
Modulation and Regulation of Canonical Transient Receptor Potential 3 (TRPC3) Channels


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
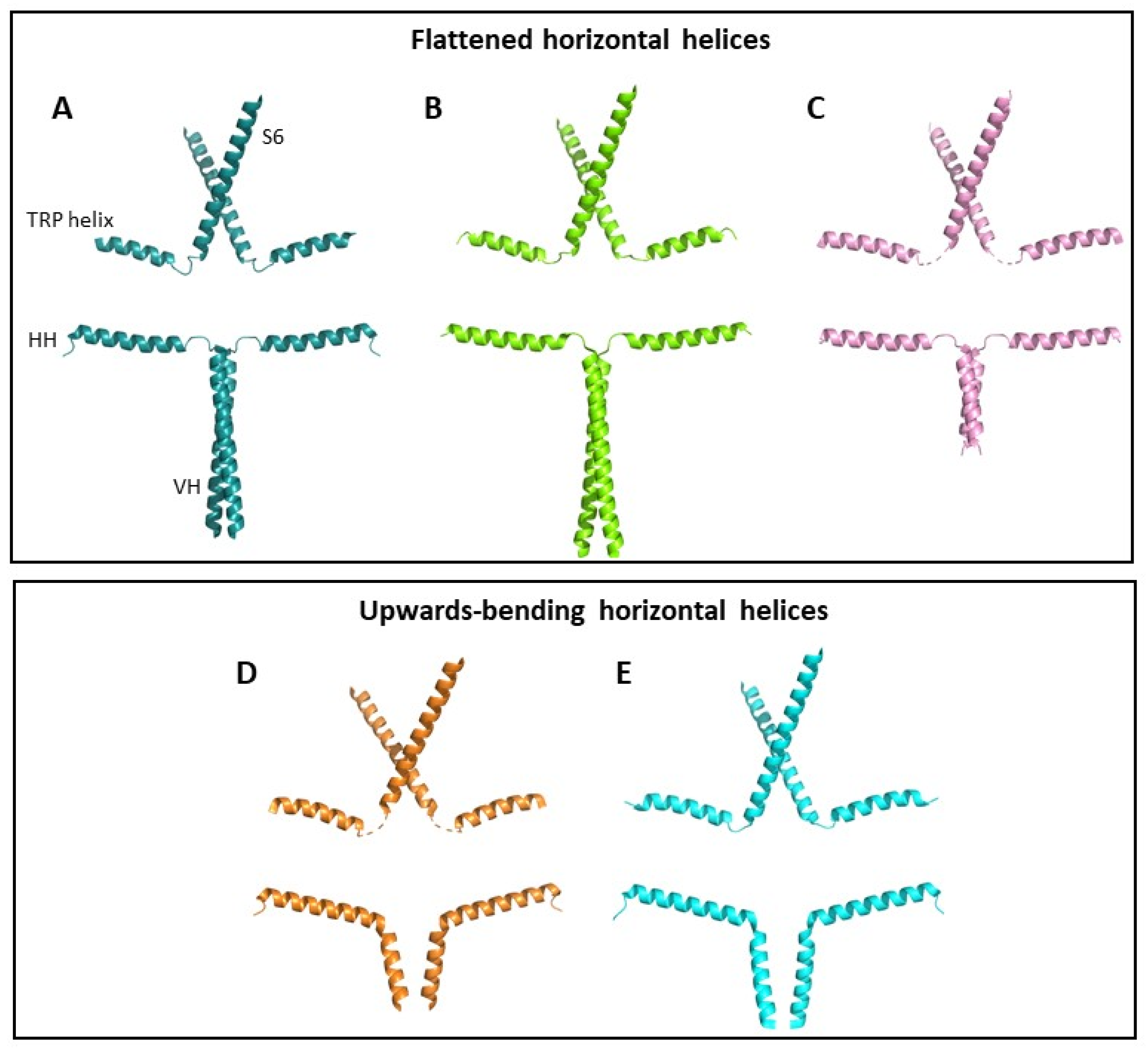
1.1. Structure and Function of TRPC3 Channels

1.2. Isoforms of TRPC3
2. The Role of TRPC3 in Normal Physiology and Human Disease
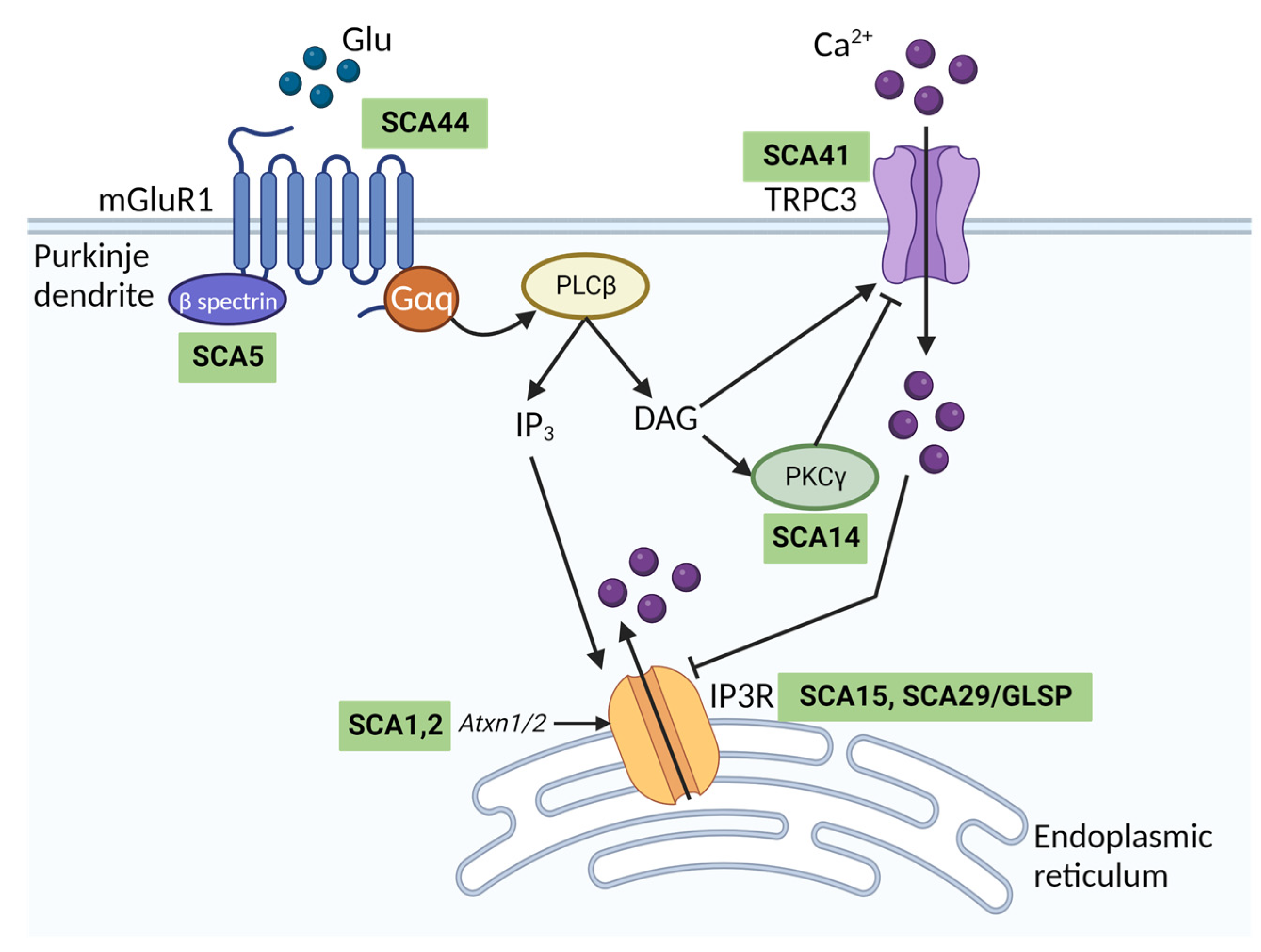
2.1. The Role of TRPC3 in the Nervous System
2.2. The Role of TRPC3 Outside of the Nervous System
3. Regulation and Modulation of TRPC3
3.1. Endogenous Modulation of TRPC3
3.1.1. TRPC3 Modulation by Lipids
3.1.2. Calcium Regulation of TRPC3
3.2. TRPC3 Activators and Their Mode of Action
3.3. TRPC3 Inhibitors and Their Mode of Action
3.3.1. Naturally Occurring TRPC3 Inhibitors
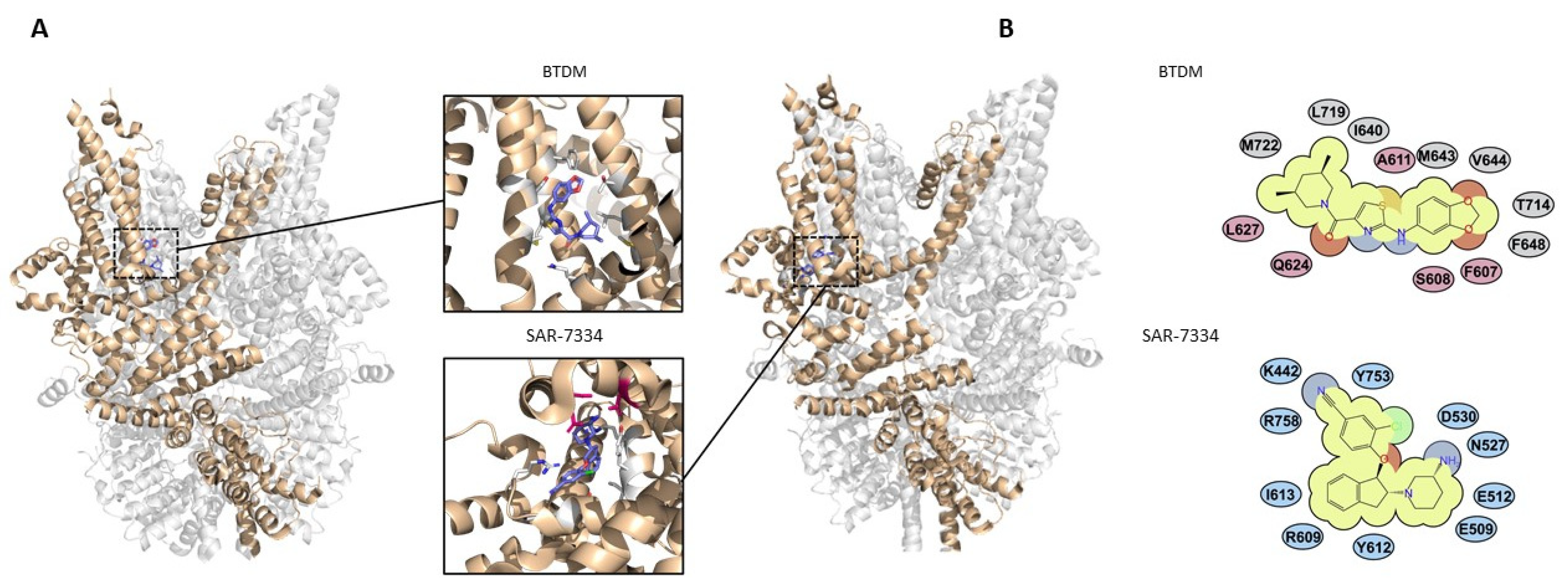
3.3.2. Pyrazole Compounds
3.3.3. 2-Anilino-Thiazole Compounds
3.3.4. Indane Derivatives
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, X.; Sooch, G.; Demaree, I.S.; White, F.A.; Obukhov, A.G. Transient Receptor Potential Canonical (TRPC) Channels: Then and Now. Cells 2020, 9, 1983. [Google Scholar] [CrossRef] [PubMed]
- Riccio, A.; Medhurst, A.D.; Mattei, C.; Kelsell, R.E.; Calver, A.R.; Randall, A.D.; Benham, C.D.; Pangalos, M.N. mRNA distribution analysis of human TRPC family in CNS and peripheral tissues. Brain Res. Mol. Brain Res. 2002, 109, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Zitt, C.; Obukhov, A.G.; Strübing, C.; Zobel, A.; Kalkbrenner, F.; Lückhoff, A.; Schultz, G. Expression of TRPC3 in Chinese hamster ovary cells results in calcium-activated cation currents not related to store depletion. J. Cell Biol. 1997, 138, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Schaefer, M.; Schultz, G.; Gudermann, T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 7461–7466. [Google Scholar] [CrossRef] [PubMed]
- Strübing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J. Biol. Chem. 2003, 278, 39014–39019. [Google Scholar] [CrossRef]
- Strübing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 2001, 29, 645–655. [Google Scholar] [CrossRef]
- Bröker-Lai, J.; Kollewe, A.; Schindeldecker, B.; Pohle, J.; Chi, V.N.; Mathar, I.; Guzman, R.; Schwarz, Y.; Lai, A.; Weißgerber, P.; et al. Heteromeric channels formed by TRPC1, TRPC4 and TRPC5 define hippocampal synaptic transmission and working memory. EMBO J. 2017, 36, 2770–2789. [Google Scholar] [CrossRef]
- Storch, U.; Forst, A.L.; Philipp, M.; Gudermann, T.; Schnitzler, M.M.Y. Transient receptor potential channel 1 (TRPC1) reduces calcium permeability in heteromeric channel complexes. J. Biol. Chem. 2012, 287, 3530–3540. [Google Scholar] [CrossRef]
- Miyagi, K.; Kiyonaka, S.; Yamada, K.; Miki, T.; Mori, E.; Kato, K.; Numata, T.; Sawaguchi, Y.; Numaga, T.; Kimura, T.; et al. A pathogenic C terminus-truncated polycystin-2 mutant enhances receptor-activated Ca2+ entry via association with TRPC3 and TRPC7. J. Biol. Chem. 2009, 284, 34400–34412. [Google Scholar] [CrossRef]
- Ma, X.; Cheng, K.T.; Wong, C.O.; O’Neil, R.G.; Birnbaumer, L.; Ambudkar, I.S.; Yao, X. Heteromeric TRPV4-C1 channels contribute to store-operated Ca2+ entry in vascular endothelial cells. Cell Calcium 2011, 50, 502–509. [Google Scholar] [CrossRef]
- Du, J.; Ma, X.; Shen, B.; Huang, Y.; Birnbaumer, L.; Yao, X. TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. FASEB J. 2014, 28, 4677–4685. [Google Scholar] [CrossRef]
- Beech, D.J.; Xu, S.Z.; McHugh, D.; Flemming, R. TRPC1 store-operated cationic channel subunit. Cell Calcium 2003, 33, 433–440. [Google Scholar] [CrossRef]
- Fan, C.; Choi, W.; Sun, W.; Du, J.; Lü, W. Structure of the human lipid-gated cation channel TRPC3. Elife 2018, 7, e36852. [Google Scholar] [CrossRef]
- Tang, Q.; Guo, W.; Zheng, L.; Wu, J.X.; Liu, M.; Zhou, X.; Zhang, X.; Chen, L. Structure of the receptor-activated human TRPC6 and TRPC3 ion channels. Cell Res. 2018, 28, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Tang, Q.; Wei, M.; Kang, Y.; Wu, J.X.; Chen, L. Structural mechanism of human TRPC3 and TRPC6 channel regulation by their intracellular calcium-binding sites. Neuron 2022, 110, 1023–1035.e1025. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Valdez, F.; Azumaya, C.M.; Romero, L.O.; Nakagawa, T.; Cordero-Morales, J.F. Structure-function analyses of the ion channel TRPC3 reveal that its cytoplasmic domain allosterically modulates channel gating. J. Biol. Chem. 2018, 293, 16102–16114. [Google Scholar] [CrossRef]
- Owsianik, G.; Talavera, K.; Voets, T.; Nilius, B. Permeation and selectivity of TRP channels. Annu. Rev. Physiol. 2006, 68, 685–717. [Google Scholar] [CrossRef]
- Poteser, M.; Schleifer, H.; Lichtenegger, M.; Schernthaner, M.; Stockner, T.; Kappe, C.O.; Glasnov, T.N.; Romanin, C.; Groschner, K. PKC-dependent coupling of calcium permeation through transient receptor potential canonical 3 (TRPC3) to calcineurin signaling in HL-1 myocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 10556–10561. [Google Scholar] [CrossRef]
- Lichtenegger, M.; Stockner, T.; Poteser, M.; Schleifer, H.; Platzer, D.; Romanin, C.; Groschner, K. A novel homology model of TRPC3 reveals allosteric coupling between gate and selectivity filter. Cell Calcium 2013, 54, 175–185. [Google Scholar] [CrossRef]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Erkan-Candag, H.; Krivic, D.; Gsell, M.A.F.; Aleksanyan, M.; Stockner, T.; Dimova, R.; Tiapko, O.; Groschner, K. Characterization of DAG Binding to TRPC Channels by Target-Dependent. Biomolecules 2022, 12, 799. [Google Scholar] [CrossRef] [PubMed]
- Erkan-Candag, H.; Clarke, A.; Tiapko, O.; Gsell, M.A.; Stockner, T.; Groschner, K. Diacylglycerols interact with the L2 lipidation site in TRPC3 to induce a sensitized channel state. EMBO Rep. 2022, 23, e54276. [Google Scholar] [CrossRef]
- Lichtenegger, M.; Tiapko, O.; Svobodova, B.; Stockner, T.; Glasnov, T.N.; Schreibmayer, W.; Platzer, D.; de la Cruz, G.G.; Krenn, S.; Schober, R.; et al. An optically controlled probe identifies lipid-gating fenestrations within the TRPC3 channel. Nat. Chem. Biol. 2018, 14, 396–404. [Google Scholar] [CrossRef]
- Svobodova, B.; Lichtenegger, M.; Platzer, D.; Di Giuro, C.M.L.; de la Cruz, G.G.; Glasnov, T.; Schreibmayer, W.; Groschner, K. A single point mutation in the TRPC3 lipid-recognition window generates supersensitivity to benzimidazole channel activators. Cell Calcium 2019, 79, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, A.; Mederos y Schnitzler, M.; Emmel, J.; Kalwa, H.; Hofmann, T.; Gudermann, T. N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J. Biol. Chem. 2003, 278, 47842–47852. [Google Scholar] [CrossRef] [PubMed]
- Dryer, S.E.; Kim, E.Y. Permeation and Rectification in Canonical Transient Receptor Potential-6 (TRPC6) Channels. Front. Physiol. 2018, 9, 1055. [Google Scholar] [CrossRef]
- Kim, Y.; Wong, A.C.; Power, J.M.; Tadros, S.F.; Klugmann, M.; Moorhouse, A.J.; Bertrand, P.P.; Housley, G.D. Alternative splicing of the TRPC3 ion channel calmodulin/IP3 receptor-binding domain in the hindbrain enhances cation flux. J. Neurosci. 2012, 32, 11414–11423. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, J.; Tikunova, S.; Johnson, J.D.; Chen, Z.; Qin, N.; Dietrich, A.; Stefani, E.; Birnbaumer, L.; Zhu, M.X. Activation of Trp3 by inositol 1,4,5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain. Proc. Natl. Acad. Sci. USA 2001, 98, 3168–3173. [Google Scholar] [CrossRef]
- Tang, J.; Lin, Y.; Zhang, Z.; Tikunova, S.; Birnbaumer, L.; Zhu, M.X. Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of trp channels. J. Biol. Chem. 2001, 276, 21303–21310. [Google Scholar] [CrossRef]
- Wedel, B.J.; Vazquez, G.; McKay, R.R.; Bird, G.S.J.; Putney, J.W. A calmodulin/inositol 1,4,5-trisphosphate (IP3) receptor-binding region targets TRPC3 to the plasma membrane in a calmodulin/IP3 receptor-independent process. J. Biol. Chem. 2003, 278, 25758–25765. [Google Scholar] [CrossRef]
- Cederholm, J.M.E.; Kim, Y.; von Jonquieres, G.; Housley, G.D. Human Brain Region-Specific Alternative Splicing of TRPC3, the Type 3 Canonical Transient Receptor Potential Non-Selective Cation Channel. Cerebellum 2019, 18, 536–543. [Google Scholar] [CrossRef]
- Tiapko, O.; Groschner, K. TRPC3 as a Target of Novel Therapeutic Interventions. Cells 2018, 7, 83. [Google Scholar] [CrossRef]
- Numaga-Tomita, T.; Oda, S.; Shimauchi, T.; Nishimura, A.; Mangmool, S.; Nishida, M. TRPC3 Channels in Cardiac Fibrosis. Front. Cardiovasc. Med. 2017, 4, 56. [Google Scholar] [CrossRef] [PubMed]
- Englisch, C.N.; Paulsen, F.; Tschernig, T. TRPC Channels in the Physiology and Pathophysiology of the Renal Tubular System: What Do We Know? Int. J. Mol. Sci. 2022, 24, 181. [Google Scholar] [CrossRef]
- Khayyat, N.H.; Tomilin, V.N.; Zaika, O.; Pochynyuk, O. Polymodal roles of TRPC3 channel in the kidney. Channels 2020, 14, 257–267. [Google Scholar] [CrossRef]
- Hartmann, J.; Dragicevic, E.; Adelsberger, H.; Henning, H.A.; Sumser, M.; Abramowitz, J.; Blum, R.; Dietrich, A.; Freichel, M.; Flockerzi, V.; et al. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 2008, 59, 392–398. [Google Scholar] [CrossRef]
- Huang, W.C.; Young, J.S.; Glitsch, M.D. Changes in TRPC channel expression during postnatal development of cerebellar neurons. Cell Calcium 2007, 42, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sekerková, G.; Kim, J.A.; Nigro, M.J.; Becker, E.B.; Hartmann, J.; Birnbaumer, L.; Mugnaini, E.; Martina, M. Early onset of ataxia in moonwalker mice is accompanied by complete ablation of type II unipolar brush cells and Purkinje cell dysfunction. J. Neurosci. 2013, 33, 19689–19694. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J. TRPC3 channel underlies cerebellar long-term depression. Cerebellum 2013, 12, 334–337. [Google Scholar] [CrossRef]
- Curcic, S.; Erkan-Candag, H.; Pilic, J.; Malli, R.; Wiedner, P.; Tiapko, O.; Groschner, K. TRPC3 governs the spatiotemporal organization of cellular Ca. Cell Calcium 2022, 108, 102670. [Google Scholar] [CrossRef]
- Brown, S.A.; Loew, L.M. Integration of modeling with experimental and clinical findings synthesizes and refines the central role of inositol 1,4,5-trisphosphate receptor 1 in spinocerebellar ataxia. Front. Neurosci. 2014, 8, 453. [Google Scholar] [CrossRef]
- Wu, B.; Blot, F.G.; Wong, A.B.; Osório, C.; Adolfs, Y.; Pasterkamp, R.J.; Hartmann, J.; Becker, E.B.; Boele, H.J.; De Zeeuw, C.I.; et al. TRPC3 is a major contributor to functional heterogeneity of cerebellar Purkinje cells. Elife 2019, 8, e45590. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lin, Z.; Voges, K.; Ju, C.; Gao, Z.; Bosman, L.W.; Ruigrok, T.J.; Hoebeek, F.E.; De Zeeuw, C.I.; Schonewille, M. Cerebellar modules operate at different frequencies. Elife 2014, 3, e02536. [Google Scholar] [CrossRef]
- Fogel, B.L.; Hanson, S.M.; Becker, E.B. Do mutations in the murine ataxia gene TRPC3 cause cerebellar ataxia in humans? Mov. Disord. 2015, 30, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.B.; Oliver, P.L.; Glitsch, M.D.; Banks, G.T.; Achilli, F.; Hardy, A.; Nolan, P.M.; Fisher, E.M.; Davies, K.E. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc. Natl. Acad. Sci. USA 2009, 106, 6706–6711. [Google Scholar] [CrossRef]
- Ibrahim, M.F.; Becker, E.B.E. Moonwalker Mouse. In Essentials of Cerebellum and Cerebellar Disorders: A Primer for Graduate Students; Gruol, D.L., Koibuchi, N., Manto, M., Molinari, M., Schmahmann, J.D., Shen, Y., Eds.; Springer International Publishing: Cham, Switzerland, 2023; pp. 441–447. [Google Scholar]
- Bai, Y.; Yu, X.; Chen, H.; Horne, D.; White, R.; Wu, X.; Lee, P.; Gu, Y.; Ghimire-Rijal, S.; Lin, D.C.; et al. Structural basis for pharmacological modulation of the TRPC6 channel. Elife 2020, 9, e53311. [Google Scholar] [CrossRef]
- Power, E.M.; Morales, A.; Empson, R.M. Prolonged Type 1 Metabotropic Glutamate Receptor Dependent Synaptic Signaling Contributes to Spino-Cerebellar Ataxia Type 1. J. Neurosci. 2016, 36, 4910–4916. [Google Scholar] [CrossRef]
- Meera, P.; Pulst, S.; Otis, T. A positive feedback loop linking enhanced mGluR function and basal calcium in spinocerebellar ataxia type 2. Elife 2017, 6, e26377. [Google Scholar] [CrossRef]
- Watson, L.M.; Bamber, E.; Schnekenberg, R.P.; Williams, J.; Bettencourt, C.; Lickiss, J.; Jayawant, S.; Fawcett, K.; Clokie, S.; Wallis, Y.; et al. Dominant Mutations in GRM1 Cause Spinocerebellar Ataxia Type 44. Am. J. Hum. Genet. 2017, 101, 866. [Google Scholar] [CrossRef]
- Becker, E.B.E. From Mice to Men: TRPC3 in Cerebellar Ataxia. Cerebellum 2017, 16, 877–879. [Google Scholar] [CrossRef] [PubMed]
- Um, K.B.; Hahn, S.; Kim, S.W.; Lee, Y.J.; Birnbaumer, L.; Kim, H.J.; Park, M.K. TRPC3 and NALCN channels drive pacemaking in substantia nigra dopaminergic neurons. Elife 2021, 10, e70920. [Google Scholar] [CrossRef]
- Zhou, F.W.; Matta, S.G.; Zhou, F.M. Constitutively active TRPC3 channels regulate basal ganglia output neurons. J. Neurosci. 2008, 28, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Phelan, K.D.; Shwe, U.T.; Cozart, M.A.; Wu, H.; Mock, M.M.; Abramowitz, J.; Birnbaumer, L.; Zheng, F. TRPC3 channels play a critical role in the theta component of pilocarpine-induced status epilepticus in mice. Epilepsia 2017, 58, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Nagib, M.M.; Zhang, S.; Yasmen, N.; Li, L.; Hou, R.; Yu, Y.; Boda, V.K.; Wu, Z.; Li, W.; Jiang, J. Inhibition of TRPC3 channels by a novel pyrazole compound confers antiseizure effects. Epilepsia 2022, 63, 1003–1015. [Google Scholar] [CrossRef]
- Zeng, C.; Zhou, P.; Jiang, T.; Yuan, C.; Ma, Y.; Feng, L.; Liu, R.; Tang, W.; Long, X.; Xiao, B.; et al. Upregulation and Diverse Roles of TRPC3 and TRPC6 in Synaptic Reorganization of the Mossy Fiber Pathway in Temporal Lobe Epilepsy. Mol. Neurobiol. 2015, 52, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Limjunyawong, N.; Narang, C.; Jamaldeen, H.; Yu, S.; Patiram, S.; Nie, H.; Caterina, M.J.; Dong, X.; Qu, L. Sensory neuron-expressed TRPC3 mediates acute and chronic itch. Pain 2023, 164, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Eder, P.; Probst, D.; Rosker, C.; Poteser, M.; Wolinski, H.; Kohlwein, S.D.; Romanin, C.; Groschner, K. Phospholipase C-dependent control of cardiac calcium homeostasis involves a TRPC3-NCX1 signaling complex. Cardiovasc. Res. 2007, 73, 111–119. [Google Scholar] [CrossRef]
- Rosker, C.; Graziani, A.; Lukas, M.; Eder, P.; Zhu, M.X.; Romanin, C.; Groschner, K. Ca2+ signaling by TRPC3 involves Na+ entry and local coupling to the Na+/Ca2+ exchanger. J. Biol. Chem. 2004, 279, 13696–13704. [Google Scholar] [CrossRef]
- Doleschal, B.; Primessnig, U.; Wölkart, G.; Wolf, S.; Schernthaner, M.; Lichtenegger, M.; Glasnov, T.N.; Kappe, C.O.; Mayer, B.; Antoons, G.; et al. TRPC3 contributes to regulation of cardiac contractility and arrhythmogenesis by dynamic interaction with NCX1. Cardiovasc. Res. 2015, 106, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.; Rainer, P.P.; Shalkey Hahn, V.; Lee, D.I.; Jo, S.H.; Andersen, A.; Liu, T.; Xu, X.; Willette, R.N.; Lepore, J.J.; et al. Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2014, 111, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, L.; Trebak, M.; Putney, J.W. Complex regulation of the TRPC3, 6 and 7 channel subfamily by diacylglycerol and phosphatidylinositol-4,5-bisphosphate. Cell Calcium 2008, 43, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lin, W.Y.; Leibow, S.R.; Morateck, A.J.; Ahuja, M.; Muallem, S. TRPC3 channel gating by lipids requires localization at the ER/PM junctions defined by STIM1. J. Cell Biol. 2022, 221, e202107120. [Google Scholar] [CrossRef]
- Graziani, A.; Rosker, C.; Kohlwein, S.D.; Zhu, M.X.; Romanin, C.; Sattler, W.; Groschner, K.; Poteser, M. Cellular cholesterol controls TRPC3 function: Evidence from a novel dominant-negative knockdown strategy. Biochem. J. 2006, 396, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Mori, E.; Mori, Y.; Mori, M.; Li, J.; Ito, Y.; Inoue, R. Multiple regulation by calcium of murine homologues of transient receptor potential proteins TRPC6 and TRPC7 expressed in HEK293 cells. J. Physiol. 2004, 561, 415–432. [Google Scholar] [CrossRef] [PubMed]
- Boulay, G. Ca2+-calmodulin regulates receptor-operated Ca2+ entry activity of TRPC6 in HEK-293 cells. Cell Calcium 2002, 32, 201–207. [Google Scholar] [CrossRef]
- Duan, J.; Li, J.; Zeng, B.; Chen, G.L.; Peng, X.; Zhang, Y.; Wang, J.; Clapham, D.E.; Li, Z.; Zhang, J. Structure of the mouse TRPC4 ion channel. Nat. Commun. 2018, 9, 3102. [Google Scholar] [CrossRef]
- Vinayagam, D.; Quentin, D.; Yu-Strzelczyk, J.; Sitsel, O.; Merino, F.; Stabrin, M.; Hofnagel, O.; Yu, M.; Ledeboer, M.W.; Nagel, G.; et al. Structural basis of TRPC4 regulation by calmodulin and pharmacological agents. Elife 2020, 9, e60603. [Google Scholar] [CrossRef]
- Song, K.; Wei, M.; Guo, W.; Quan, L.; Kang, Y.; Wu, J.X.; Chen, L. Structural basis for human TRPC5 channel inhibition by two distinct inhibitors. Elife 2021, 10, e63429. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tóth, B.; Szollosi, A.; Chen, J.; Csanády, L. Structure of a TRPM2 channel in complex with Ca. Elife 2018, 7, e36409. [Google Scholar] [CrossRef]
- Wijerathne, T.; Lin, W.Y.; Cooray, A.; Muallem, S.; Lee, K.P. Hydrophobic interactions within the C terminus pole helices tunnel regulate calcium-dependent inactivation of TRPC3 in a calmodulin-dependent manner. Cell Calcium 2023, 109, 102684. [Google Scholar] [CrossRef]
- Tiapko, O.; Shrestha, N.; Lindinger, S.; de la Cruz, G.G.; Graziani, A.; Klec, C.; Butorac, C.; Graier, W.F.; Kubista, H.; Freichel, M.; et al. Lipid-independent control of endothelial and neuronal TRPC3 channels by light. Chem. Sci. 2019, 10, 2837–2842. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.L.; Li, X.H.; Wang, J.; Ma, X.F.; Zhou, B.Y.; Jiao, Y.F.; Wang, W.H.; Cao, P.; Zhu, M.X.; Li, P.W.; et al. GSK1702934A and M085 directly activate TRPC6 via a mechanism of stimulating the extracellular cavity formed by the pore helix and transmembrane helix S6. J. Biol. Chem. 2021, 297, 101125. [Google Scholar] [CrossRef]
- Urban, N.; Schaefer, M. Direct Activation of TRPC3 Channels by the Antimalarial Agent Artemisinin. Cells 2020, 9, 202. [Google Scholar] [CrossRef]
- Liu, C.; Reese, R.; Vu, S.; Rougé, L.; Shields, S.D.; Kakiuchi-Kiyota, S.; Chen, H.; Johnson, K.; Shi, Y.P.; Chernov-Rogan, T.; et al. A Non-covalent Ligand Reveals Biased Agonism of the TRPA1 Ion Channel. Neuron 2021, 109, 273–284.e4. [Google Scholar] [CrossRef]
- Qu, C.; Ding, M.; Zhu, Y.; Lu, Y.; Du, J.; Miller, M.; Tian, J.; Zhu, J.; Xu, J.; Wen, M.; et al. Pyrazolopyrimidines as Potent Stimulators for Transient Receptor Potential Canonical 3/6/7 Channels. J. Med. Chem. 2017, 60, 4680–4692. [Google Scholar] [CrossRef] [PubMed]
- Bon, R.S.; Beech, D.J. In pursuit of small molecule chemistry for calcium-permeable non-selective TRPC channels—Mirage or pot of gold? Br. J. Pharmacol. 2013, 170, 459–474. [Google Scholar] [CrossRef]
- Hu, H.Z.; Gu, Q.; Wang, C.; Colton, C.K.; Tang, J.; Kinoshita-Kawada, M.; Lee, L.Y.; Wood, J.D.; Zhu, M.X. 2-aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J. Biol. Chem. 2004, 279, 35741–35748. [Google Scholar] [CrossRef]
- Maruyama, T.; Kanaji, T.; Nakade, S.; Kanno, T.; Mikoshiba, K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J. Biochem. 1997, 122, 498–505. [Google Scholar] [CrossRef]
- Lievremont, J.P.; Bird, G.S.; Putney, J.W. Mechanism of inhibition of TRPC cation channels by 2-aminoethoxydiphenylborane. Mol. Pharmacol. 2005, 68, 758–762. [Google Scholar] [CrossRef]
- Liu, B.; Freyer, A.M.; Hall, I.P. Bradykinin activates calcium-dependent potassium channels in cultured human airway smooth muscle cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2007, 292, L898–L907. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Jiang, M.; Birnbaumer, L. Receptor-activated Ca2+ influx via human Trp3 stably expressed in human embryonic kidney (HEK)293 cells. Evidence for a non-capacitative Ca2+ entry. J. Biol. Chem. 1998, 273, 133–142. [Google Scholar] [CrossRef]
- Singh, A.; Hildebrand, M.E.; Garcia, E.; Snutch, T.P. The transient receptor potential channel antagonist SKF96365 is a potent blocker of low-voltage-activated T-type calcium channels. Br. J. Pharmacol. 2010, 160, 1464–1475. [Google Scholar] [CrossRef]
- Rychkov, G.; Barritt, G.J. TRPC1 Ca2+-permeable channels in animal cells. Handb. Exp. Pharmacol. 2007, 179, 23–52. [Google Scholar] [CrossRef]
- Miehe, S.; Crause, P.; Schmidt, T.; Löhn, M.; Kleemann, H.W.; Licher, T.; Dittrich, W.; Rütten, H.; Strübing, C. Inhibition of diacylglycerol-sensitive TRPC channels by synthetic and natural steroids. PLoS ONE 2012, 7, e35393. [Google Scholar] [CrossRef] [PubMed]
- Majeed, Y.; Amer, M.S.; Agarwal, A.K.; McKeown, L.; Porter, K.E.; O’Regan, D.J.; Naylor, J.; Fishwick, C.W.; Muraki, K.; Beech, D.J. Stereo-selective inhibition of transient receptor potential TRPC5 cation channels by neuroactive steroids. Br. J. Pharmacol. 2011, 162, 1509–1520. [Google Scholar] [CrossRef]
- Urban, N.; Wang, L.; Kwiek, S.; Rademann, J.; Kuebler, W.M.; Schaefer, M. Identification and Validation of Larixyl Acetate as a Potent TRPC6 Inhibitor. Mol. Pharmacol. 2016, 89, 197–213. [Google Scholar] [CrossRef]
- Kiyonaka, S.; Kato, K.; Nishida, M.; Mio, K.; Numaga, T.; Sawaguchi, Y.; Yoshida, T.; Wakamori, M.; Mori, E.; Numata, T.; et al. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc. Natl. Acad. Sci. USA 2009, 106, 5400–5405. [Google Scholar] [CrossRef]
- Schleifer, H.; Doleschal, B.; Lichtenegger, M.; Oppenrieder, R.; Derler, I.; Frischauf, I.; Glasnov, T.N.; Kappe, C.O.; Romanin, C.; Groschner, K. Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated Ca2+ entry pathways. Br. J. Pharmacol. 2012, 167, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Romero, L.O.; Deng, S.; Wang, J.; Li, Y.; Yang, L.; Hamilton, D.J.; Miller, D.D.; Liao, F.F.; Cordero-Morales, J.F.; et al. Discovery of a Highly Selective and Potent TRPC3 Inhibitor with High Metabolic Stability and Low Toxicity. ACS Med. Chem. Lett. 2021, 12, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Washburn, D.G.; Holt, D.A.; Dodson, J.; McAtee, J.J.; Terrell, L.R.; Barton, L.; Manns, S.; Waszkiewicz, A.; Pritchard, C.; Gillie, D.J.; et al. The discovery of potent blockers of the canonical transient receptor channels, TRPC3 and TRPC6, based on an anilino-thiazole pharmacophore. Bioorg. Med. Chem. Lett. 2013, 23, 4979–4984. [Google Scholar] [CrossRef]
- Gao, Y.; Cao, E.; Julius, D.; Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 2016, 534, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.E.T.; Lodowski, D.T.; Huynh, K.W.; Yazici, A.; Del Rosario, J.; Kapoor, A.; Basak, S.; Samanta, A.; Han, X.; Chakrapani, S.; et al. Structural basis of TRPV5 channel inhibition by econazole revealed by cryo-EM. Nat. Struct. Mol. Biol. 2018, 25, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Follmann, M.; Hessler, G.; Kleemann, H.W.; Hachtel, S.; Fuchs, B.; Weissmann, N.; Linz, W.; Schmidt, T.; Löhn, M.; et al. Discovery and pharmacological characterization of a novel potent inhibitor of diacylglycerol-sensitive TRPC cation channels. Br. J. Pharmacol. 2015, 172, 3650–3660. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cole, B.A.; Becker, E.B.E. Modulation and Regulation of Canonical Transient Receptor Potential 3 (TRPC3) Channels. Cells 2023, 12, 2215. https://doi.org/10.3390/cells12182215
Cole BA, Becker EBE. Modulation and Regulation of Canonical Transient Receptor Potential 3 (TRPC3) Channels. Cells. 2023; 12(18):2215. https://doi.org/10.3390/cells12182215
Chicago/Turabian StyleCole, Bethan A., and Esther B. E. Becker. 2023. "Modulation and Regulation of Canonical Transient Receptor Potential 3 (TRPC3) Channels" Cells 12, no. 18: 2215. https://doi.org/10.3390/cells12182215