J. Leukoc. Biol. 2017, 102, 775–781. [Google Scholar] [CrossRef]
- Chow, O.A.; von Köckritz-Blickwede, M.; Bright, A.T.; Hensler, M.E.; Zinkernagel, A.S.; Cogen, A.L.; Gallo, R.L.; Monestier, M.; Wang, Y.; Glass, C.K.; et al. Statins Enhance Formation of Phagocyte Extracellular Traps. Cell Host Microbe 2010, 8, 445–454. [Google Scholar] [CrossRef]
- Yipp, B.G.; Kubes, P. NETosis: How Vital Is It? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable Neutrophils Release Mitochondrial DNA to Form Neutrophil Extracellular Traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Z.; Li, X.; Dong, G.; Zhang, M.; Xu, Z.; Yang, J. Neutrophil Extracellular Traps: Signaling Properties and Disease Relevance. Mediators Inflamm. 2020, 2020, 9254087. [Google Scholar] [CrossRef]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.D.; Lucas, C.D.; MacKellar, A.; Li, F.; Hiersemenzel, K.; Haslett, C.; Davidson, D.J.; Rossi, A.G. Activation of Conventional Protein Kinase C (PKC) Is Critical in the Generation of Human Neutrophil Extracellular Traps. J. Inflamm. 2013, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Vorobjeva, N.V.; Vakhlyarskaya, S.S.; Chernyak, B.V. The Role of Protein Kinase C Isoforms in the Formation of Neutrophil Extracellular Traps. Moscow Univ. Biol. Sci. Bull. 2022, 77, 112–118. [Google Scholar] [CrossRef]
- Vorobjeva, N.; Dagil, Y.; Pashenkov, M.; Pinegin, B.; Chernyak, B. Protein Kinase C Isoforms Mediate the Formation of Neutrophil Extracellular Traps. Int. Immunopharmacol. 2023, 114, 109448. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Knackstedt, S.L.; Abu Abed, U.; Deigendesch, N.; Harbort, C.J.; Caffrey, B.E.; Brinkmann, V.; Heppner, F.L.; Hinds, P.W.; Zychlinsky, A. Cell-Cycle Proteins Control Production of Neutrophil Extracellular Traps. Dev. Cell 2017, 43, 449–462.e5. [Google Scholar] [CrossRef]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK Pathway Is Required for Neutrophil Extracellular Trap Formation. Nat. Chem. Biol. 2011, 7, 75–77. [Google Scholar] [CrossRef]
- Vorobjeva, N.V. Participation of Non-Receptor Src-Family Tyrosine Kinases in the Formation of Neutrophil Extracellular Traps. Mosc. Univ. Biol. Sci. Bull. 2023, 78, 8–13. [Google Scholar] [CrossRef]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus Aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef]
- Parker, H.; Dragunow, M.; Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Requirements for NADPH Oxidase and Myeloperoxidase in Neutrophil Extracellular Trap Formation Differ Depending on the Stimulus. J. Leukoc. Biol. 2012, 92, 841–849. [Google Scholar] [CrossRef]
- Arai, Y.; Nishinaka, Y.; Arai, T.; Morita, M.; Mizugishi, K.; Adachi, S.; Takaori-Kondo, A.; Watanabe, T.; Yamashita, K. Uric Acid Induces NADPH Oxidase-Independent Neutrophil Extracellular Trap Formation. Biochem. Biophys. Res. Commun. 2014, 443, 556–561. [Google Scholar] [CrossRef]
- Pieterse, E.; Rother, N.; Yanginlar, C.; Gerretsen, J.; Boeltz, S.; Munoz, L.E.; Herrmann, M.; Pickkers, P.; Hilbrands, L.B.; van der Vlag, J. Cleaved N-Terminal Histone Tails Distinguish between NADPH Oxidase (NOX)-Dependent and NOX-Independent Pathways of Neutrophil Extracellular Trap Formation. Ann. Rheum. Dis. 2018, 77, 1790–1798. [Google Scholar] [CrossRef]
- de Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, Complement, and Coagulation: A Triangular Relationship. Cell. Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Tatsiy, O.; McDonald, P.P. Physiological Stimuli Induce PAD4-Dependent, ROS-Independent NETosis, With Early and Late Events Controlled by Discrete Signaling Pathways. Front. Immunol. 2018, 9, 2036. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Nishi, H.; Travers, R.; Tsuboi, N.; Martinod, K.; Wagner, D.D.; Stan, R.; Croce, K.; Mayadas, T.N. Endocytosis of Soluble Immune Complexes Leads to Their Clearance by FcγRIIIB but Induces Neutrophil Extracellular Traps via FcγRIIA In Vivo. Blood 2012, 120, 4421–4431. [Google Scholar] [CrossRef]
- Kenny, E.F.; Herzig, A.; Krüger, R.; Muth, A.; Mondal, S.; Thompson, P.R.; Brinkmann, V.; von Bernuth, H.; Zychlinsky, A. Diverse Stimuli Engage Different Neutrophil Extracellular Trap Pathways. Elife 2017, 6, e24437. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil Extracellular Traps Enriched in Oxidized Mitochondrial DNA Are Interferogenic and Contribute to Lupus-like Disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Vorobjeva, N.V.; Chernyak, B.V. NADPH Oxidase Modulates Ca2+-Dependent Formation of Neutrophil Extracellular Traps. Mosc. Univ. Biol. Sci. Bull. 2020, 75, 104–109. [Google Scholar] [CrossRef]
- Dunham-Snary, K.J.; Surewaard, B.G.; Mewburn, J.D.; Bentley, R.E.; Martin, A.Y.; Jones, O.; Al-Qazazi, R.; Lima, P.A.; Kubes, P.; Archer, S.L. Mitochondria in Human Neutrophils Mediate Killing of Staphylococcus Aureus. Redox Biol. 2022, 49, 102225. [Google Scholar] [CrossRef]
- Ueki, S.; Melo, R.C.N.; Ghiran, I.; Spencer, L.A.; Dvorak, A.M.; Weller, P.F. Eosinophil Extracellular DNA Trap Cell Death Mediates Lytic Release of Free Secretion-Competent Eosinophil Granules in Humans. Blood 2013, 121, 2074–2083. [Google Scholar] [CrossRef]
- Germic, N.; Fettrelet, T.; Stojkov, D.; Hosseini, A.; Horn, M.P.; Karaulov, A.; Simon, D.; Yousefi, S.; Simon, H.-U. The Release Kinetics of Eosinophil Peroxidase and Mitochondrial DNA Is Different in Association with Eosinophil Extracellular Trap Formation. Cells 2021, 10, 306. [Google Scholar] [CrossRef]
- Germic, N.; Stojkov, D.; Oberson, K.; Yousefi, S.; Simon, H.-U. Neither Eosinophils nor Neutrophils Require ATG5-Dependent Autophagy for Extracellular DNA Trap Formation. Immunology 2017, 152, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Rådmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase, a Key Enzyme for Leukotriene Biosynthesis in Health and Disease. Biochim. Biophys. Acta 2015, 1851, 331–339. [Google Scholar] [CrossRef]
- Funk, C.D. Prostaglandins and Leukotrienes: Advances in Eicosanoid Biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef]
- Ford-Hutchinson, A.W.; Bray, M.A.; Doig, M.V.; Shipley, M.E.; Smith, M.J. Leukotriene B, a Potent Chemokinetic and Aggregating Substance Released from Polymorphonuclear Leukocytes. Nature 1980, 286, 264–265. [Google Scholar] [CrossRef] [PubMed]
- Yokomizo, T.; Shimizu, T. The Leukotriene B4 Receptors BLT1 and BLT2 as Potential Therapeutic Targets. Immunol. Rev. 2023, 317, 30–41. [Google Scholar] [CrossRef]
- Afonso, P.V.; Janka-Junttila, M.; Lee, Y.J.; McCann, C.P.; Oliver, C.M.; Aamer, K.A.; Losert, W.; Cicerone, M.T.; Parent, C.A. LTB4 Is a Signal-Relay Molecule during Neutrophil Chemotaxis. Dev. Cell 2012, 22, 1079–1091. [Google Scholar] [CrossRef]
- Lämmermann, T.; Afonso, P.V.; Angermann, B.R.; Wang, J.M.; Kastenmüller, W.; Parent, C.A.; Germain, R.N. Neutrophil Swarms Require LTB4 and Integrins at Sites of Cell Death In Vivo. Nature 2013, 498, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Gresnigt, M.S.; Werz, O.; Hube, B.; Garscha, U. Candida Albicans-Induced Leukotriene Biosynthesis in Neutrophils Is Restricted to the Hyphal Morphology. FASEB J. 2021, 35, e21820. [Google Scholar] [CrossRef]
- Golenkina, E.A.; Galkina, S.I.; Pletjushkina, O.; Chernyak, B.; Gaponova, T.V.; Romanova, Y.M.; Sud’ina, G.F. Gram-Negative Bacteria Salmonella Typhimurium Boost Leukotriene Synthesis Induced by Chemoattractant fMLP to Stimulate Neutrophil Swarming. Front. Pharmacol. 2021, 12, 814113. [Google Scholar] [CrossRef]
- Lee, E.K.S.; Gillrie, M.R.; Li, L.; Arnason, J.W.; Kim, J.H.; Babes, L.; Lou, Y.; Sanati-Nezhad, A.; Kyei, S.K.; Kelly, M.M.; et al. Leukotriene B4-Mediated Neutrophil Recruitment Causes Pulmonary Capillaritis during Lethal Fungal Sepsis. Cell Host Microbe 2018, 23, 121–133.e4. [Google Scholar] [CrossRef]
- Gaudreault, E.; Thompson, C.; Stankova, J.; Rola-Pleszczynski, M. Involvement of BLT1 Endocytosis and Yes Kinase Activation in Leukotriene B4-Induced Neutrophil Degranulation. J. Immunol. 2005, 174, 3617–3625. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Chen, Y.; Cai, Q. The Role of the LTB4-BLT1 Axis in Health and Disease. Pharmacol. Res. 2020, 158, 104857. [Google Scholar] [CrossRef] [PubMed]
- Pal, K.; Feng, X.; Steinke, J.W.; Burdick, M.D.; Shim, Y.M.; Sung, S.-S.; Teague, W.G.; Borish, L. Leukotriene A4 Hydrolase Activation and Leukotriene B4 Production by Eosinophils in Severe Asthma. Am. J. Respir. Cell Mol. Biol. 2019, 60, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.S.; Rokach, J. Targeting the OXE Receptor as a Potential Novel Therapy for Asthma. Biochem. Pharmacol. 2020, 179, 113930. [Google Scholar] [CrossRef] [PubMed]
- Weller, P.F.; Lee, C.W.; Foster, D.W.; Corey, E.J.; Austen, K.F.; Lewis, R.A. Generation and Metabolism of 5-Lipoxygenase Pathway Leukotrienes by Human Eosinophils: Predominant Production of Leukotriene C4. Proc. Natl. Acad. Sci. USA 1983, 80, 7626–7630. [Google Scholar] [CrossRef]
- Warner, J.A.; Peters, S.P.; Lichtenstein, L.M.; Hubbard, W.; Yancey, K.B.; Stevenson, H.C.; Miller, P.J.; MacGlashan, D.W., Jr. Differential Release of Mediators from Human Basophils: Differences in Arachidonic Acid Metabolism Following Activation by Unrelated Stimuli. J. Leukoc. Biol. 1989, 45, 558–571. [Google Scholar] [CrossRef]
- Samuelsson, B. Leukotrienes: Mediators of Immediate Hypersensitivity Reactions and Inflammation. Science 1983, 220, 568–575. [Google Scholar] [CrossRef]
- Li, C.-T.; Zhang, W.-P.; Lu, Y.-B.; Fang, S.-H.; Yuan, Y.-M.; Qi, L.-L.; Zhang, L.-H.; Huang, X.-J.; Zhang, L.; Chen, Z.; et al. Oxygen-Glucose Deprivation Activates 5-Lipoxygenase Mediated by Oxidative Stress through the p38 Mitogen-Activated Protein Kinase Pathway in PC12 Cells. J. Neurosci. Res. 2009, 87, 991–1001. [Google Scholar] [CrossRef]
- Song, Z.; Huang, G.; Chiquetto Paracatu, L.; Grimes, D.; Gu, J.; Luke, C.J.; Clemens, R.A.; Dinauer, M.C. NADPH Oxidase Controls Pulmonary Neutrophil Infiltration in the Response to Fungal Cell Walls by Limiting LTB4. Blood 2020, 135, 891–903. [Google Scholar] [CrossRef]
- McGill, K.A.; Busse, W.W. Zileuton. Lancet 1996, 348, 519–524. [Google Scholar] [CrossRef]
- Wan, M.; Tang, X.; Stsiapanava, A.; Haeggström, J.Z. Biosynthesis of Leukotriene B4. Semin. Immunol. 2017, 33, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Figueroa, E.; Álvarez-Carrasco, P.; Ortega, E.; Maldonado-Bernal, C. Neutrophils: Many Ways to Die. Front. Immunol. 2021, 12, 631821. [Google Scholar] [CrossRef] [PubMed]
- McCracken, J.M.; Allen, L.-A.H. Regulation of Human Neutrophil Apoptosis and Lifespan in Health and Disease. J. Cell Death 2014, 7, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Burdon, P.C.E.; Bridger, G.; Gutierrez-Ramos, J.C.; Williams, T.J.; Rankin, S.M. Chemokines Acting via CXCR2 and CXCR4 Control the Release of Neutrophils from the Bone Marrow and Their Return Following Senescence. Immunity 2003, 19, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Henson, P.M. Cell Removal: Efferocytosis. Annu. Rev. Cell Dev. Biol. 2017, 33, 127–144. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and Apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Brunelle, J.K.; Letai, A. Control of Mitochondrial Apoptosis by the Bcl-2 Family. J. Cell Sci. 2009, 122, 437–441. [Google Scholar] [CrossRef]
- Jeng, P.S.; Inoue-Yamauchi, A.; Hsieh, J.J.; Cheng, E.H. BH3-Dependent and Independent Activation of BAX and BAK in Mitochondrial Apoptosis. Curr. Opin. Physiol. 2018, 3, 71–81. [Google Scholar] [CrossRef]
- Murphy, B.M.; O’Neill, A.J.; Adrain, C.; Watson, R.W.G.; Martin, S.J. The Apoptosome Pathway to Caspase Activation in Primary Human Neutrophils Exhibits Dramatically Reduced Requirements for Cytochrome C. J. Exp. Med. 2003, 197, 625–632. [Google Scholar] [CrossRef]
- Moulding, D.A.; Quayle, J.A.; Hart, C.A.; Edwards, S.W. Mcl-1 Expression in Human Neutrophils: Regulation by Cytokines and Correlation with Cell Survival. Blood 1998, 92, 2495–2502. [Google Scholar] [CrossRef]
- Weinmann, P.; Gaehtgens, P.; Walzog, B. Bcl-Xl– and Bax-□–Mediated Regulation of Apoptosis of Human Neutrophils Via Caspase-3. Blood 1999, 93, 3106–3115. [Google Scholar] [CrossRef] [PubMed]
- Leuenroth, S.J.; Grutkoski, P.S.; Ayala, A.; Simms, H.H. Suppression of PMN Apoptosis by Hypoxia Is Dependent on Mcl-1 and MAPK Activity. Surgery 2000, 128, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.B.; Buridi, A.; Coxon, P.Y.; Rane, M.J.; Manning, T.; Kettritz, R.; McLeish, K.R. Role of Extracellular Signal-Regulated Kinase and Phosphatidylinositol-3 Kinase in Chemoattractant and LPS Delay of Constitutive Neutrophil Apoptosis. Cell. Signal. 2001, 13, 335–343. [Google Scholar] [CrossRef]
- Derouet, M.; Thomas, L.; Cross, A.; Moots, R.J.; Edwards, S.W. Granulocyte Macrophage Colony-Stimulating Factor Signaling and Proteasome Inhibition Delay Neutrophil Apoptosis by Increasing the Stability of Mcl-1. J. Biol. Chem. 2004, 279, 26915–26921. [Google Scholar] [CrossRef]
- Rollet-Labelle, E.; Grange, M.J.; Elbim, C.; Marquetty, C.; Gougerot-Pocidalo, M.A.; Pasquier, C. Hydroxyl Radical as a Potential Intracellular Mediator of Polymorphonuclear Neutrophil Apoptosis. Free Radic. Biol. Med. 1998, 24, 563–572. [Google Scholar] [CrossRef]
- Kasahara, Y.; Iwai, K.; Yachie, A.; Ohta, K.; Konno, A.; Seki, H.; Miyawaki, T.; Taniguchi, N. Involvement of Reactive Oxygen Intermediates in Spontaneous and CD95 (Fas/APO-1)-Mediated Apoptosis of Neutrophils. Blood 1997, 89, 1748–1753. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.W.; Redmond, H.P.; Wang, J.H.; Condron, C.; Bouchier-Hayes, D. Neutrophils Undergo Apoptosis Following Ingestion of Escherichia Coli. J. Immunol. 1996, 156, 3986–3992. [Google Scholar] [CrossRef]
- Fadeel, B.; Ahlin, A.; Henter, J.I.; Orrenius, S.; Hampton, M.B. Involvement of Caspases in Neutrophil Apoptosis: Regulation by Reactive Oxygen Species. Blood 1998, 92, 4808–4818. [Google Scholar] [CrossRef]
- Conus, S.; Perozzo, R.; Reinheckel, T.; Peters, C.; Scapozza, L.; Yousefi, S.; Simon, H.-U. Caspase-8 Is Activated by Cathepsin D Initiating Neutrophil Apoptosis during the Resolution of Inflammation. J. Exp. Med. 2008, 205, 685–698. [Google Scholar] [CrossRef]
- Watson, R.W.G. Redox Regulation of Neutrophil Apoptosis. Antioxid. Redox Signal. 2002, 4, 97–104. [Google Scholar] [CrossRef]
- Asehnoune, K.; Strassheim, D.; Mitra, S.; Kim, J.Y.; Abraham, E. Involvement of Reactive Oxygen Species in Toll-like Receptor 4-Dependent Activation of NF-Kappa B. J. Immunol. 2004, 172, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-de-Souza, P.; Canetti, C.; Barja-Fidalgo, C.; Arruda, M.A. Leukotriene B4 Inhibits Neutrophil Apoptosis via NADPH Oxidase Activity: Redox Control of NF-κB Pathway and Mitochondrial Stability. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2012, 1823, 1990–1997. [Google Scholar] [CrossRef] [PubMed]
- Andreev-Andrievskiy, A.A.; Kolosova, N.G.; Stefanova, N.A.; Lovat, M.V.; Egorov, M.V.; Manskikh, V.N.; Zinovkin, R.A.; Galkin, I.I.; Prikhodko, A.S.; Skulachev, M.V.; et al. Efficacy of Mitochondrial Antioxidant Plastoquinonyl-Decyl-Triphenylphosphonium Bromide (SkQ1) in the Rat Model of Autoimmune Arthritis. Oxid. Med. Cell. Longev. 2016, 2016, 8703645. [Google Scholar] [CrossRef]
- Willson, J.A.; Arienti, S.; Sadiku, P.; Reyes, L.; Coelho, P.; Morrison, T.; Rinaldi, G.; Dockrell, D.H.; Whyte, M.K.B.; Walmsley, S.R. Neutrophil HIF-1α Stabilization Is Augmented by Mitochondrial ROS Produced via the Glycerol 3-Phosphate Shuttle. Blood 2022, 139, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Nakamura, Y.; Inoue, F.; Yamamoto, K.; Saito, K.; Furusawa, S. Glucocorticoid-Induced Apoptotic Pathways in Eosinophils: Comparison with Glucocorticoid-Sensitive Leukemia Cells. Int. J. Hematol. 2000, 71, 340–349. [Google Scholar]
- Ilmarinen-Salo, P.; Moilanen, E.; Kinnula, V.L.; Kankaanranta, H. Nitric Oxide-Induced Eosinophil Apoptosis Is Dependent on Mitochondrial Permeability Transition (mPT), JNK and Oxidative Stress: Apoptosis Is Preceded but Not Mediated by Early mPT-Dependent JNK Activation. Respir. Res. 2012, 13, 73. [Google Scholar] [CrossRef]
- Martinez-Losa, M.; Cortijo, J.; Juan, G.; Ramón, M.; Sanz, M.J.; Morcillo, E.J. Modulatory Effects of N-Acetyl-L-Cysteine on Human Eosinophil Apoptosis. Eur. Respir. J. 2007, 30, 436–442. [Google Scholar] [CrossRef]
- Gardai, S.J.; Hoontrakoon, R.; Goddard, C.D.; Day, B.J.; Chang, L.Y.; Henson, P.M.; Bratton, D.L. Oxidant-Mediated Mitochondrial Injury in Eosinophil Apoptosis: Enhancement by Glucocorticoids and Inhibition by Granulocyte-Macrophage Colony-Stimulating Factor. J. Immunol. 2003, 170, 556–566. [Google Scholar] [CrossRef]
- Lee, Y.A.; Shin, M.H. Mitochondrial Respiration Is Required for Activation of ERK1/2 and Caspase-3 in Human Eosinophils Stimulated with Hydrogen Peroxide. J. Investig. Allergol. Clin. Immunol. 2009, 19, 188–194. [Google Scholar]
- Groth, C.; Weber, R.; Lasser, S.; Özbay, F.G.; Kurzay, A.; Petrova, V.; Altevogt, P.; Utikal, J.; Umansky, V. Tumor Promoting Capacity of Polymorphonuclear Myeloid-Derived Suppressor Cells and Their Neutralization. Int. J. Cancer 2021, 149, 1628–1638. [Google Scholar] [CrossRef]
- Schmielau, J.; Finn, O.J. Activated Granulocytes and Granulocyte-Derived Hydrogen Peroxide Are the Underlying Mechanism of Suppression of T-Cell Function in Advanced Cancer Patients. Cancer Res. 2001, 61, 4756–4760. [Google Scholar] [PubMed]
- Bhardwaj, V.; Ansell, S.M. Modulation of T-Cell Function by Myeloid-Derived Suppressor Cells in Hematological Malignancies. Front. Cell Dev. Biol. 2023, 11, 1129343. [Google Scholar] [CrossRef]
- Yazdani, H.O.; Roy, E.; Comerci, A.J.; van der Windt, D.J.; Zhang, H.; Huang, H.; Loughran, P.; Shiva, S.; Geller, D.A.; Bartlett, D.L.; et al. Neutrophil Extracellular Traps Drive Mitochondrial Homeostasis in Tumors to Augment Growth. Cancer Res. 2019, 79, 5626–5639. [Google Scholar] [CrossRef]
- Cristinziano, L.; Modestino, L.; Antonelli, A.; Marone, G.; Simon, H.-U.; Varricchi, G.; Galdiero, M.R. Neutrophil Extracellular Traps in Cancer. Semin. Cancer Biol. 2022, 79, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Rice, C.M.; Davies, L.C.; Subleski, J.J.; Maio, N.; Gonzalez-Cotto, M.; Andrews, C.; Patel, N.L.; Palmieri, E.M.; Weiss, J.M.; Lee, J.-M.; et al. Tumour-Elicited Neutrophils Engage Mitochondrial Metabolism to Circumvent Nutrient Limitations and Maintain Immune Suppression. Nat. Commun. 2018, 9, 5099. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a Therapeutic Target for Common Pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Morosanova, M.A.; Pevzner, I.B.; Zorova, L.D.; Manskikh, V.N.; Pulkova, N.V.; Galkina, S.I.; Skulachev, V.P.; Zorov, D.B. Protective Effect of Mitochondria-Targeted Antioxidants in an Acute Bacterial Infection. Proc. Natl. Acad. Sci. USA 2013, 110, E3100–E3108. [Google Scholar] [CrossRef]
- Zakharova, V.V.; Pletjushkina, O.Y.; Zinovkin, R.A.; Popova, E.N.; Chernyak, B.V. Mitochondria-Targeted Antioxidants and Uncouplers of Oxidative Phosphorylation in Treatment of the Systemic Inflammatory Response Syndrome (SIRS). J. Cell. Physiol. 2017, 232, 904–912. [Google Scholar] [CrossRef]
- Bakeeva, L.E.; Barskov, I.V.; Egorov, M.V.; Isaev, N.K.; Kapelko, V.I.; Kazachenko, A.V.; Kirpatovsky, V.I.; Kozlovsky, S.V.; Lakomkin, V.L.; Levina, S.B.; et al. Mitochondria-Targeted Plastoquinone Derivatives as Tools to Interrupt Execution of the Aging Program. 2. Treatment of Some ROS- and Age-Related Diseases (heart Arrhythmia, Heart Infarctions, Kidney Ischemia, and Stroke). Biochemistry 2008, 73, 1288–1299. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Pevzner, I.B.; Zorova, L.D.; Chernikov, V.P.; Prusov, A.N.; Kireev, I.I.; Silachev, D.N.; Skulachev, V.P.; Zorov, D.B. Mitochondrial Damage and Mitochondria-Targeted Antioxidant Protection in LPS-Induced Acute Kidney Injury. Antioxidants 2019, 8, 176. [Google Scholar] [CrossRef]
- Neroev, V.V.; Archipova, M.M.; Bakeeva, L.E.; Fursova, A.Z.; Grigorian, E.N.; Grishanova, A.Y.; Iomdina, E.N.; Ivashchenko, Z.N.; Katargina, L.A.; Khoroshilova-Maslova, I.P.; et al. Mitochondria-Targeted Plastoquinone Derivatives as Tools to Interrupt Execution of the Aging Program. 4. Age-Related Eye Disease. SkQ1 Returns Vision to Blind Animals. Biochemistry 2008, 73, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Zernii, E.Y.; Gancharova, O.S.; Baksheeva, V.E.; Golovastova, M.O.; Kabanova, E.I.; Savchenko, M.S.; Tiulina, V.V.; Sotnikova, L.F.; Zamyatnin, A.A., Jr.; Philippov, P.P.; et al. Mitochondria-Targeted Antioxidant SkQ1 Prevents Anesthesia-Induced Dry Eye Syndrome. Oxid. Med. Cell. Longev. 2017, 2017, 9281519. [Google Scholar] [CrossRef]
- Brzheskiy, V.V.; Efimova, E.L.; Vorontsova, T.N.; Alekseev, V.N.; Gusarevich, O.G.; Shaidurova, K.N.; Ryabtseva, A.A.; Andryukhina, O.M.; Kamenskikh, T.G.; Sumarokova, E.S.; et al. Results of a Multicenter, Randomized, Double-Masked, Placebo-Controlled Clinical Study of the Efficacy and Safety of Visomitin Eye Drops in Patients with Dry Eye Syndrome. Adv. Ther. 2015, 32, 1263–1279. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P.; Vyssokikh, M.Y.; Chernyak, B.V.; Averina, O.A.; Andreev-Andrievskiy, A.A.; Zinovkin, R.A.; Lyamzaev, K.G.; Marey, M.V.; Egorov, M.V.; Frolova, O.J.; et al. Mitochondrion-Targeted Antioxidant SkQ1 Prevents Rapid Animal Death Caused by Highly Diverse Shocks. Sci. Rep. 2023, 13, 4326. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vorobjeva, N.V.; Chelombitko, M.A.; Sud’ina, G.F.; Zinovkin, R.A.; Chernyak, B.V. Role of Mitochondria in the Regulation of Effector Functions of Granulocytes. Cells 2023, 12, 2210. https://doi.org/10.3390/cells12182210
Vorobjeva NV, Chelombitko MA, Sud’ina GF, Zinovkin RA, Chernyak BV. Role of Mitochondria in the Regulation of Effector Functions of Granulocytes. Cells. 2023; 12(18):2210. https://doi.org/10.3390/cells12182210
Chicago/Turabian StyleVorobjeva, Nina V., Maria A. Chelombitko, Galina F. Sud’ina, Roman A. Zinovkin, and Boris V. Chernyak. 2023. "Role of Mitochondria in the Regulation of Effector Functions of Granulocytes" Cells 12, no. 18: 2210. https://doi.org/10.3390/cells12182210
APA StyleVorobjeva, N. V., Chelombitko, M. A., Sud’ina, G. F., Zinovkin, R. A., & Chernyak, B. V. (2023). Role of Mitochondria in the Regulation of Effector Functions of Granulocytes. Cells, 12(18), 2210. https://doi.org/10.3390/cells12182210